Identifying Phlorofucofuroeckol-A as a Dual Inhibitor of Amyloid-β25-35 Self-Aggregation and Insulin Glycation: Elucidation of the Molecular Mechanism of Action



Abstract
:1. Introduction
2. Results
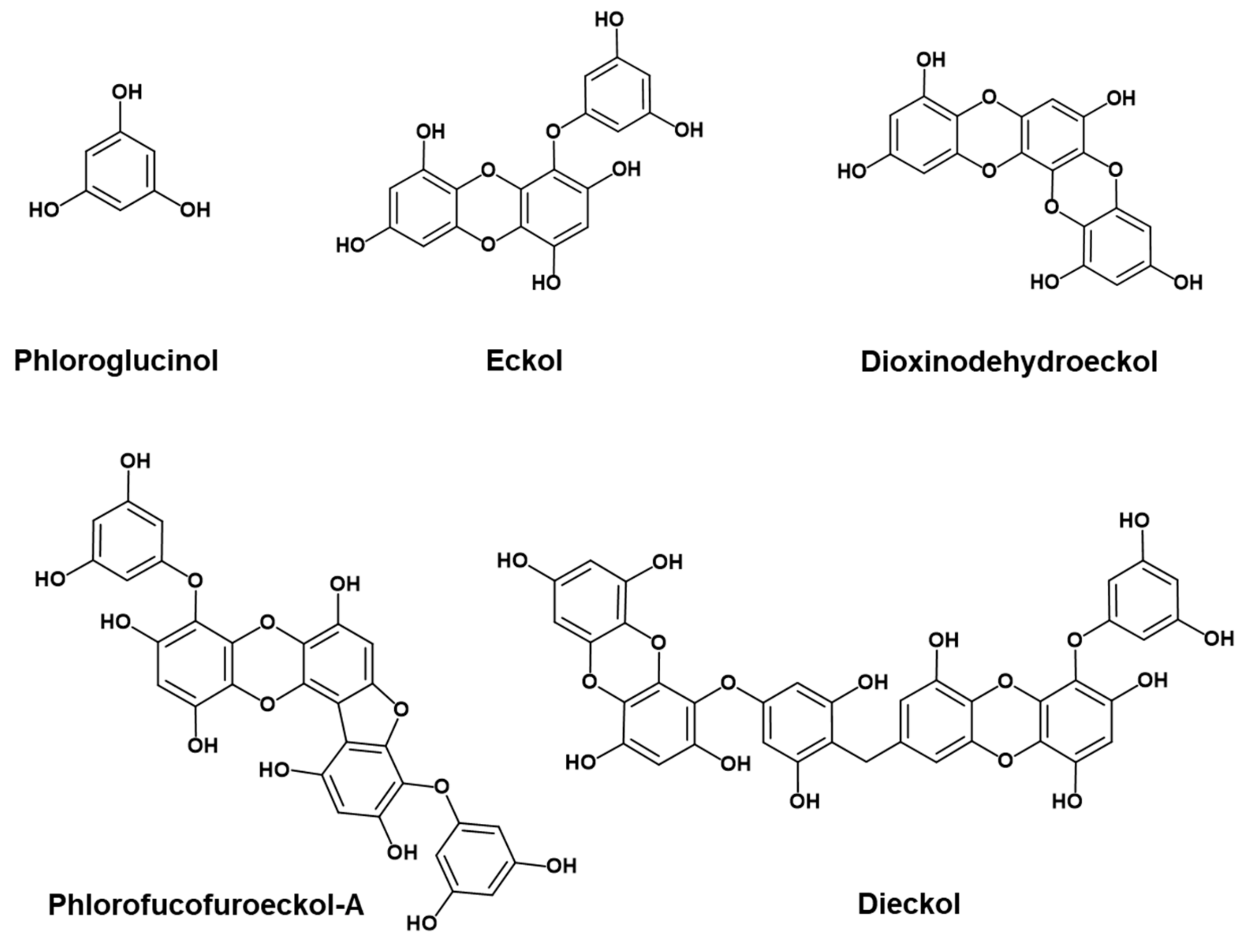
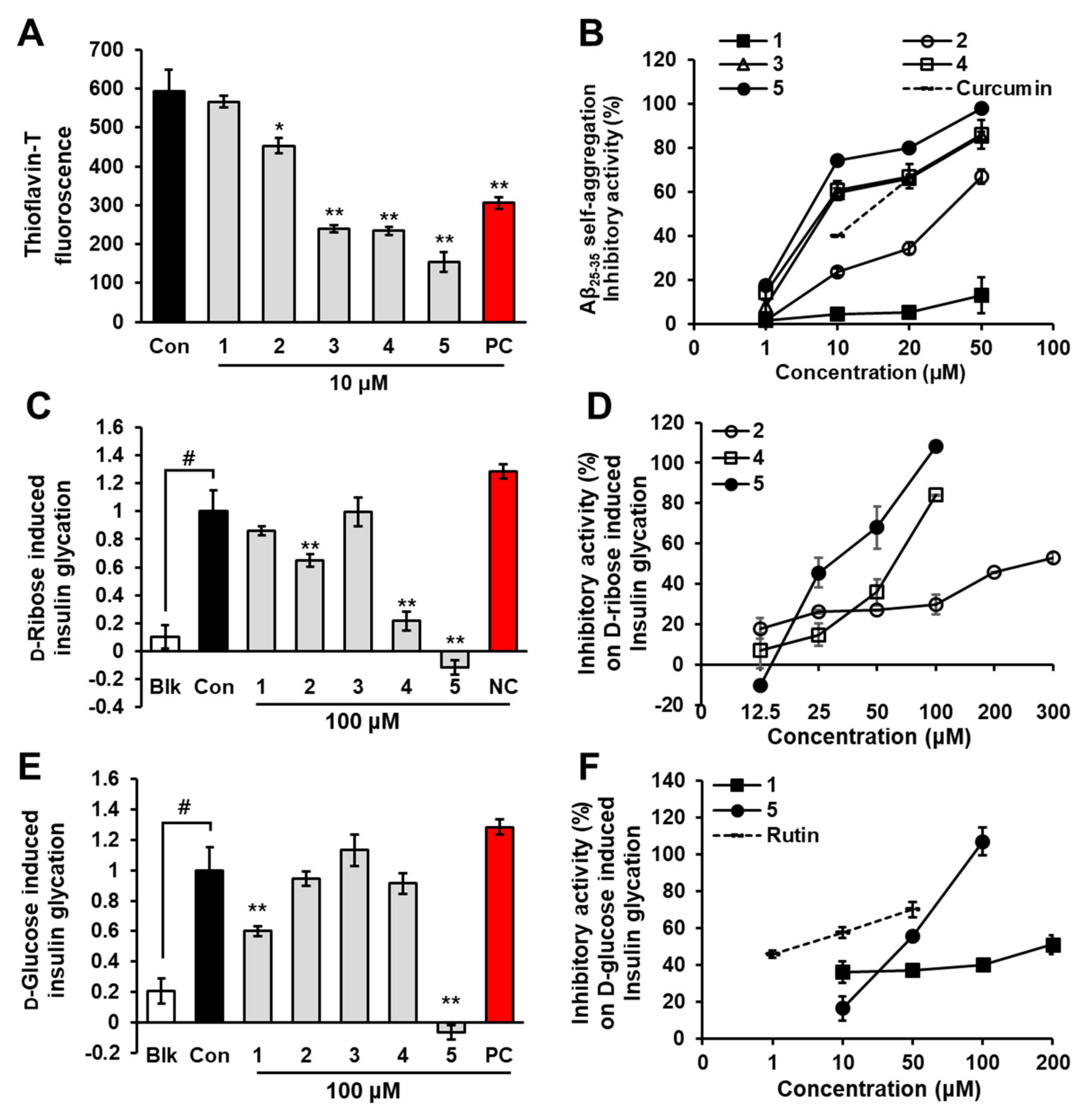
2.1. Inhibition of Aβ25-35 