Cytotoxic Scalarane Sesterterpenes from the Sponge Hyrtios erectus
by
 , ,
, ,
Oh-Seok Kwon
1, 
Donghwa Kim
1,
Chang-Kwon Kim
1,
Jeongyoon Sun
2,
Chung J. Sim
3,
Dong-Chan Oh
1,
Sang Kook Lee
1,
Ki-Bong Oh
2,* and
Jongheon Shin
1,* 1
Natural Products Research Institute, College of Pharmacy, Seoul National University, San 56-1, Sillim, Gwanak, Seoul 151-742, Korea
2
Department of Agricultural Biotechnology, College of Agriculture and Life Science, Seoul National University, San 56-1, Sillim, Gwanak, Seoul 151-921, Korea
3
Department of Biological Sciences, College of Life Science and Nano Technology, Hannam University, 461-6 Jeonmin, Yuseong, Daejeon 305-811, Korea
*
Authors to whom correspondence should be addressed.
Mar. Drugs 2020, 18(5), 253; https://doi.org/10.3390/md18050253
Submission received: 7 April 2020
/
Revised: 8 May 2020
/
Accepted: 11 May 2020
/
Published: 13 May 2020
(This article belongs to the Special Issue Terpenoids from Marine Organisms II)
Abstract
:Twelve new sesterterpenes along with eight known sesterterpenes were isolated from the marine sponge Hyrtios erectus collected off the coast of Chuuk Island, the Federated State of Micronesia. Based upon a combination of spectroscopic and computational analyses, these compounds were determined to be eight glycine-bearing scalaranes (1–8), a 3-keto scalarane (9), two oxidized-furan-bearing scalaranes (10 and 11), and a salmahyrtisane (12). Several of these compounds exhibited weak antiproliferation against diverse cancer cell lines as well as moderate anti-angiogenesis activities. The antiproliferative activity of new compound 4 was found to be associated with G0/G1 arrest in the cell cycle.
1. Introduction
Marine scalaranes are a well-known class of sesterterpenes that are exclusively distributed in sponges and their predator nudibranch mollusks [1,2]. This carbon skeleton is composed of a 6/6/6/6-tetracyclic fused ring system. Among the wide structural variations in both the ring systems and the substituents, the terminal isoprene unit typically forms a cyclopentane or an equivalent structure, thus establishing a 6/6/6/6/5-pentacyclic system. Other noticeable variations are nor-, homo- and bishomo-scalaranes, which are generated by either removal or incorporation of one or two methyl groups on the terminal framework [3]. Since the first members of this family were identified from Cacospongia scalaris in the early 1970s [4,5], scalarane sesterterpenes have been the most frequently encountered sesterterpenes in sponges, and particularly in the order Dictyoceratida [6], and several hundred unique scalarane sesterterpenes have been reported to date [7]. These compounds exhibit diverse bioactivities such as cytotoxic, antimicrobial, anti-inflammatory, anti-HIV, anti-tubercular, and anti-feedant activities [8,9,10]. Overall, scalaranes are widely recognized as a representative class of terpenes from sponges [1,2,8,9,10]. Despite their immense structural diversity, however, the incorporation of heteroatoms is rather rare, and only less than 13 nitrogeneous scalaranes have been reported to date [11,12,13,14,15,16,17,18].
The genus Hyrtios (Family Thorectidae) is a chemically interesting group of Dictyoceratida sponges. Widely distributed in tropical oceans, these animals are the prolific sources of structurally unique and biologically active compounds. As comprehensively covered in a recent review, approximately 150 compounds have been isolated from Hyrtios sponges [19]. The majority of Hyrtios natural products are sesquiterpenes, sesterterpenes, meroterpenes, and indole- and β-carboline -bearing alkaloids [20]. Several of these compounds, in particular, those from the extensively studied H. erectus, exhibit various bioactivities, including broad spectrum of cytotoxic and antimicrobial activities [19].
During the course of our search for bioactive compounds from marine sponges, we encountered specimens of the black encrusting Hyrtios erectus off the coast of Chuuk Island, the Federated State of Micronesia. The intriguing LC/ESI-MS profile and significant brine-shrimp lethality (LC50 84 ppm) of the crude extract were indicative of the presence of bioactive compounds, prompting an extensive chemical investigation. Here, we report the isolation of twelve new sesterterpenes along with eight known compounds. Based upon the results of a combination of spectroscopic and computational analyses, the new compounds were determined to be eight glycine-bearing scalaranes (1–8), one 3-keto scalarane (9), two oxidized furan-containing scalaranes (10 and 11), and a salmahyrtisane (12), thus further contributing to the chemical diversity of Hyrtios sponges. These were designated hyrtioscalarins A-H (1–8), 12-deacetyl-3-oxoscalarin (9), 17(R),18(S)-dihydroxy-19(R),20(S)-dimethoxysesterstatin 5 (10), 17(R),18(S)-dihydroxy-19(R),20(R) -dimethoxysesterstatin 5 (11), and salmahyrtisol B (12), respectively (Chart 1). Several of these compounds exhibited weak antiproliferation against various human cancer cell lines as well as moderate anti-angiogenesis activities. The antiproliferative effect of new compound 4 was found to be associated with G0/G1 arrest in the cell cycle.
2. Results and Discussion
2.1. Structure Elucidation
The molecular formula of compound 1 was deduced to be C27H39NO8, corresponding to 9 degrees of unsaturation, by HRFABMS analysis ([M-H2O-H]− m/z 486.2491, calcd for C27H36NO7, 486.2492). The 13C NMR data of this compound showed signals indicative of four carbonyl carbons (δC 204.2, 177.8, 170.4, and 168.7), two oxygenated and non-protonated carbons (δC 73.3 and 68.7) and one oxymethine carbon (δC 74.7) (Table 1). The remaining 20 carbons were all aliphatic (four non-protonated, three methine, eight methylene, and five methyl carbons). Therefore, 1 was thought to be a pentacyclic compound. The 1H NMR spectra also showed five singlet methyl signals, revealing a terpene or related structure. In conjunction with the mass data and inherent degrees of unsaturation, our preliminary interpretation of the 1-D NMR data suggested that 1 was a highly oxygenated pentacyclic sesterterpene with a nitrogen-containing functionality.
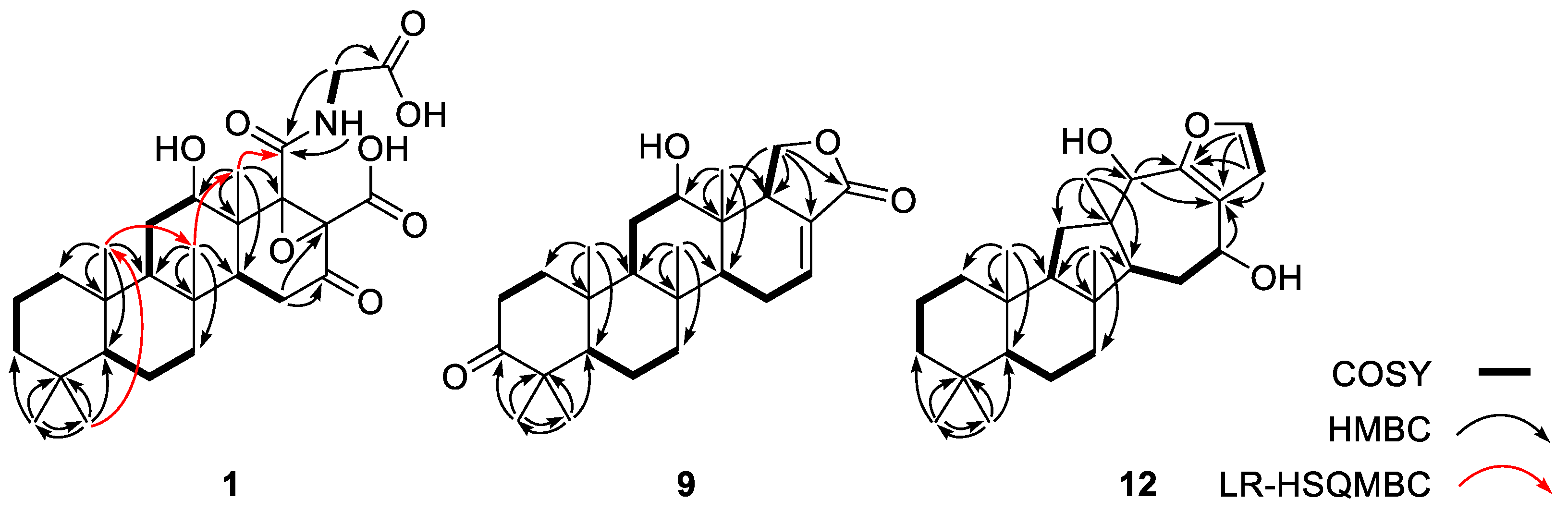
The planar structure of 1 was determined by combined 2-D NMR analyses (Figure 1). After the initial matching of the protons with their bearing carbons by the HSQC data, three aliphatic proton spin systems were deduced from the 1H-1H COSY data. These were consisted of three methylenes (C-1-C-3), one methine and two methylenes (C-5-C-7), and two methines and one methylene (C-9, C-11 and C-12), respectively. Subsequently, these linear systems were assembled by using the HMBC correlations with bridgehead singlet methyl protons (H3-21-H3-25): H3-21/C-3, C-4 and C-5, H3-22/C-3, C-4 and C-5, H3-23/C-1,C-5, C-9, and C-10, H3-24/C-7, C-8, C-9, and C-14, and H3-25/ C-12, C-13 and C-14. Then these HMBC-based assemblies were confirmed by the LR-HSQMBC correlations between the methyl groups: H3-22/C-23, H3-23/C-24 and H3-24/C-25. Overall 1 was found to possess rings A-C as a 6/6/6 system frequently seen in scalaranes [1,3].
Based on the COSY data, the C-14 methine group was directly connected to a methylene (δC/δH 35.4/2.35 and 2.29, C-15) that showed no further proton–proton couplings (Figure 1). The HMBC cross peaks of these protons placed a ketone (δC 204.2, C-16) and a non-protonated carbon (δC 68.7, C-17) at the neighboring positions: H-14/C-16 and H2-15/C-16 and C-17. Similarly, another non-protonated carbon (δC 73.3, C-18) was placed based on its HMBC cross peaks to previously assigned protons: H-12/C-18 and H3-25/C-18. The assignments of C-17 and C-18 were supported by the LR-HSQMBC data [21], a variant of an HMBC experiment, thus constructing a cyclohexanone (ring D): H-14/C-18 and H2-15/C-17 (Supporting Information Figure S8). The chemical shifts of these non-protonated carbons at δC 73.3 and 68.7 indicated not only the attachments of oxygen but also the presence of an epoxide moiety involving these carbons, accounting for the last degree of unsaturation required by the mass data.
The remaining NMR signals were those of an exchangeable NH (δH 7.88), a methylene (δC/δH 43.9/4.15 and 3.33) and three carbonyl carbons (δC 177.8, 170.4 and 168.7). The NH and methylene were directly connected to each other based on their vicinal coupling constants (J = 7.4, 3.2 Hz) and COSY data. This spin system was expanded to an N-carboglycine moiety by the HMBC cross peaks with the carbonyl carbons at δC 177.8 and 168.7. A crucial long-range correlation of the latter carbon with H3-25 by LR-HSQMBC data linked the N-carboglycine moiety to C-18, assigning the carbonyl carbons at C-2’ and C-19, respectively (Figure 1). Although no NMR correlations were observed to the carbonyl carbon at δC 170.4, this carbon (C-20) must be attached at C-18, the only open position in the main framework. Of the eight oxygens indicated by the mass data of 1, the four carbonyl groups at C-16, C-19, C-20, and C-2’ and the epoxide at C-17-C-18 only accounted for five. Therefore, the three remaining oxygens must be OH groups at C-12, C-19 and C-20, forming a hydroxyl moiety and two carboxylic acids. Thus, the planar structure of 1 was determined to be a glycine- and epoxide-bearing scalarane sesterterpene.
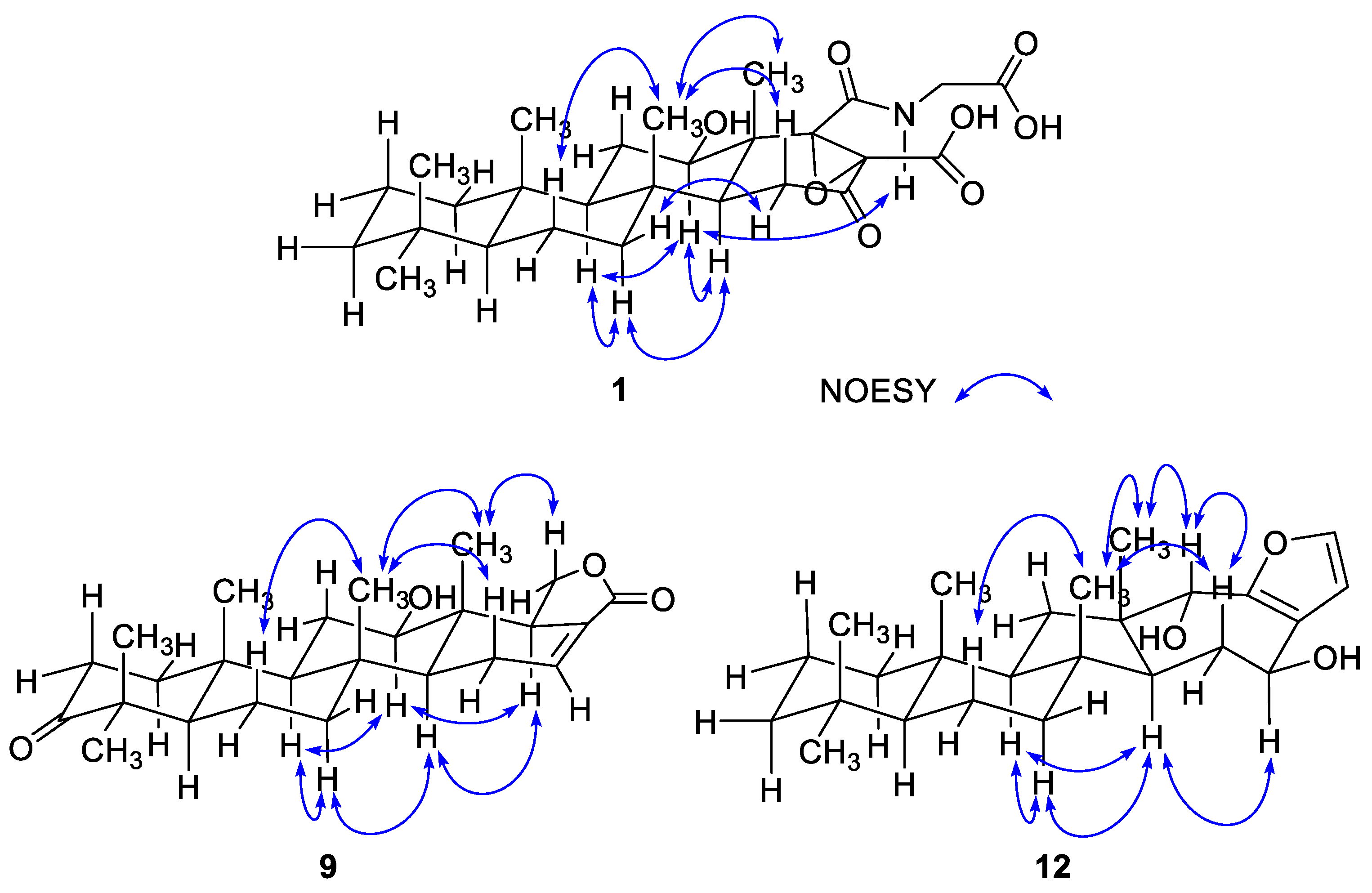
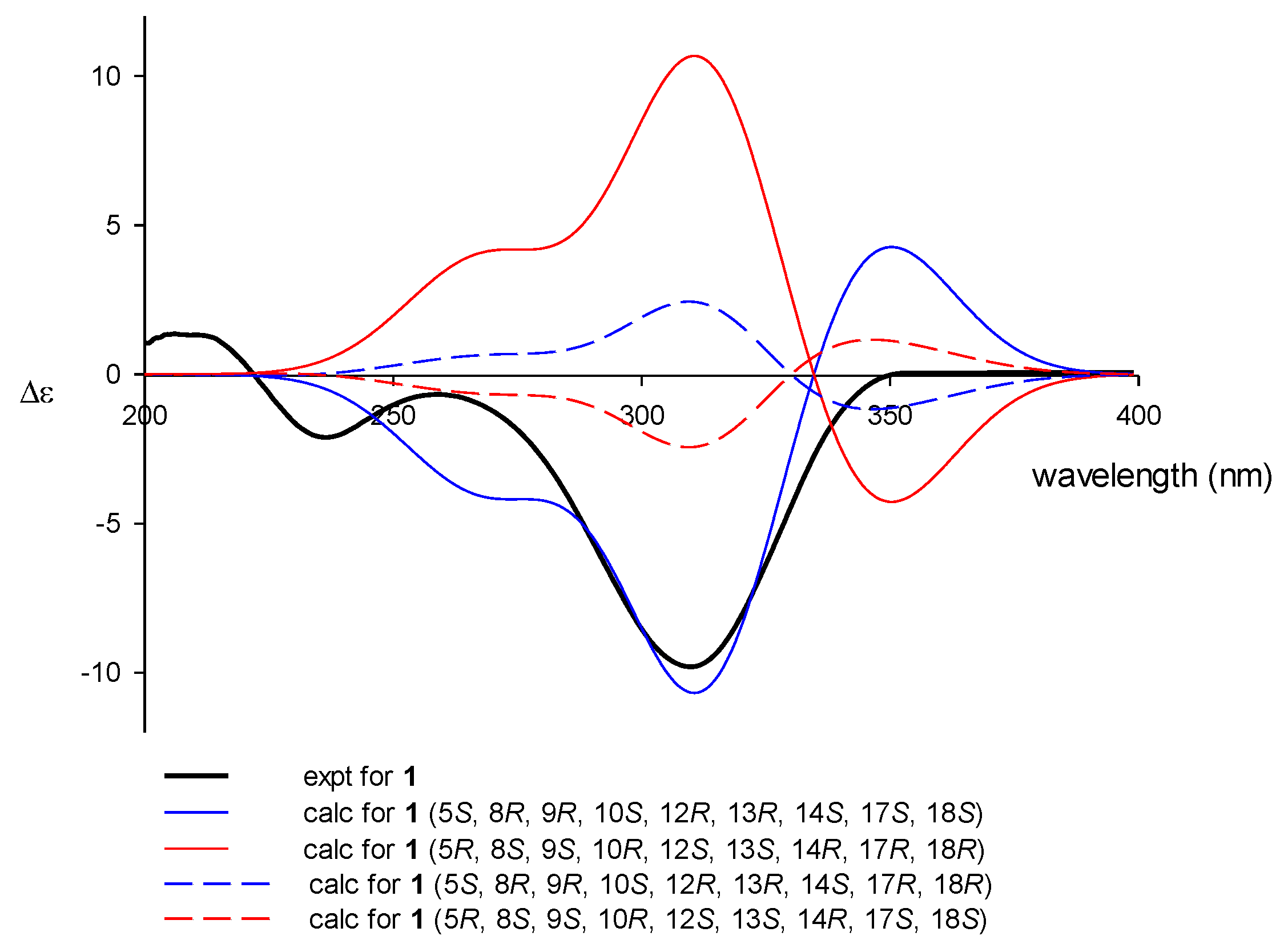
The configurations at the stereogenic centers of 1 were assigned based on NOESY data, which readily showed all trans junctures for rings A–D, which are typical of scalaranes and similar sesterterpenes (Figure 2). This interpretation was also supported by the characteristic carbon chemical shifts of the bridgehead methines and methyl groups. The β-orientation (12R* configuration) was assigned based on the NOESY cross peak for H-12/H-14 and its vicinal coupling constants (J = 11.0, 4.4 Hz) with H2-11. For the C-17-C-18 epoxide, which does not have any bound hydrogens, severe steric crowding with the neighboring C-25 methyl group indicated that the olefinic precursor underwent α-oriented attack by the oxygen. This interpretation was confirmed by ECD calculations (Figure 3). Given the all trans ring junctures and 12R* configuration, the calculated ECD profile for the 17S and 18S configurations matched well with the observed profile in both the intensity and wavelength of the signals. In this way, the absolute configurations of the ring junctures and C-12 were also satisfactorily assigned as (5S, 8R, 9R, 10S, 12R, 13R, 14S, 17S, 18S). Thus, compound 1, designated hyrtioscalarin A, was determined to be a new glycine-bearing scalarane.
The molecular formula of compound 2 was deduced to be C27H39NO5 by HRFABMS analysis ([M-H]− m/z 456.2760, calcd for C27H38NO5, 456.2750), which corresponds to 9 degrees of unsaturation. The spectroscopic data of this compound were highly reminiscent of those of 1, suggesting they shared the same glycine-bearing scalarane core. Detailed examination of the 1H and 13C NMR and HSQC data, however, revealed remarkable differences, the most noticeable of which were the replacement of the C-16 ketone and C-17-C-18 epoxide of 1 with a methylene group (δC/δH 23.4/2.53 and 2.26) and two non-protonated carbons (δC 150.8 and 144.1) in 2 (Table 1).
The structural differences were defined by a combination of 2-D NMR data (Supporting Information Figure S1). First, the A–C rings of 1 were found to be intact in 2 based on their COSY, HSQC and HMBC data. Then, tracing the proton spin system using the COSY data revealed that the C-14 methine group (δC/δH 56.4/1.18) was connected to the methylene at C-15 (δC/δH 17.8/1.98 and 1.67), which was connected in turn to the methylene at C-16 (δC/δH 23.4/2.53 and 2.26). Subsequently, two non-protonated carbons were placed at C-17 (δC 144.1) and C-18 (δC 150.8), constructing ring D as a cyclohexene moiety, based on the HMBC cross peaks of H2-15/C-17, H2-16/C-17, and H3-25/C-18.
The partial formula of the A-D ring portion of 2 was C23H36O, leaving a C4H3NO4 unit for the remainder of the molecule. The 1H NMR and HSQC data of 2 revealed the remaining fragment was an isolated methylene (δC/δH 40.2/4.19 (2H), C-1’) with HMBC cross peaks with three carbonyl carbons (δC 175.1, 171.8 and 171.5) (Supporting Information Figure S1). Among these, the carbon at δC 171.5 was placed at C-20 based on its additional HMBC cross peak with H2-16. Therefore, one of the remaining carbonyl carbons must be located at C-19 and attached at C-18, the last open end of the ring system, while the other was a free carboxylic acid carbon at C-2’. The noticeably small and broad shape of the carbon signal at δC 171.8 in the 13C NMR data secured the assignment of this group at C-2’, leaving the signal at δC 175.1 as C-19. Therefore, 2 must possess a glycine-derived succinimide group (C-17-C-20), fulfilling the remaining 4 degrees of unsaturation required by the mass data. The configurations of stereogenic centers in the A–C rings were also assigned to be identical to those of 1 by the NOESY data (Supporting Information Figure S2) and biogenetic consideration with 1. Thus, hyrtioscalarin B (2) was determined to be a scalarane sesterterpene bearing a succinimide group.
Compounds 3 and 4 were isolated as pale-yellow amorphous solids, and they were found to have the same molecular formula (C28H41NO6) by HRFABMS analyses. The spectroscopic data of these compounds were very similar to each other and highly reminiscent of those of 2, suggesting they shared the same succinimide-bearing scalarane structure. The most noticeable differences in the 13C, 1H and HSQC NMR data of 3 and 4 from those of 2 were the replacement of a methylene with a methine (δC/δH 3: 73.6/4.18, 4: 68.4/4.10) and a methoxy group (δC/δH 3: 58.1/3.55, 4: 57.8/3.45) (Supporting Information Tables S1 and S2).
These compounds were defined as 16-methoxy derivatives of 2 based on their combined 1-D and 2-D NMR data, including the crucial HMBC H3-OCH3/C-16 cross peaks in the spectra of both compounds (Supporting Information Figure S1). The relative configurations at the epimeric C-16 stereogenic center were also determined from the NOESY data. In addition to the same all trans ring junctures and 12R configurations as seen in 1 and 2 derived from the NOESY data and biogenetic consideration, additional NOESY cross peaks of H-14/H-16, H-15α (δH 2.32)/H-16, H-15β (δH 1.65)/H3-25, and H-15β/H3-24 assigned the 16S configuration for 3 (Supporting Information Figure S2). In contrast, the opposite 16R configuration was assigned for 4 based on the cross peaks of H-14/H3-16OCH3, H-15α (δH 2.12)/H3-16OCH3 and H-15β/H3-25. The absolute configurations of this compound were further supported the ECD calculations (Supporting Information Figure S79). Thus, compounds 3 and 4, designated hyrtioscalarin C and D, respectively, were defined as epimeric 16-methoxy derivatives of hyrtioscalarin B (2).