Self-Aggregation by Phlorotannins
2.2. Inhibition of Insulin Glycation by Phlorotannins
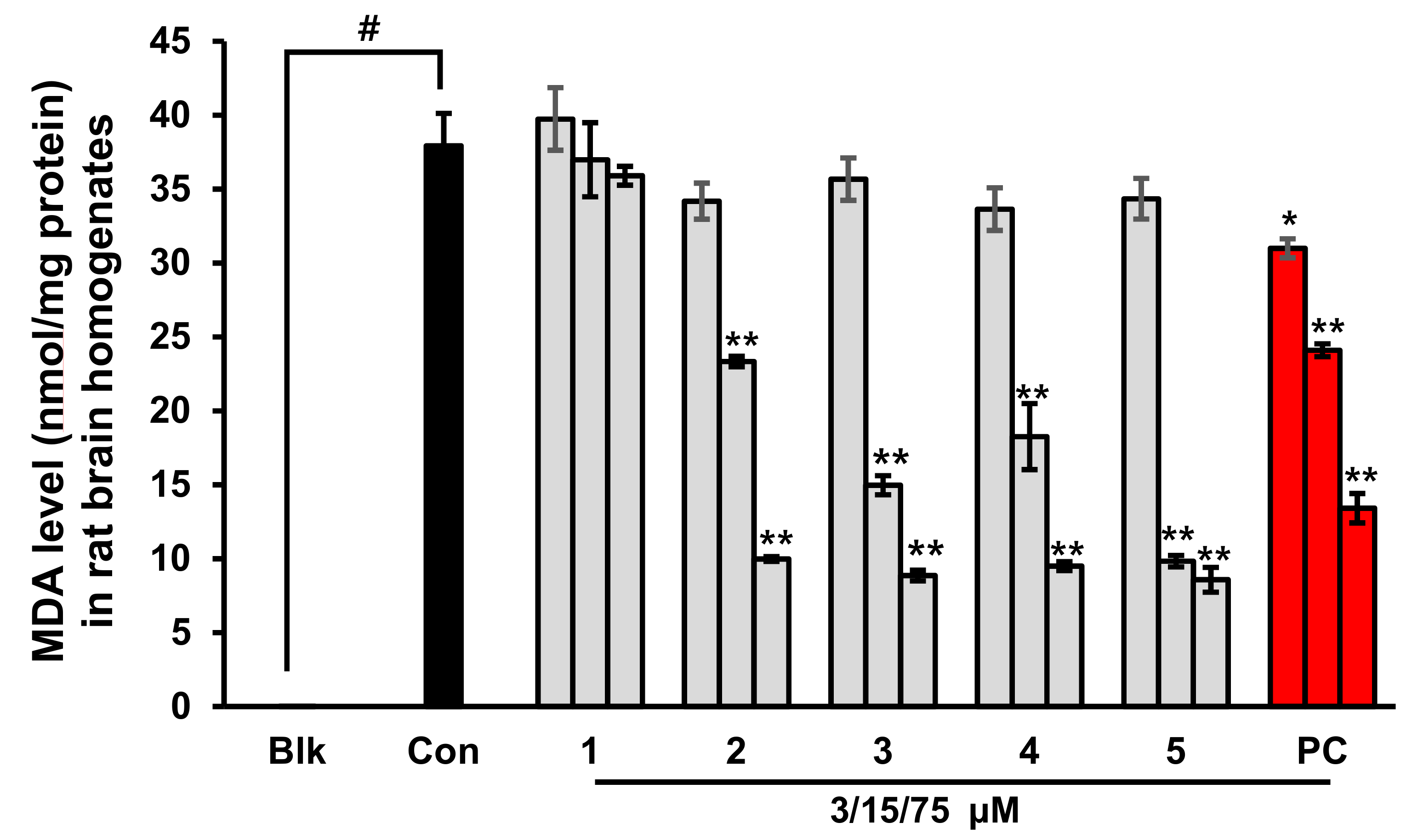
2.3. Prevention of Lipid