Compounds 5 and 6 were obtained as another pair of epimeric pale-yellow solids and were found to have the formula C29H45NO6 by HRFABMS analyses. The 13C and 1H NMR data of these compounds were similar to those of 3 and 4, indicating they shared the same glycine-incorporated scalarane structures. Aided by the HSQC data, the most conspicuous differences in the 13C and 1H NMR data were the replacement of a carbonyl carbon with a methoxy and a methine group (δC/δH 5: 84.8/5.75 and 50.3/3.09, 6: 84.5/5.69 and 50.1/3.12) (Supporting Information Tables S1 and S2). The remarkable differentiation of the C-17 and C-18 olefinic carbons between compounds 3/4 and 5/6 suggested that structural differences were present in compounds 5/6 around the amide carbons at C-19 and C-20. This interpretation was confirmed by the COSY and HMBC data of 5, which assigned a methoxy-methine group at C-19 based on the proton-carbon correlations of H2-15/C-17; H-16/C-17; H-19/C-17, C-18 and C-20; H3-25/C-18; H2-1’/C-19, C-20, and C-2’; H3-16OCH3/C-16; and H3-19OCH3/C-19 (Supporting Information Figure S1). Possibly due to the small amounts of materials isolated, the HMBC data of 6 were less informative than those of 5 (Supporting Information Table S4). However, in conjunction with a comparison of their 13C and 1H NMR data, the planar structure of 6 was defined to be identical to that of 5 based on the key HMBC cross peaks of H2-1’/C-19, C-20, and C-2’; H3-16OCH3/C-16; and H3-19OCH3/C-19. Overall, these compounds possessed γ-methoxy-α,β-unsaturated butyrolactam rings as a common structural motif (ring E).
Compounds 5 and 6 each possessed two methoxy-bearing stereogenic centers, C-16 and C-19. The relative configurations at these centers were determined from their NOESY data as well as the biogenetic consideration with other scalaranes. For 5, the 16S and 19S configurations were assigned based on the cross peaks of H-12/H-14, H-12/H3-19OCH3, H-14/H-16, H-15β (δH 1.65)/H3-25, H-19/H3-25, and H-19/H-1’ (δH 3.65) (Supporting Information Figure S2). Similarly, the 16R and 19S configurations were assigned for 6 based on the cross peaks of H-12/H-14, H-12/H3-19OCH3, H-15α (δH 2.04)/H3-16OCH3, H-15β (δH 1.53)/H3-25, H-19/H3-25, and H-19/H-1’ (δH 3.69). These results clearly indicated that 5 and 6 were epimeric at the methoxy-bearing C-16 position. Overall, hyrtioscalarins E (5) and F (6) are epimeric scalaranes bearing a glycine-derived γ-methoxy-α,β-unsaturated butyrolactam moiety.
The molecular formula of compound 7 was established as C30H45NO7 based on its HRFABMS data ([M-H]− m/z 530.3112, calcd for C30H44NO7, 530.3118). The spectroscopic data of this compound were similar to those of 5 and 6, suggesting an analogous structure. The most noticeable differences in the 13C and 1H NMR data were the appearance of an acetyl group (δC/δH 173.4 and 22.2/2.03) and the lack of a methoxy group (Supporting Information Tables S1 and S2). Based upon the extensive 2-D NMR analyses, the structural differences were readily found to be the 12-acetyl and 19-hydroxy groups (Supporting Information Figure S1). After assigning the same ring junctures and 12R configurations using the NOESY data and biogenetic consideration, the 16R and 19S configurations were also assigned in a similar manner based on the cross peaks of H-12/H-14, H-14/H3-16OCH3, H-15α (δH 2.09)/H3-16OCH3, H-15β (δH 1.71)/H3-25, H-19/H3-25, H-19/H-1’ (δH 3.88), and H-19/H3-12OCOCH3 (Supporting Information Figure S2). Thus, hyrtioscalarin G (7) was defined as a new scalarane bearing a γ-hydroxy-α,β-unsaturated butyrolactam group.
The molecular formula of compound 8 was established to be C29H45NO6, identical to that of 5 and 6, by HRFABMS analysis ([M-H]− m/z 502.3169, calcd for C29H44NO6, 502.3169). Although the spectroscopic data of this compound were also very similar to those of 5 and 6, a detailed examination of its 13C and 1H NMR data revealed significant differences among the carbons and protons of the butyrolactam (ring E) and the adjacent positions (Supporting Information Tables S1 and S2). Although the 2-D NMR data indicated the same A–D rings as in the other compounds, the butyrolactam moiety of 8 was revealed to have a distinct substitution mode. That is, key HMBC cross peaks were found for H2-15/C-17; H-16/C-17, C-18 and C-19; H2-1’/C-19, C-20 and C-2’; and H3-20OCH3/C-19 (Supporting Information Figure S1). Therefore, methylation must have occurred at the C-20 lactam carbon instead of C-19. After assigning the all trans ring junctures and 12R configurations from the NOESY data and biogenetic consideration, the 16R and 20S configurations of the methoxy-bearing stereogenic centers were also determined from the NOESY cross peaks of H-15α (δH 2.16)/H3-16OCH3, H-15β (δH 1.71)/H3-25, H-16/H3-20OCH3, H-20/H3-16OCH3, and H2-1’ (δH 3.87)/H3-20OCH3 (Supporting Information Figure S2). This interpretation was further supported by the ECD calculation (Supporting Information Figure S79). In this way, compound 8, designated hyrtioscalarin H, was determined to be a new butyrolactam-bearing scalarane.
Hyrtioscalarins A-H (1–8) all possess glycine-derived nitrogenous functionalities. A literature study showed that among the numerous scalarane sesterterpenes reported to date, few are nitrogenous. To the best of our knowledge, the only examples are molliorins A–C from Cacospongia mollior [11,12,13], an unnamed compound (14, a congener in this work) from Hyatella sp. [14], petrosaspongiolactams A–C from Petrosaspongia sp. [15], scalalactam A–D from Spongia sp. [16], a pyrrole containing scalarane from Scalarispongia sp. [17], and 24-methoxypetrosaspongia C from Hyrtios erectus [18]. Moreover, only the one compound from Hyatella sp. possesses the same glycine-derived moiety as seen in 1–8, emphasizing the unusual structural features of these new compounds. Indeed, our finding of diverse nitrogenous derivatives suggests that these compounds are important structural variants of scalaranes.
In addition to glycine-bearing scalaranes 1–8, a number of non-nitrogenous analogs were also isolated. The molecular formula of compound 9 was deduced to be C25H36O4, corresponding to 8 degrees of unsaturation, from its HRFABMS data ([M-H]− m/z 399.2531, calcd for C25H35O4, 399.2535). The combined 13C, 1H and HSQC NMR data of this compound showed the presence of two carbonyl carbons (δC 220.3 and 172.4) and two olefinic carbons (δC/δH 137.2/6.81 and 128.6) in the deshielded region, which together account for 3 degrees of unsaturation (Table 2). Signals for an oxymethine (δC/δH 81.4/3.41) and an oxymethylene (δC/δH 70.8/4.50 and 4.20) were also conspicuous in the NMR data. Other signals in the shielded region of the 13C NMR spectra were those of four non-protonated, four methine, six methylene, and five methyl carbons, which matched well with the corresponding protons based on the HSQC data. Altogether, these data indicated that 9 is pentacyclic and likely to be a typical scalarane sesterterpene.
The planar structure of 9 was elucidated from its 2-D NMR data (Figure 1). First, the COSY data revealed the presence of a spin system involving two methylenes (δC/δH 40.2/1.98 and 1.58 and 34.8/2.53 and 2.47, C-1 and C-2) and requiring a non-protonated carbon at C-3. Based on the HMBC data, a ketone carbon (δC 220.3) was placed at this position based on its correlations with neighboring protons at H2-1, H2-2, H3-21, and H3-22. Similarly, the A–C ring portion was defined as the same 6/6/6 tricyclic moiety seen in 1-8 based on the COSY and HMBC data, including crucial long-range correlations between the five bridgehead methyl protons and their neighboring carbons. Similar to other scalaranes, the chemical shifts (δC/δH 81.4/3.41), vicinal coupling constants (J = 11.4, 3.4 Hz) and HMBC cross peaks to the C-25 methyl group (H-12/C-25, H3-25/C-12, H-9/C-12, and H2-11/C-12) placed a β-hydroxy group at C-12.
The COSY data located a double bond at C-16 based on the proton–proton correlations of H-14 with H-16 via H2-15 (δH 1.34, 2.39 and 2.27, and 6.81, respectively). Due to the small amount of material obtained, no HMBC data were available to directly support this interpretation. However, the chemical shifts of the C-18 methine group (δC/δH 51.6/2.92), which showed long-range coupling with H3-25, were definitive enough to place an olefinic carbon at C-17 (δC 128.6). Then, the placement of an oxymethylene (δC/δH 70.8/4.50 and 4.20) at C-19 was accomplished based on direct proton–proton couplings with H-18. Finally, the HMBC cross peaks of H2-19 with the neighboring carbons (H2-19/C-13, C-17, C-18, and C-20) not only confirmed its location but also constructed a five-membered lactone as ring E.
The configuration at the newly constructed C-18 center was assigned as R based on the NOESY cross peaks of H-18 with the α-oriented H-12 and H-14 protons as well as the all trans ring junctures derived from the NOESY data and biogenetic relation with other scalaranes (Figure 2). Thus, the structure of 9, designated 12-deacetyl-3-oxoscalarin (following the previously reported analog scalarin), was determined to be a scalarane lactone bearing a 3-keto group. A literature study showed that scalarane sesterterpenes possessing a 3-keto group are quite rare. To the best of our knowledge, the only four reported examples are two scalaranes from H. erectus [22] and H. erecta [23], a norscalarane from the mollusk Dorisprismatica (= Glossodoris) atromarginata [24] and a scalarane with acetoxy group at C-3 position from H. erecta [25].
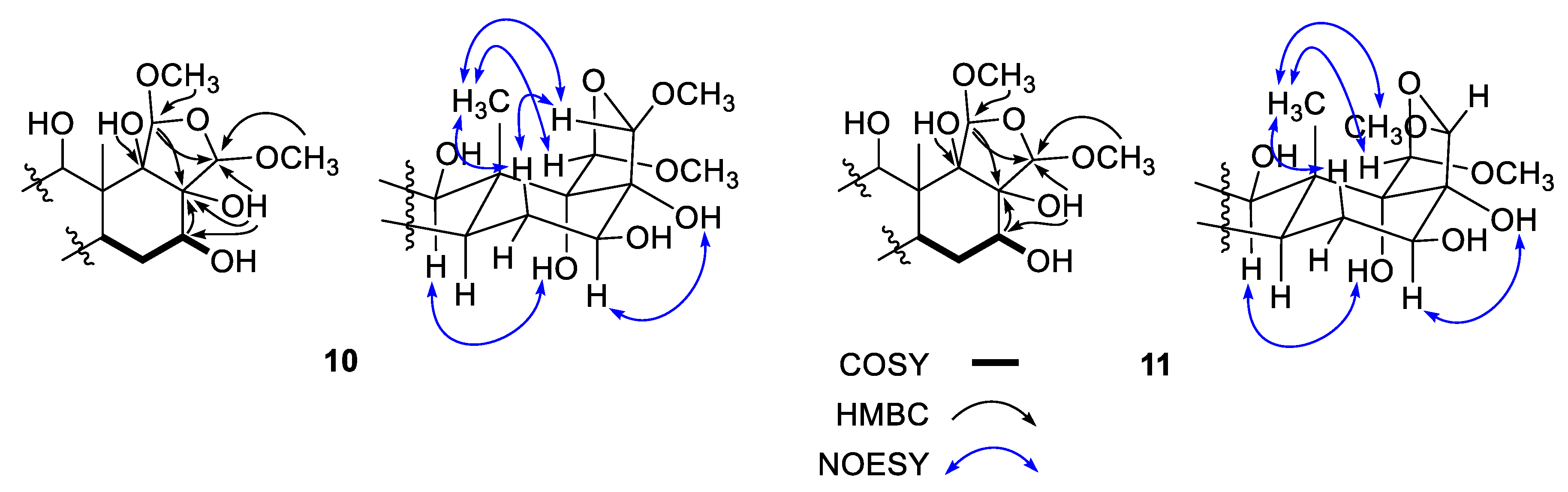
An oxidized-furan-bearing scalarane (10) was isolated as a pale-yellow solid, and it was found to have the molecular formula C27H46O7, corresponding to 5 degrees of unsaturation, by HRFABMS ([M-H]− m/z 481.3168, calcd for C27H45O7, 481.3165). With the aid of the HSQC data, the 13C and 1H NMR spectra of this compound showed signals for two oxygen-bearing non-protonated carbons (δC 83.1 and 81.0), four methines (δC/δH 106.1/5.22, 103.2/4.99, 74.4/4.34, and 74.2/3.72) and two methyl groups (δC/δH 56.0/3.56 and 55.6/3.40) (Supporting Information Tables S1 and S2). Since all the other carbon and proton signals were observed in the shielded regions and showed no evidence of unsaturation, 10 was thought to be a pentacyclic sesterterpene.
Based upon the results of 2-D NMR analyses and comparison with other scalaranes, 10 was readily found to possess the same A-C rings, including the 12-βOH group seen in the compounds described above, and most of the oxygenated functionalities of 10 were located on rings D and E. The HMBC data, including the correlations of hydroxy protons, were crucial to defining the remaining structure (Figure 4). That is, starting at H-14 (δH 1.32), the COSY correlations placed a hydroxy methine at C-16 (δC/δH 74.2/3.72), which was confirmed by the HMBC cross peaks of this methine carbon with H-14 and H2-15 (δH 1.79 and 1.44). The neighboring carbon, C-17 (δC 81.0), was similarly assigned as being a hydroxy-bearing non-protonated carbon based on the correlations of H2-15/C-17, H-16/C-17, and 17-OH (δH 2.32)/C-16 and C-17. Another hydroxy group was also placed at C-18 (δC 83.1) by the HMBC correlations of H3-25/C-18 and of 18-OH (δH 3.87)/C-13 and C-18.
In addition, two methoxy groups (δC/δH 55.6/3.40 and 56.0/3.56) were directly attached to oxymethines (δC/δH 106.1/5.22 and 103.2/4.99, C-19 and C-20) by diagnostic 3-bond HMBC interactions. The deshielded chemical shifts of these oxymethines in conjunction with the remaining oxygen from the mass data indicated these were part of an acetal. This interpretation was confirmed, and a tetrahydrofuran moiety was constructed by a series of HMBC cross peaks, namely, H-19/C-13, C-17 and C-20, H-20/C-17, and 18-OH (δH 3.47)/C-20. Thus, the planar structure of 10 was defined as a polyoxygenated scalarane.
Compound 10 possessed stereogenic centers at the D/E ring juncture and adjacent sites, and their relative configurations were assigned based on the NOESY data (Figure 4). The H-14/H-16 cross peak defined a β-orientation for the 16-OH group, which was in good agreement with the vicinal coupling constants (J = 12.0, 5.7 Hz) of H-16. In addition, the H3-25 protons showed cross peaks with H-15β (δH 1.44), H-19 and H-20. To achieve sufficient spatial proximity with the bridgehead H3-25 protons, not only a cis D/E ring juncture but also β-orientations of both H-19 and H-20 in ring E are required. Overall, aided by the biogenetic consideration, the configurations of the stereogenic centers were assigned as 12R, 16S, 17R, 18S, 19R, and 20S. Thus, 17(R),18(S)-dihydroxy-19(R),20(S)-dimethoxysesterstatin 5 (10), was determined to be a tetrahydrofuran-bearing scalarane sesterterpene.
The molecular formula of compound 11 was deduced to be C27H46O7, identical to that of 10, based on HRFABMS analysis ([M-H]− m/z 481.3172, calcd for C27H45O7, 481.3165). The spectroscopic data of this compound were also very similar to those of 10, implying the same planar structure, which was confirmed by 1-D and 2-D NMR analyses. However, a detailed comparison of its 13C and 1H NMR data revealed remarkable differences among the carbons and protons of ring E and the adjacent positions, suggesting that 10 and 11 are epimers (Supporting Information Tables S1 and S2). To confirm this, the NOESY data of 11 showed a cross peak for H-14/H-16, indicating the presence of the same 16β-OH group as in 10. Additionally, cross peaks of H3-25 with H-15β (δH 1.65), H-19 and H3-20OCH3 were observed. To generate these signals, the D/E ring juncture must be cis, identical to that in 10, but an opposite orientation was required at the C-20 stereogenic center, indicating a 20R configuration (Figure 4). Thus, 17(R),18(S)-dihydroxy-19(R),20(R)-dimethoxysesterstatin 5 (11) was defined as another tetrahydrofuran-bearing scalarane.
In addition to scalaranes, the H. erectus extract contained a sesterterpene with a different skeleton. The molecular formula of compound 12 was established as C25H38O3, corresponding to 7 degrees of unsaturation, by HRFABMS analysis ([M-H]− m/z 385.2738, calcd for C25H37O3, 385.2743). The 13C NMR data of this compound showed four olefinic carbons (δC 150.2, 141.4, 124.2, and 111.5) (Table 2). The chemical shifts (δH 7.30 and 6.50) and coupling constants (J = 1.7 Hz for both) of the corresponding protons in the 1H NMR data were indicative of a disubstituted furan moiety. Aided by the HSQC data, two oxymethines (δC/δH 76.1/4.46 and 69.8/4.66) were also identified from the NMR data. The other signals were those of four non-protonated, three methine, seven methylene and five methyl groups. Accordingly, 12 must be a pentacyclic sesterterpene.
A detailed comparison of the 1-D NMR data with those of other compounds revealed remarkable differences in the chemical shifts of several carbons and protons, prompting extensive 2-D NMR analyses (Figure 1). First, the COSY data revealed the presence of two linear chains of three protonated carbons (C-1-C-3 and C-5-C-7). Subsequently, several long-range correlations from these carbons to the protons of four singlet methyl groups (H3-19, H3-20, H3-21 and H3-22) secured a 6/6 bicyclic moiety (rings A and B), the same as its congeners.
In the process of determining the structures of rings A and B, the C-9 methine group (δC/δH 61.4/1.30) was confidently assigned based on its HMBC cross peaks with the H3-21 and H3-22 protons. In contrast to the scalaranes, however, the proton spin system involving H-9 terminated at the C-11 methylene (δC/δH 34.9/1.89 and 1.37), placing a non-protonated carbon at neighboring C-12. This was confirmed by the HMBC cross peaks of C-11 and C-12 (δC 43.9) with a common methyl proton (δH 1.01, H3-23). This methyl proton showed an additional HMBC cross peak with a methine group (δC/δH 49.3/1.82, C-13). Then, a crucial 3-bond correlation with H3-21 confirmed the attachment of C-13 to C-8 of ring B, establishing a five-membered ring (C-8, C-9, C-11-C-13, ring C) (Figure 1).
The COSY data revealed a linear spin system of H-13 (δH 1.82), H2-14 (δH 1.90 and 1.70) and H-15 (δH 4.66), analogous to the H-14-H-16 spin system in the scalarane congeners. The assignment of an oxygenated methine at C-15 (δC 69.8) is also reminiscent of several previously reported compounds. This three-carbon unit was then extended by two additional non-protonated carbons (δC 124.2 and 150.2, C-16 and C-17, respectively) by the HMBC cross peaks of H2-14/C-16 and of H-15/C-16 and C-17. Similarly, another hydroxy-bearing methine (δC/δH 76.1/4.46, C-18) was attached to C-12 by the HMBC cross peaks of H-18/C-12 and C-14 and of H3-23/C-18. Then, C-18 was linked to C-17 by the similar correlations of H-18 with C-16 and C-17, establishing a seven-membered ring (C-12-C-18, ring D).
The preliminary examination of the 13C and 1H NMR data revealed the presence of a disubstituted furan moiety that required the linkage of C-16 and C-17 to the remaining carbons at C-24 (δC/δH 141.4/7.30) and C-25 (δC/δH 111.5/6.50). This confirmed by the small proton–proton coupling constants (J = 1.7 Hz) between H-24 and H-25 as well as their HMBC cross peaks with C-16 and C-17 (Figure 1). Thus, a 1,2-disubstituted furan moiety was deduced as the final structural motif (ring E). Overall, the planar structure of 12 was determined to be that of a salmahyrtisane-type sesterterpene [25,26,27].
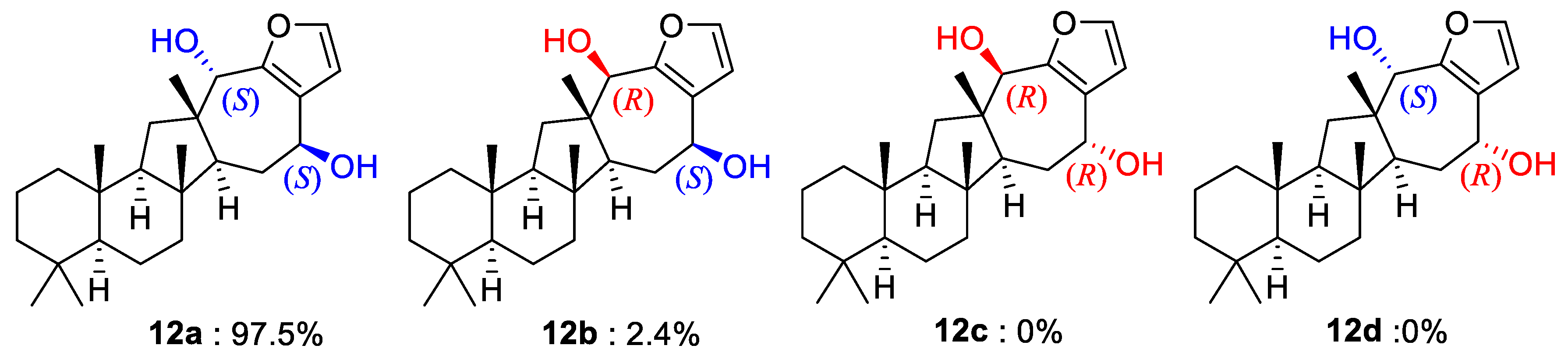
In addition to the ring junctures, compound 12 possessed two hydroxy-bearing stereogenic centers at C-15 and C-18. The NOESY data showed several diagnostic 1,3-diaxial cross peaks between the bridgehead methyl protons and spatially adjacent protons, assigning all trans ring junctures (Figure 2). The orientations of the hydroxy groups were assigned as 15β and 18α by a number of NOESY cross peaks for protons on ring D and in adjacent positions (H-13/H-15, H-14α (δH 1.90)/H-15, H-14β (δH 1.70)/H3-21 and 23, and H-18/H3-23). Therefore, the 15S and 18S configurations were assigned. The NOESY-based configurations were further supported by the DP4 calculations in which these configurations (12a) matched the NMR data better than did any other configurational combination (12b–12d) (Figure 5 and Supporting Information Figure S77). Thus, compound 12, designated sarmahyrtisol B, was elucidated as a new sesterterpene.
The salmahyrtisane skeleton of 12 is rare, and to date, only three compounds with this skeleton have been reported: salmahyrtisol A from the Red Sea sponge H. erecta (= H. erectus) [25], similan A from a Thai specimen of H. gumininae [26], and hippospongide A from a Taiwanese Hippospongia sp [27]. The structures of these compounds were closely related to each other by the possession of oxygenated functionalities at C-15 and C-18 on an identical furan-bearing carbon framework. Despite the weak or no cytotoxicity of these compounds, the unusual carbon framework and co-isolation with scalaranes and other sesterterpenes makes the biosynthesis of sarmahyrtisanes of great interest. Accordingly, salmahyrsitol A and hippospongide A were recently prepared by biomimetic synthesis [28]. Notably, 12 has been proposed as a biosynthetic intermediate but was isolated for the first time as a natural product in this work. Both our finding of a sarmahyrtisane from the Hyrtios sponge and its co-isolation with scalaranes coincides well with previous works [25,26,27].
Several new compounds, such as 3/4, 5/6, and 10/11, were isolated as configurational pairs at their methoxy-bearing centers. In addition, 10 and 11 showed typical oxidized furans for ring E. These findings suggest that these compounds are abiotic products derived from natural precursors, the results of solvolysis and Michael additions, either during prolonged storage or the isolation process [29]. Even considering this, however, the quantities, diversity and unusual structural features of these new compounds demonstrate both the immense structural variation in sponge sesterterpenes and the chemical prosperity of Hyrtios sponges.
In addition to 1-12, the extract of H. erectus contained several known scalaranes (Supporting Information Figure S78). Based upon the results of 1-D and 2-D NMR and mass spectroscopy, these were identified as heteronemin (13, known from various sponges) [30,31,32], a glycine-bearing derivative (14, known from a Hyatella sp.) [14], sesterstatins 5 and 6 (15 and 16, known from H. erecta (= H. erectus)) [33,34], 16-hydroxyscalarolide (17, known from H. erectus) [35], 12-O-deacetyl-12-epi-scalarin (18, known from a Spongia sp. [36] and H. erectus [37]), and hyrtiosin A and 16-O-deacetyl-16-epi-scalarolbutenolide (19 and 20, known from H. erecta) [38,39]. The spectroscopic data of these compounds were in good accordance with those in the literature. Compound 13 was the major constituent, far exceeding all the known and new compounds isolated in this work.
2.2. Biological Activity
The bioactivities of new compounds 1–12 and the major metabolite 13 were extensively studied. Anti-angiogenesis activity of compounds 1–13 was evaluated in HUVEC cells. Compounds 1–12 showed moderate anti-angiogenesis activity without cytotoxicity, while strong anti-angiogenesis activity of 13 was derived from overt cytotoxicity in HUVEC cells. In tests against various human cancer cell lines, compounds 1–12 exhibited weak to no antiproliferation activity. Among the scalaranes, 4 and 7 were generally more antiproliferative than other new compounds. In contrast, 13 displayed significant inhibition, comparable to that of etoposide, the positive control, suggesting it may be responsible for the remarkable brine-shrimp lethality of the crude extract (Table 3).
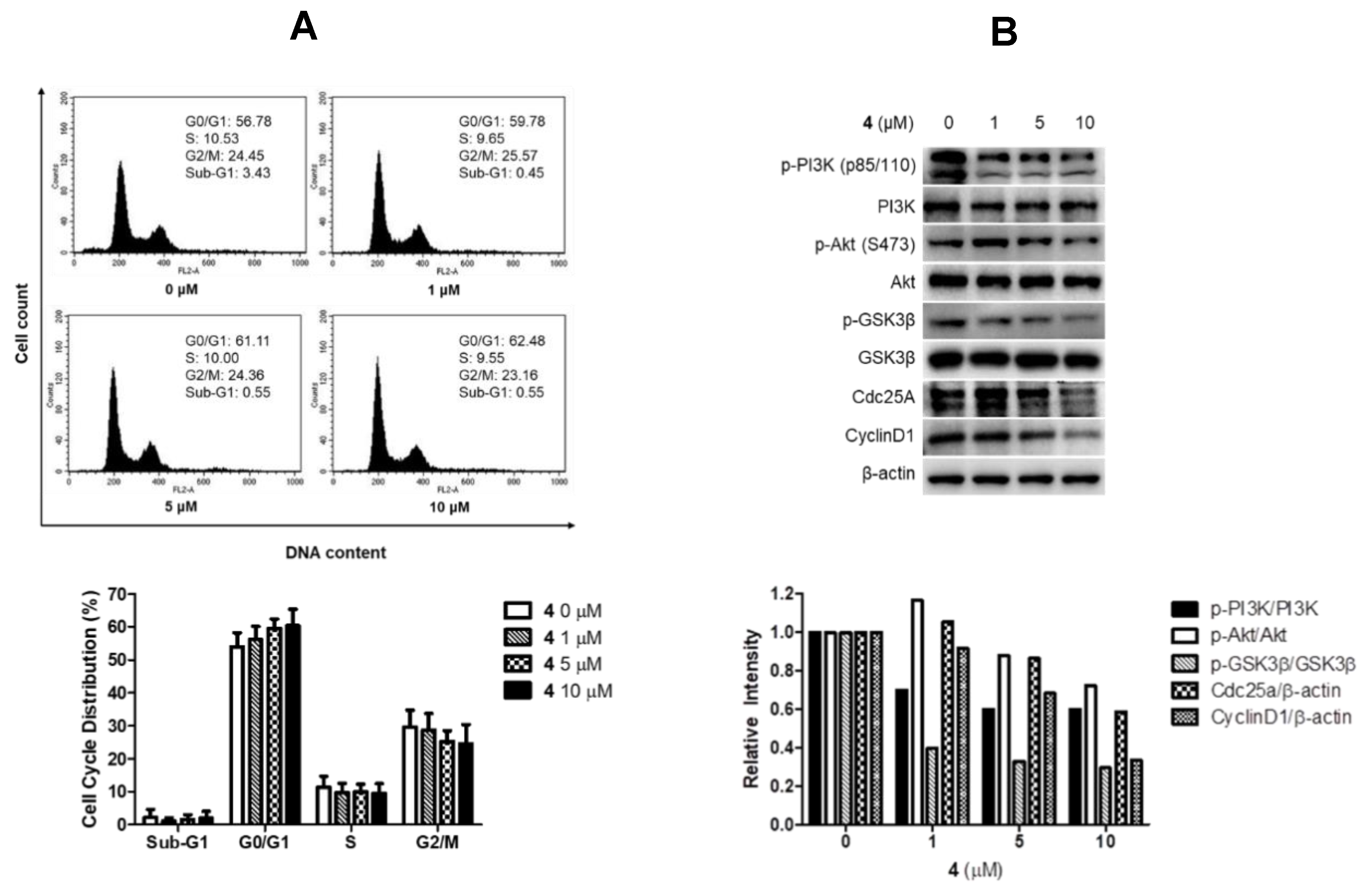
Compound 12, a salmahyrstane derivative, also showed weak but consistent inhibition toward the cancer strains. The antiproliferative activity of 4 was further studied in PC3 human prostate cancer cells which were insensitive to etoposide treatment. Treatment with 4 slightly increased the G0/G1 cell population (Figure 6A). Therefore, the effects of 4 on proteins that are associated with G0/G1 arrest were further evaluated using Western blot analyses. Consistent with the cell cycle analysis results, treatment of 4 suppressed the expressions of Cdc25A and CyclinD1 in a concentration dependent manner (Figure 6B). Furthermore, treatment of 4 decreased the expressions of p-PI3K, p-Akt, and p-GSK3β without affecting their total protein expression level (Figure 6B). These data suggested that the antiproliferative activities of these compounds would be associated with G0/G1 arrest and PI3K/Akt/GSK-3β pathway. In the anti-angiogenesis assay, several compounds moderately inhibited tube formation at nontoxic concentrations (>10 μM). These compounds also showed weak to no antibacterial activities against various Gram-positive and Gram-negative strains (Supporting Information Table S3). In related assays against microbial enzymes, no compounds significantly inhibited the action of sortase A (SrtA) or isocitrate lyase (ICL) (IC50 > 128 g/mL).
In summary, twelve new sesterterpenes along with eight known sesterterpenes were isolated from the tropical sponge Hyrtios erectus. Based upon a combination of spectroscopic and computational analyses, these compounds were determined to be eight glycine-bearing scalaranes (1–8), a 3-keto scalarane (9), two oxidized furan-containing scalaranes (10 and 11), and a salmahyrtisane derivative (12). Several of these compounds exhibited weak antiproliferation against diverse cancer cell lines as well as moderate anti-angiogenesis activities. The antiproliferative activities were found to be associated with G0/G1 arrest in the cell cycle. This work demonstrates both the structural diversity of sponge-derived sesterterpenes and the prosperity of Hyrtios erectus as their source.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were measured using a JASCO P-1020 polarimeter (Jasco, Tokyo, Japan) with a 1-cm cell. CD spectra were obtained using an Applied Photophysics Ltd. Chirascan Plus spectrometer (Leatherhead, Surrey, UK). UV spectra were acquired using a Hitachi U-3010 spectrophotometer (Hitachi High-Technologies, Tokyo, Japan). IR spectra were recorded on a JASCO 4200 FT-IR spectrometer (Jasco, Tokyo, Japan) using a ZnSe cell. NMR spectra were recorded in MeOH-d3, MeOH-d4 and CDCl3 with the solvent peaks (δH 3.30/δc 47.5 and δH 7.26/δc 77.0) as internal standards on a Bruker Avance 600 MHz spectrometer (Billerica, MA, USA). Proton and carbon NMR spectra were measured at 600 and 150 MHz, respectively. High-resolution FABMS data were obtained at the National Center for Inter-university Research Facilities (NCIRF), Seoul National University, and acquired using a JEOL JMS 700 mass spectrometer (Jeol, Tokyo, Japan) with 6 keV-energy, emission current 5.0 mA, xenon as the inert gas, and meta-nitrobenzyl alcohol (NBA) as the matrix. HPLC separations were performed on a SpectraSYSTEM p2000 equipped with a refractive index detector (SpectraSYSTEM RI-150 (Waltham, MA, USA)) and a UV-Vis detector (Gilson UV-Vis-151 (Middleton, WI, USA)). All solvents used were of spectroscopic grade or were distilled prior to use.
3.2. Animal Material
Specimens of Hyrtios erectus sponge (sample number 163CH-111) were collected by hand using SCUBA offshore of Chuuk Island in the Federated States of Micronesia at a depth of 15 m on 7 March 2012. The thick, black encrusting sponge formed finger-like projections that were 3 mm thick and up to 14 cm long. The surface of the sponge retained a distinctly conulose appearance. The specimens also showed a wide variety of encrusting masses with finger-like projections. Sand was present in the skeletal fibers of this sponge, and the fibers were easily broken. The texture of the sponge was quite firm and brittle. These gross morphological features, including the skeletal structures, were consistent with those of H. erectus (Heteronema erecta Keller, 1889) [40]. A voucher specimen (registry no. spo. 81) was deposited in the MABIK (Marine Biodiversity Institute of Korea), Seocheon, Korea.
3.3. Extraction and Isolation
Freshly collected specimens were immediately frozen and stored at −25 °C until use. Lyophilized specimens (225.8 g) were macerated and repeatedly extracted with MeOH (3 × 2 L) and CH2Cl2 (3 × 2 L). The combined extracts (48.7 g) were sequentially partitioned between H2O (33.2 g) and n-BuOH (14.7 g); the latter layer was repartitioned between H2O-MeOH (15:85, 8.15 g) and n-hexane (6.30 g). Then, the H2O-MeOH layer was separated by C18 reversed-phase flash chromatography using sequential mixtures of MeOH and H2O as the eluent (six fractions in an H2O-MeOH gradient, from 50:50 to 0:100) followed by acetone and finally EtOAc.
Based on the 1H NMR spectra, the fractions eluted with 40:60 H2O-MeOH (0.36 g), 20:80 H2O-MeOH (0.42 g), 10:90 H2O-MeOH (0.56 g) and 0:100 H2O-MeOH (1.80 g) were chosen for further separation. The 40:60 H2O-MeOH fraction was separated by semipreparative reversed-phase HPLC (YMC-ODS column, 10 × 250 mm; 2.0 mL/min; H2O-MeOH, 65:35) to yield compound 1 (tR = 44.2 min). This compound was further purified by analytical HPLC (YMC-ODS column, 4.6 × 250 mm; 0.7 mL/min; H2O-MeCN gradient (from 90:10 to 40:60 in 40 min); tR = 21.1 min).
The 20:80 H2O-MeOH fraction was separated by semipreparative reversed-phase HPLC (H2O-MeOH, 30:70 with 0.1% TFA), yielding, in order of elution, compounds 9 (tR = 8.2 min), 5 (tR = 10.1 min), 7 (tR = 12.5 min), 6 (tR = 16.7 min), 12 (tR = 17.9 min), 8 (tR = 20.0 min), 3 (tR = 20.5 min), 4 (tR = 21.9 min), 2 (tR = 23.0 min), 10 (tR = 35.1 min), and 11 (tR = 38.2 min). The former nine compounds were purified by analytical HPLC (H2O-MeCN gradient from 70:30 to 0:100 with 0.1% TFA in 50 min); tR = 42.9, 44.4, 46.0, 37.0, 40.8, 42.0, 46.6, 47.7, and 48.1 min for 9, 5, 7, 6, 12, 8, 3, 4, and 2, respectively).
The 10:90 H2O-MeOH fraction was separated by semipreparative reversed-phase HPLC (H2O-MeOH, 25:65), yielding, in order of elution, compounds 16 (tR = 18.0 min), 18 (tR = 22.8 min), 15 (tR = 28.6 min), 20 (tR = 38.3 min), 14 (tR = 44.1 min), 17 (tR = 46.9 min), and 19 (tR = 50.4 min). Compounds 13, 16–18, and 20 were further purified by analytical HPLC (H2O-MeCN gradient (from 50:50 to 0:100 in 40 min); tR = 29.0, 22.1, 35.5, 34.3, and 30.0 min, for 16, 18, 20, 19, and 17, respectively). The MeOH fraction from flash chromatography was also separated by semipreparative reversed-phase HPLC (H2O-MeOH, 10:65) to afford compound 13 (tR = 23.0 min) as a pure compound. The purified metabolites were isolated in the following amounts: 2.8, 6.1, 7.0, 2.2, 4.6, 1.9, 1.8, 3.0, 0.8, 5.2, 3.7, 2.3, 110, 1.8, 9.8, 0.9, 1.2, 1.6, 4.7, and 2.2 mg for 1–20, respectively.
Hyrtioscalarin A (1): pale yellow, amorphous solid; [α] −14 (c 0.20, MeOH); UV (MeOH) λmax (log ε) 203 (3.77) nm; IR (ZnSe) νmax 3399, 2981, 1718, 1710, 1666, 1397 cm−1; 1H and 13C NMR data, Table 1; HRFABMS m/z 486.2491 [M-H2O-H]− (calcd for C27H36NO7, 486.2492).
Hyrtioscalarin B (2): pale yellow, amorphous solid; [α] −5 (c 0.25, MeOH); UV (MeOH) λmax (log ε) 207 (3.74); IR (ZnSe) νmax 3525, 2969, 1717, 1701, 1384 cm−1; 1H and 13C NMR data, Table 1; HRFABMS m/z 456.2760 [M-H]− (calcd for C27H38NO5, 456.2750).
Hyrtioscalarin C (3): pale yellow, amorphous solid; [α] −8 (c 0.25, MeOH); UV (MeOH) λmax (log ε) 207 (3.74) nm; IR (ZnSe) νmax 3526, 2969, 1717, 1702, 1680, 1395 cm−1; 1H and 13C NMR data, Supporting Information Tables S1 and S2; HRFABMS m/z 486.2859 [M-H]− (calcd for C28H41NO6, 486.2856).
Hyrtioscalarin D (4): pale yellow, amorphous solid; [α] −21 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 206 (3.69) nm; IR (ZnSe) νmax 3525, 2933, 1714, 1701, 1674, 1391 cm−1; 1H and 13C NMR data, Supporting Information Tables S1 and S2; HRFABMS m/z 486.2850 [M-H]− (calcd for C28H41NO6, 486.2856).
Hyrtioscalarin E (5): pale yellow, amorphous solid; [α] +3 (c 0.25, MeOH); UV (MeOH) λmax (log ε) 207 (3.85), 250 (3.05) nm; IR (ZnSe) νmax 3525, 2938, 1720, 1711, 1674, 1316 cm−1; 1H and 13C NMR data, Supporting Information Tables S1 and S2; HRFABMS m/z 502.3167 [M-H]− (calcd for C29H44NO6, 502.3169).
Hyrtioscalarin F (6): pale yellow, amorphous solid; [α] +2 (c 0.20, MeOH); UV (MeOH) λmax (log ε) 206 (3.79), 247 (3.10) nm; IR (ZnSe) νmax 3525, 2939, 1720, 1709, 1678, 1316 cm−1; 1H and 13C NMR data, Supporting Information Tables S1 and S2; HRFABMS m/z 502.3169 [M-H]− (calcd for C29H44NO6, 502.3169).
Hyrtioscalarin G (7): pale yellow, amorphous solid; [α] −9 (c 0.30, MeOH); UV (MeOH) λmax (log ε) 207 (3.88), 249 (3.11) nm; IR (ZnSe) νmax 3535, 2939, 1723, 1710, 1678, 1374 cm−1; 1H and 13C NMR data, Supporting Information Tables S1 and S2; HRFABMS m/z 530.3112 [M-H]− (calcd for C30H44NO7, 530.3118).
Hyrtioscalarin H (8): pale yellow, amorphous solid; [α] −4 (c 0.25, MeOH); UV (MeOH) λmax (log ε) 206 (3.81), 242 (3.07) nm; IR (ZnSe) νmax 3525, 2968, 1724, 1709, 1680, 1395 cm−1; 1H and 13C NMR data, Supporting Information Tables S1 and S2; HRFABMS m/z 502.3169 [M-H]− (calcd for C29H44NO6, 502.3169).
12-Deacetyl-3-oxoscalarin (9): pale yellow, amorphous solid; [α] −2 (c 0.30, MeOH); UV (MeOH) λmax (log ε) 203 (3.68) nm; IR (ZnSe) νmax 3422, 2940, 1719, 1678, 1640, 1399 cm−1; 1H and 13C NMR data, Table 2; HRFABMS m/z 399.2531 [M-H]− (calcd for C25H35O4, 399.2535).
17(R),18(S)-Dihydroxy-19(R),20(S)-dimethoxysesterstatin 5 (10): pale yellow, amorphous solid; [α] −9 (c 0.25, MeOH); IR (ZnSe) νmax 3524, 2936 cm−1; 1H and 13C NMR data, Supporting Information Tables S1 and S2; HRFABMS m/z 481.3168 [M-H]- (calcd for C27H45O7, 481.3165).
17(R),18(S)-Dihydroxy-19(R),20(R)-dimethoxysesterstatin 5 (11): pale yellow, amorphous solid; [α] −22 (c 0.30, MeOH); IR (ZnSe) νmax 3522, 2941 cm−1; 1H and 13C NMR data, Supporting Information Tables S1 and S2; HRFABMS m/z 481.3172 [M-H]− (calcd for C27H45O7, 481.3165).
Sarmahyrtisol B (12): pale yellow, amorphous solid; [α] −19 (c 0.25, MeOH); UV (MeOH) λmax (log ε) 215 (3.68) nm; IR (ZnSe) νmax 3397, 2927, 1761, 1714, 1463, 1384 cm−1; 1H and 13C NMR data, Table 2; HRFABMS m/z 385.2738 [M-H]− (calcd for C25H37O3, 385.2743).
3.4. ECD Calculations
All conformational searches were performed using Macromodel (Version 9.9, Schrodinger LLC., NY, USA) software with “mixed torsional/low mode sampling” in the Merck molecular force field (MMFF). The searches were conducted in the gas phase with a 50 kJ/mol energy window limit and a maximum of 10,000 steps to thoroughly examine all low-energy conformers. The Polak-Ribiere conjugate gradient (PRCG) method was utilized for minimization processes with a maximum of 10,000 iterations and a 0.001 kJ (mol Å)−1 convergence threshold on the RMS gradient. Conformers within 10 kJ/mol of each global minimum for compound 1 were used for ECD calculations with TmoleX Version 4.2.1 (COSMOlogic GmbH and Co. KG, Leverkusen, Germany) at the B3LYP/6-31G(d,p) level in the gas phase. The CD spectra were simulated by overlapping each transition, where σ is the width of the band at 1/e height. ΔEi and Ri are the excitation energies and rotatory strengths, respectively, for transition i. In the current work, the value was 0.10 eV.
3.5. DP4 Probability Method
All conformational searches were performed using the MacroModel (Version 9.9, Schrodinger LLC., NY, USA) program with “Mixed torsional/Low-mode sampling” in the MMFF force field. To thoroughly examine all low-energy conformers, the searches were implemented in the gas phase with a 10 kJ/mol energy window limit and 10,000 as the maximum number of steps. The Polak-Ribiere conjugate gradient (PRCG) method was utilized for minimization processes with 10,000 maximum iterations and a 0.001 kJ (mol Å)−1 convergence threshold on the RMS gradient. Conformers within 10 kJ/mol of each global minimum of 12a, 12b, 12c, and 12d were used for gauge-independent atomic orbital (GIAO) shielding constant calculations without geometry optimization employing TmoleX Version 4.2.1 (COSMOlogic GmbH and Co. KG, Leverkusen, Germany) at the B3LYP/6-31G(d,p) level in the gas phase. Calculated chemical shift values were obtained based on the following equation: in which is the calculated chemical shift value for nucleus x (e.g., 1H or 13C) and and are the calculated isotropic constants for nucleus x and tetramethylsilane (TMS), respectively. These calculated chemical shifts of 12a, 12b, 12c, and 12d were averaged based on their Boltzmann populations and used for calculations of the DP4 analysis by employing an applet available at http://www-jmg.ch.cam.ac.uk/tools/nmr/.
3.6. Cell Proliferation Assay and Antibacterial Activity Assay
The antiproliferative activities of the compounds against cancer cells were measured using the sulforhodamine B (SRB) method as described previously [41]. HUVEC growth was assessed by the MTT assay as described previously [42]. Antimicrobial and enzyme-inhibition assays were performed according to a method described previously [43].
3.7. Tube Formation Assay
The tube formation assay was conducted as described previously [42]. Briefly, HUVECs in the presence of various concentrations of the test compounds and VEGF (50 ng/mL) were seeded in each well of a Matrigel-coated 96-well plate. The cells were further incubated for 6 h and visualized under an inverted microscope (Olympus Optical Co. Ltd., Tokyo, Japan). The tube formation activity was calculated using the following equation: tube formation (%) = 100 × (average tube number of sample—average tube number of VEGF-control)/(average tube number of VEGF+control—average tube number of VEGF-control). The length of tubular structures was analyzed using the Angiogenesis Image Analyzer in ImageJ software (Bethesda, MD, USA).
3.8. Western Blotting
PC3 cells were treated with indicated concentrations of compound 4 for 24 h. Western blotting was conducted as described previously [41].
3.9. Cell Cycle Analysis
PC3 cells were seeded and incubated for 24 h and then starved in serum-free medium overnight. The medium was replenished, and compound 4 were treated for 18 h. After the cells were collected and fixed with 70% ethanol, they were stained with propidium iodide (PI) mixed with RNase. The stained cells were analyzed by a FACSCalibur flow cytometry, and the results are presented as histograms using CELLQuest 3.0.1 software (BD Biosciences, CA, USA).
4. Conclusions
Twelve new sesterterpenes were isolated and structurally elucidated from the tropical sponge Hyrtios erectus. The configurations of stereogenic centers were determined by combined spectroscopic (NOESY) and computational analyses (ECD, DP4). The new compounds exhibited weak antiproliferation against diverse cancer cell lines. In addition, the mechanistic study showed that the antiproliferative activity of compound 4 was related to G0/G1 arrest in cell cycle.
Supplementary Materials
The following are available online at https://www.mdpi.com/1660-3397/18/5/253/s1, Tables S1–S2: The NMR table of compounds 3–8, 10, and 11; Table S3: Results of antibacterial tests; Table S4: Isolated amount of each compound; Figures S1–S76: 1-D and 2-D NMR spectra of 1-12; Figure S77: The DP4 analyses of 12; Figure S78: Isolated known compounds from Hyrtios erectus; Figure S79: Calculated and experimental ECD spectra of 4, 8 and 13.
Author Contributions
O.-S.K. and C.-K.K. carried out the isolation and structural elucidation; D.K. and S.K.L. performed antiproliferation bioassays; J.S. and K.-B.O. performed antimicrobial bioassays; C.J.S. identified the sponge; J.S. and D.-C.O. reviewed and evaluated all data; J.S. and K.-B.O. supervised the research work and prepared the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This researcch was supported by the National Research Foundation (NRF, grant No. 2018R1A4A1021703) funded by the Ministry of Science, ICT and Future Planning, Korea.
Acknowledgments
We thank the National Center for Inter-university Research Facilities (NCIRF), Korea, for providing mass spectrometric data. Particular thanks are given to the Department of Marine Resources, State of Chuuk, Federated States of Micronesia, for allowing this marine organism research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Liu, Y.; Wang, L.; Jung, J.H.; Zhang, S. Sesterterpenoids. Nat. Prod. Rep. 2007, 24, 1401–1429. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, B.; Lin, X.P.; Zhou, X.F.; Liu, Y. Sesterterpenoids. Nat. Prod. Rep. 2013, 30, 455–473. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.A. Scalarane sesterterpenoids. Curr. Bioact. Compd. 2010, 6, 178–206. [Google Scholar] [CrossRef] [Green Version]
- Fattorusso, E.; Magno, S.; Santacroce, C.; Sica, D. Scalarin, a new pentacyclic C-25 terpenoid from the sponge. Cacospongia scalaris. Tetrahedron 1972, 28, 5993–5997. [Google Scholar] [CrossRef]
- Cimino, G.; De Stefano, S.; Minale, L. Scalaradial, a third sesterterpene with the tetracarbocyclic skeleton of calarin, from the sponge. Cacoscpongia mollior. Experientia 1974, 30, 846–847. [Google Scholar] [CrossRef]
- Bergquist, R.R.; Wells, R.J. Marine Natural Products: Chemical and Biological Perspectives; Scheuer, P.J., Ed.; Academic Press: London, UK, 1983; Chapter 1; Volume 5, pp. 1–50. [Google Scholar]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Natural product reports. Nat. Prod. Rep. 2019, 36, 122–173. [Google Scholar] [CrossRef] [Green Version]
- Evidente, A.; Kornienko, A.; Lefranc, F.; Cimmino, A.; Dasari, R.; Evidente, M.; Mathieu, V.; Kiss, R. Sesterterpenoids with anticancer activity. Curr. Med. Chem. 2015, 22, 3502–3522. [Google Scholar] [CrossRef] [Green Version]
- Mehbub, M.F.; Perkins, M.V.; Zhang, W.; Franco, C.M.M. New marine natural products from sponges (porifera) of the order Dictyoceratida (2001 to 2012); a promising source for drug discovery, exploration and future prospects. Biotechnol. Adv. 2016, 34, 473–491. [Google Scholar] [CrossRef]
- Maximo, P.; Lourenco, A. Marine sesterterpenes: An overview. Curr. Org. Chem. 2018, 22, 2381–2393. [Google Scholar] [CrossRef]
- Cafieri, F.; De Napoli, L.; Fattorusso, E.; Santacroce, C.; Sica, D. Molliorin-A unique scalarin-like pyrroloterpene from the sponge. Cacospongia mollior. Tetrahedron Lett. 1977, 18, 477–480. [Google Scholar] [CrossRef]
- Cafieri, F.; De Napoli, L.; Fattorusso, E.; Santacroce, C. Molliorin-B, a second scalarin-like pyrroloterpene from the sponge. Cacospongia mollior. Experientia 1977, 33, 994–995. [Google Scholar] [CrossRef] [PubMed]
- Cafieri, F.; De Napoli, L.; Iengo, A.; Santacroce, C. Molliorin-c, a further pyrroloterpene present in the sponge Cacospongia mollior. Experientia 1978, 34, 300–301. [Google Scholar] [CrossRef]
- Jeon, J.e.; Bae, J.; Lee, K.J.; Oh, K.-B.; Shin, J. Scalarane sesterterpenes from the sponge Hyatella sp. J. Nat. Prod. 2011, 74, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Festa, C.; Cassiano, C.; D’Auria, M.V.; Debitus, C.; Monti, M.C.; De Marino, S. Scalarane sesterterpenes from Thorecidae sponges as inhibitors of TDP-43 nuclear factor. Org. Biomol. Chem. 2014, 12, 8646–8655. [Google Scholar] [CrossRef]
- Yang, I.; Lee, J.; Lee, J.; Hahn, D.; Chin, J.; Won, D.H.; Ko, J.; Choi, H.; Hong, A.; Nam, S.J.; et al. Scalalactams A-D, scalarane sesterterpenes with a γ-lactam moiety from a Korean Spongia sp. marine sponge. Molecules 2018, 23, 3187. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Kim, S.H.; Choi, H.; Lee, H.S.; Lee, J.S.; Shin, H.J.; Lee, J. Cytotoxic furan- and pyrrole-contatining scalarane sesterterpenoids isolated from the sponge. Scalarispongia sp. Molecules 2019, 24, 840. [Google Scholar] [CrossRef] [Green Version]
- Elhady, S.S.; El-Halawany, A.M.; Alahdal, A.M.; Hassanean, H.A.; Ahmed, S.A. A new bioactive metabolite isolated from the red sea marine sponge Hyrios erectus. Molecules 2016, 21, 82. [Google Scholar] [CrossRef] [Green Version]
- Shady, N.H.; El-Hossary, E.M.; Fouad, M.A.; Gulder, T.A.M.; Kamel, M.S.; Abdelmohsen, U.R. Bioactive natural products of marine sponges from the genus Hyrtios. Molecules 2017, 22, 781. [Google Scholar] [CrossRef]
- Novanna, M.; Ethiraj, K.R.; Kannadasan, S. An overview of synthesis of indole alkaloids and biological activities of secondary metabolites isolated from Hyrtios species. Mini. Rev. Med. Chem. 2019, 19, 194–205. [Google Scholar] [CrossRef]
- Williamson, R.T.; Buevich, A.V.; Martin, G.E.; Parella, T.J. LR-HSQMBC: A sensitive NMR technique to probe very long-range heteronuclear coupling pathways. J. Org. Chem. 2014, 79, 3887–3894. [Google Scholar] [CrossRef]
- Sato, A.; Sugano, M.; Morishita, T.; Hatake, T.; Sasagawa, K.; Kobayashi, T. New Scalarane Derivative. JP 7,305,595A, 15 October 1996. [Google Scholar]
- Tsuchiya, N.; Sato, A.; Hata, T.; Sato, N.; Sasagawa, K.; Kobayashi, T. Cytotoxic scalarane sesterterpenes from a sponge, Hyrtios erecta. J. Nat. Prod. 1998, 61, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Yong, K.W.L.; Mudianta, I.W.; Cheney, K.L.; Mollo, E.; Blanchfield, J.T.; Garson, M.J. Isolation of norsesterterpenes and spongian diterpenes from Dorisprismatica (= Glossodoris) atromarginata. J. Nat. Prod. 2015, 78, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Youssef, D.T.A.; Yamaki, R.K.; Kelly, M.; Scheuer, P.J. Salmahyrtisol A, a Novel cytotoxic sesterterpne from the red sea sponge Hyrtios erecta. J. Nat. Prod. 2002, 65, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Mahidol, C.; Prawat, H.; Sangpetsiripan, S.; Ruchirawat, S. Bioactive scalaranes from the thai sponge Hyrtios gumminae. J. Nat. Prod. 2009, 72, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Tseng, S.W.; Liu, L.L.; Chou, Y.; Ho, Y.S.; Lu, M.C.; Su, J.H. Cytotoxic sesterterpenoids from a sponge Hippospongia sp. Mar. Drugs 2012, 10, 987–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M.; Urosa, A.; Marcos, I.S.; Diez, D.; Padron, J.M.; Basabe, P. Biomimetic synthesis of two salmahyrtisanes: Salmahyrtisol A and hippospongide A. J. Org. Chem. 2015, 80, 4566–4572. [Google Scholar] [CrossRef]
- Capon, R.J. Extracting value: Mechanistic insights into the formation of natural product artifacts—Case studies in marine natural products. Nat. Prod. Rep. 2020, 37, 55–79. [Google Scholar] [CrossRef]
- Kazlauskas, R.; Murphy, P.T.; Quinn, R.J.; Wells, R.J. Heteronemin, a new scalarin type sesterterpene from the sponge Heteronema erecta. Tetrahedron Lett. 1976, 30, 2631–2634. [Google Scholar] [CrossRef]
- Kashman, Y.; Rudi, A. The 13C-NMR spectrum and stereochemistry of heteronemin. Tetrahedron 1977, 33, 2997–2998. [Google Scholar] [CrossRef]
- Ledroit, V.; Debitus, C.; Ausseil, F.; Raux, R.; Menou, J.-L.; Hill, B. Heteronemin as a protein farnesyl transferase inhibitor. Pharm. Biol. 2004, 42, 454–456. [Google Scholar] [CrossRef]
- Pettit, G.R.; Tan, R.; Melody, N.; Cichacz, Z.A.; Herald, D.L.; Hoard, M.S.; Pettit, R.K.; Chapuis, J.-C. Antineoplastic agents 397: Isolation and structure of sesterstations 4 and 5 from Hyrtios erecta (The republic of maldives). Bioorg. Med. Chem. Lett. 1998, 8, 2093–2098. [Google Scholar] [CrossRef]
- Pettit, G.R.; Tan, R.; Cichacz, Z.A. Antineoplastic agents. 542. Isolation and structure of sesterstatin 6 from the indian ocean sponge Hyrtios erecta. J. Nat. Prod. 2005, 68, 1253–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyaoka, H.; Nishijima, S.; Mitome, H.; Yamada, Y. Three new scalarane sesterterpenoids from the okinawan sponge Hyrtios erectus. J. Nat. Prod. 2000, 63, 1369–1372. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, S.; Miura, S.; van Soest, R.W.M.; Ohta, T. Three new cytotoxic sesterterpenes from a marine sponge Spongia sp. J. Nat. Prod. 2003, 66, 438–440. [Google Scholar] [CrossRef] [PubMed]
- Kaweetripob, W.; Mahidol, C.; Tuntiwachwuttikul, P.; Ruchirawat, S.; Prawat, H. Cytotoxic sesterterpenes from thai marine sponge Hyrtios erectus. Mar. Drugs 2018, 16, 474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.-G.; Bi, K.-S.; Guo, Y.-W. Hyrtiosins A-E, five new scalarane sesterterpenes from the south china sea sponge Hyrtios erecta. Helv. Chim. Acta. 2005, 88, 1004–1009. [Google Scholar] [CrossRef]
- Ryu, G.; Matsunaga, S.; Fusetani, N. Three new cytotoxic sesterterpenes from the marine sponge Hyrtios cf. erectus. J. Nat. Prod. 1996, 59, 515–517. [Google Scholar] [CrossRef]
- Keller, C. Die Spongienfauna des rothen Meeres (I. Hälfte). Z. wiss. Zool. 1889, 48, 311–405. [Google Scholar]
- Kim, D.; Wang, C.Y.; Hu, R.; Lee, J.Y.; Luu, T.-T.-T.; Park, H.-J.; Lee, S.K. Antitumor activity of vanicoside B isolated from Persicaria dissitiflora by targeting CDK8 in triple-negative breast cancer cells. J. Nat. Prod. 2019, 82, 3140–3149. [Google Scholar] [CrossRef]
- Yu, S.; Oh, J.; Li, F.; Kwon, Y.; Cho, H.; Shin, J.; Lee, S.K.; Kim, S. New scaffold for angiogenesis inhibitors discovered by targeted chemical transformations of wondonin batural products. ACS Med. Chem. Lett. 2017, 8, 1066–1071. [Google Scholar] [CrossRef]
- Kim, C.-K.; Woo, J.-K.; Kim, S.-H.; Cho, E.; Lee, Y.-J.; Lee, H.-S.; Sim, C.J.; Oh, D.-C.; Oh, K.-B.; Shin, J. Meroterpenoids from a Tropical Dysidea sp. sponge. J. Nat. Prod. 2015, 78, 2814–2821. [Google Scholar] [CrossRef] [PubMed]
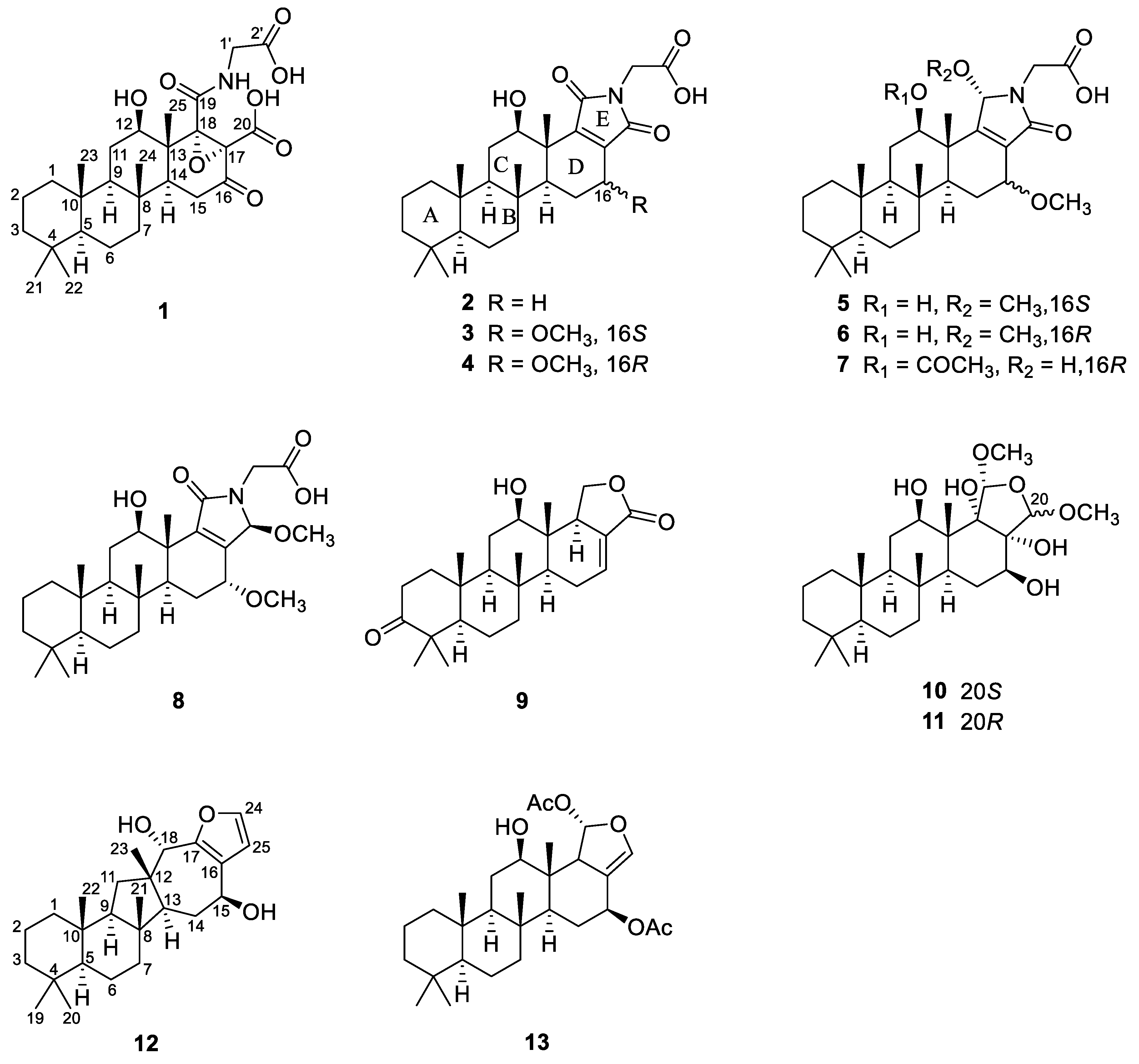
Chart 1.
Chemical structures of compounds 1–13.

Figure 1.
Key correlations of COSY (bold), HMBC (arrows) and LR-HSQMBC (red arrows) experiments for compounds 1, 9 and 12.
Figure 1.
Key correlations of COSY (bold), HMBC (arrows) and LR-HSQMBC (red arrows) experiments for compounds 1, 9 and 12.

Figure 2.
Key correlations of NOESY (blue arrows) experiments for compounds 1, 9 and 12.

Figure 3.
Calculated and experimental ECD spectra of 1.

Figure 4.
Key correlations of COSY (bold), HMBC (arrows), and NOESY (blue arrows) experiments for compounds 10 and 11.
Figure 4.
Key correlations of COSY (bold), HMBC (arrows), and NOESY (blue arrows) experiments for compounds 10 and 11.

Figure 5.
The results of DP4 probability analysis for compound 12.

Figure 6.
Effect of 4 on the regulation of cell cycle in PC3 cells. (A) Cell-cycle regulation measured by flow cytometry. The data are expressed as the mean values ± SD (n = 4) and are representative of four separate experiments; (B) Western blotting assay for PI3K/Akt/GSK-3β pathway and G0/G1 cell-cycle regulatory proteins. Relative intensity of indicated proteins was quantified using NIH ImageJ software.
Figure 6.
Effect of 4 on the regulation of cell cycle in PC3 cells. (A) Cell-cycle regulation measured by flow cytometry. The data are expressed as the mean values ± SD (n = 4) and are representative of four separate experiments; (B) Western blotting assay for PI3K/Akt/GSK-3β pathway and G0/G1 cell-cycle regulatory proteins. Relative intensity of indicated proteins was quantified using NIH ImageJ software.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
13C (150 MHz), 1H (600 MHz) NMR Assignments for Compounds 1 a and 2 b.
| Position | 1 | 2 | ||
|---|---|---|---|---|
| ppm, Type | δ, mult (J in Hz) | ppm, type | δ, mult (J in Hz) | |
| 1α | 41.5, CH2 | 0.86, m | 41.4, CH2 | 0.86, m |
| 1β | 1.71, br d (12.7) | 1.73, br d (12.1) | ||
| 2α | 20.1, CH2 | 1.43, m | 20.1, CH2 | 1.44, m |
| 2β | 1.66, ddd (13.6, 3.4, 3.4) | 1.67, m | ||
| 3α | 43.8, CH2 | 1.18, ddd (13.5, 13.4, 3.8) | 43.7, CH2 | 1.16, m |
| 3β | 1.37, br d (12.9) | 1.38, br d (13.0) | ||
| 4 | 34.7, C | 34.7, C | ||
| 5 | 58.3, CH | 0.85, m | 58.3, CH | 0.87, m |
| 6α | 19.7, CH2 | 1.55, m | 19.9, CH2 | 1.61, m |
| 6β | 1.41, m | 1.48, m | ||
| 7α | 42.9, CH2 | 0.89, m | 43.1, CH2 | 0.97, m |
| 7β | 1.61, ddd (12.8, 2.9, 2.9) | 1.88, ddd (12.6, 2.9, 2.9) | ||
| 8 | 38.8, C | 39.0, C | ||
| 9 | 59.8, CH | 0.96, dd (12.7, 1.2) | 59.7, CH | 0.95, m |
| 10 | 38.9, C | 39.0, C | ||
| 11α | 29.0, CH2 | 1.77, ddd (13.1, 4.5, 1.2) | 26.8, CH2 | 1.81, ddd (13.1, 4.1, 1.7) |
| 11β | 1.54, m | 1.51, m | ||
| 12 | 74.7, CH | 3.96, dd (11.0, 4.4) | 76.8, CH | 3.71, dd (11.0, 4.5) |
| 13 | 45.6, C | 45.2, C | ||
| 14 | 47.3, CH | 1.80, dd (12.9, 4.8) | 56.4, CH | 1.18, m |
| 15a | 35.4, CH2 | 2.29, dd (18.5, 4.8) | 17.8, CH2 | 1.98, dd (13.3, 7.1) |
| 15β | 2.35, dd (18.5, 13.0) | 1.67, m | ||
| 16α | 204.2, C | 23.4, CH2 | 2.26, m | |
| 16β | 2.53, dd (19.8, 5.5) | |||
| 17 | 68.7, C | 144.1, C | ||
| 18 | 73.3, C | 150.8, C | ||
| 19 | 168.7, C | 175.1, C | ||
| 20 | 170.4, C | 171.5, C | ||
| 21 | 34.3, CH3 | 0.85, s | 34.2, CH3 | 0.86, s |
| 22 | 22.3, CH3 | 0.83, s | 22.2, CH3 | 0.84, s |
| 23 | 17.2, CH3 | 0.87, s | 17.1, CH3 | 0.90, s |
| 24 | 17.6, CH3 | 0.90, s | 18.3, CH3 | 0.93, s |
| 25 | 12.9, CH3 | 1.43, s | 17.8, CH3 | 1.15, s |
| 1’ | 43.9, CH2 | 4.15, dd (17.0, 7.4) 3.33, dd (17.0, 4.3) | 40.2, CH2 | 4.19, s |
| 2’ | 177.8, C | 171.8, C | ||
| 19-NH | 7.88, dd (7.4, 3.2) | |||
a,b Data were measured at MeOH-d3 and MeOH-d4, respectively.
Table 2.
13C (150 MHz), 1H NMR (600 MHz) Assignments for Compounds 9 a and 12 b.
| Position | 9 | 12 | ||
|---|---|---|---|---|
| ppm, Type | δ, mult (J in Hz) | ppm, type | δ, mult (J in Hz) | |
| 1α | 40.2, CH2 | 1.50, m | 42.8, CH2 | 1.17, ddd (13.6, 13.3, 3.4) |
| 1β | 1.98, m | 1.37, m | ||
| 2α | 34.8, CH2 | 2.47, m | 19.0, CH2 | 1.44, m |
| 2β | 2.53, m | 1.68, m | ||
| 3α | 220.3, C | 40.4, CH2 | 1.12, ddd (12.4, 12.4, 3.7) | |
| 3β | 1.41, m | |||
| 4 | 48.5, C | 33.3, C | ||
| 5 | 55.7, CH | 1.49, m | 58.0, CH | 0.93, dd (12.3, 2.4) |
| 6α | 20.1, CH2 | 1.62, m | 18.6, CH2 | 1.57, m |
| 6β | 1.52, m | 1.40, m | ||
| 7α | 41.6, CH2 | 1.02, m | 40.3, CH2 | 0.98, m |
| 7β | 1.81, br d (12.8) | 1.71, m | ||
| 8 | 38.2, C | 45.2, C | ||
| 9 | 58.9, CH | 1.06, m | 61.4, CH | 1.30, dd (13.4, 6.4) |
| 10 | 37.8, C | 37.1, C | ||
| 11α | 28.5, CH2 | 1.67, m | 34.9, CH2 | 1.89, m |
| 11β | 1.55, m | 1.37, m | ||
| 12 | 81.4, CH | 3.41, dd (11.4, 3.4) | 43.9, C | |
| 13 | 41.3, C | 49.3, CH | 1.82, br d (11.3) | |
| 14 | 54.3, CH | 1.34, dd (11.3, 5.7) | 32.9, CH2 | 1.90, m 1.70, m |
| 15a | 24.9, CH2 | 2.39, m | 69.8, CH | 4.66, dd (10.9, 6.1) |
| 15β | 2.27, m | |||
| 16 | 137.2, CH | 6.81, dd (6.8, 3.3) | 124.2, C | |
| 17 | 128.6, C | 150.2, C | ||
| 18 | 51.6, CH | 2.92, m | 76.1, CH | 4.46, s |
| 19 | 70.8, CH2 | 4.50, dd (9.5, 9.5)4.20, dd (9.5, 9.5) | 33.8, CH3 | 0.85, s |
| 20 | 172.4, C | 21.5, CH3 | 0.84, s | |
| 21 | 27.1, CH3 | 1.07, s | 16.8, CH3 | 0.81, s |
| 22 | 21.3, CH3 | 1.04, s | 15.5, CH3 | 0.85, s |
| 23 | 16.9, CH3 | 0.98, s | 25.5, CH3 | 1.01, s |
| 24 | 16.5, CH3 | 1.00, s | 141.4, C | 7.30, d (1.7) |
| 25 | 8.3, CH3 | 0.77, s | 111.5, C | 6.50, d (1.7) |
a, b Data were measured at MeOH-d4 and CDCl3, respectively.
Table 3.
Results of Antiproliferation and Anti-Angiogenesis Tests.
| Compound | IC50 (μM) | |||||||
|---|---|---|---|---|---|---|---|---|
| Antiproliferation a | Anti-Angiogenesis b | |||||||
| A549 | SNU638 | HCT116 | MDA- MB-231 | SK- Hep-1 | PC3 | Tube Formation | Cell Viability | |
| 1 | >20 | >20 | >20 | >20 | >20 | >20 | 5.4 | >10 |
| 2 | 18 | 12 | 18 | >20 | >20 | 14 | 4.9 | >10 |
| 3 | >20 | >20 | >20 | >20 | >20 | >20 | 7.2 | >10 |
| 4 | 5.4 | 5.5 | 9.7 | >20 | 13 | 5.9 | 2.9 | >10 |
| 5 | >20 | 16 | 19 | 18 | >20 | >20 | 12 | >10 |
| 6 | 18 | 19 | >20 | >20 | >20 | 19 | 6.2 | >10 |
| 7 | 6.9 | 8.9 | 12 | >20 | 11 | 5.2 | 4.3 | >10 |
| 8 | >20 | >20 | 16 | >20 | >20 | >20 | 9 | >10 |
| 9 | 10 | 5.8 | 10 | >20 | 8.1 | >20 | 17 | >10 |
| 10 | 9.8 | 5 | 8.9 | >20 | 18 | 6.7 | 13 | >10 |
| 11 | 12 | 11 | 14 | 17 | 17 | 15 | 14 | >10 |
| 12 | 7.1 | 5.5 | 4.5 | 7.5 | 11 | 7.8 | 6.9 | >10 |
| 13 | 0.48 | 0.3 | 0.41 | 0.17 | 0.46 | 0.25 | 0.27 | 0.59 |
| etoposide | 0.76 | 0.13 | 0.54 | 4 | 0.18 | >20 | ||
| sunitinib | 1.7 | 6 | ||||||
a A549 (human lung cancer cell), SNU638 (human stomach cancer cell), HCT116 (human colon cancer cell), and MDA-MB-231 (human breast cancer cell), SK-Hep1 (human liver cancer cell), and PC3 (human prostate cancer cell); b Measured for PC3 cell line. Tube formation and viability were measured at 6 and 24 h, respectively.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kwon, O.-S.; Kim, D.; Kim, C.-K.; Sun, J.; Sim, C.J.; Oh, D.-C.; Lee, S.K.; Oh, K.-B.; Shin, J. Cytotoxic Scalarane Sesterterpenes from the Sponge Hyrtios erectus. Mar. Drugs 2020, 18, 253. https://doi.org/10.3390/md18050253
AMA Style
Kwon O-S, Kim D, Kim C-K, Sun J, Sim CJ, Oh D-C, Lee SK, Oh K-B, Shin J. Cytotoxic Scalarane Sesterterpenes from the Sponge Hyrtios erectus. Marine Drugs. 2020; 18(5):253. https://doi.org/10.3390/md18050253
Chicago/Turabian StyleKwon, Oh-Seok, Donghwa Kim, Chang-Kwon Kim, Jeongyoon Sun, Chung J. Sim, Dong-Chan Oh, Sang Kook Lee, Ki-Bong Oh, and Jongheon Shin. 2020. "Cytotoxic Scalarane Sesterterpenes from the Sponge Hyrtios erectus" Marine Drugs 18, no. 5: 253. https://doi.org/10.3390/md18050253
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.