Peroxidation in Whole Rat Brain Homogenates by Phlorotannins
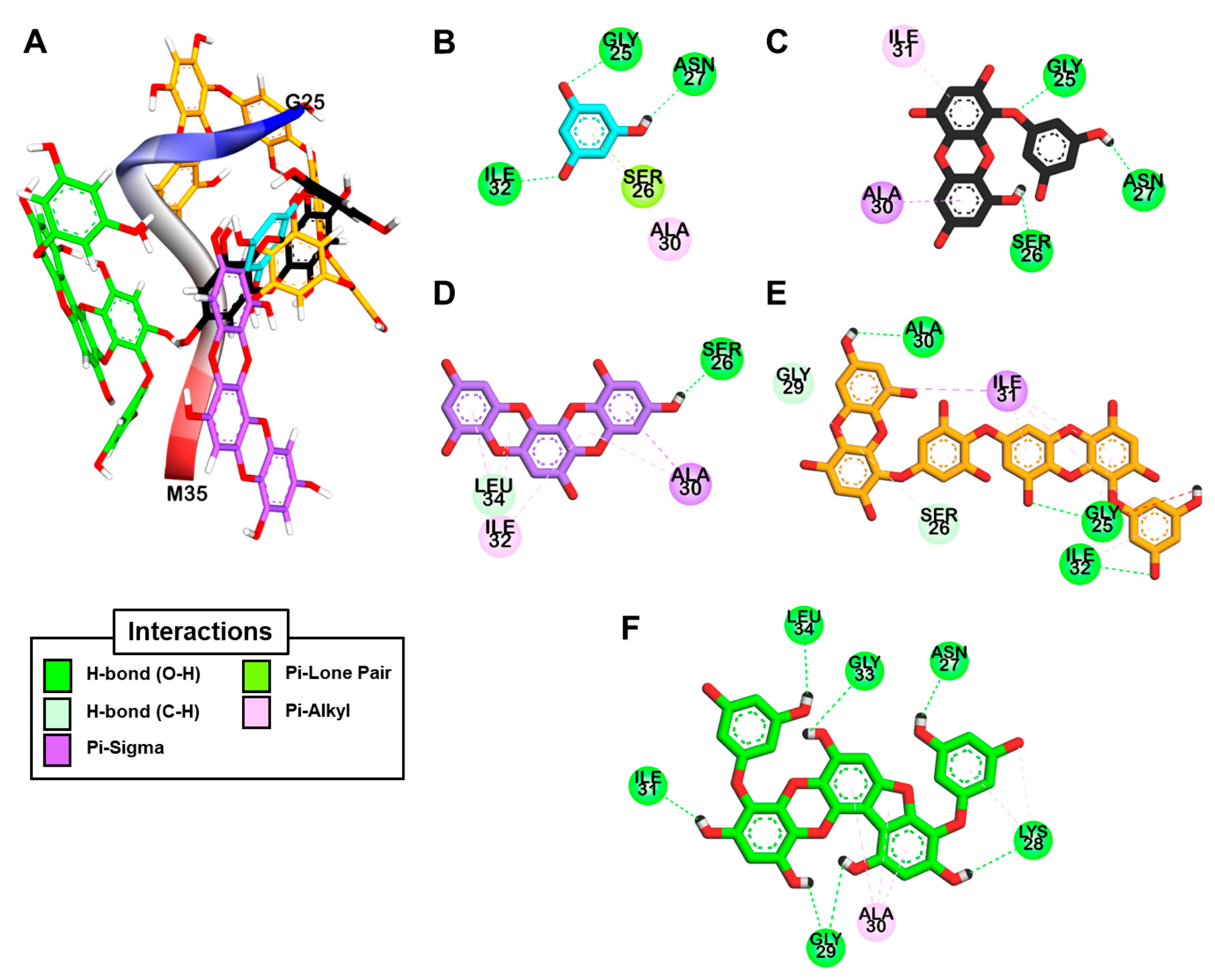
2.4. Docking Simulation for Phlorotannins on Aβ25-35
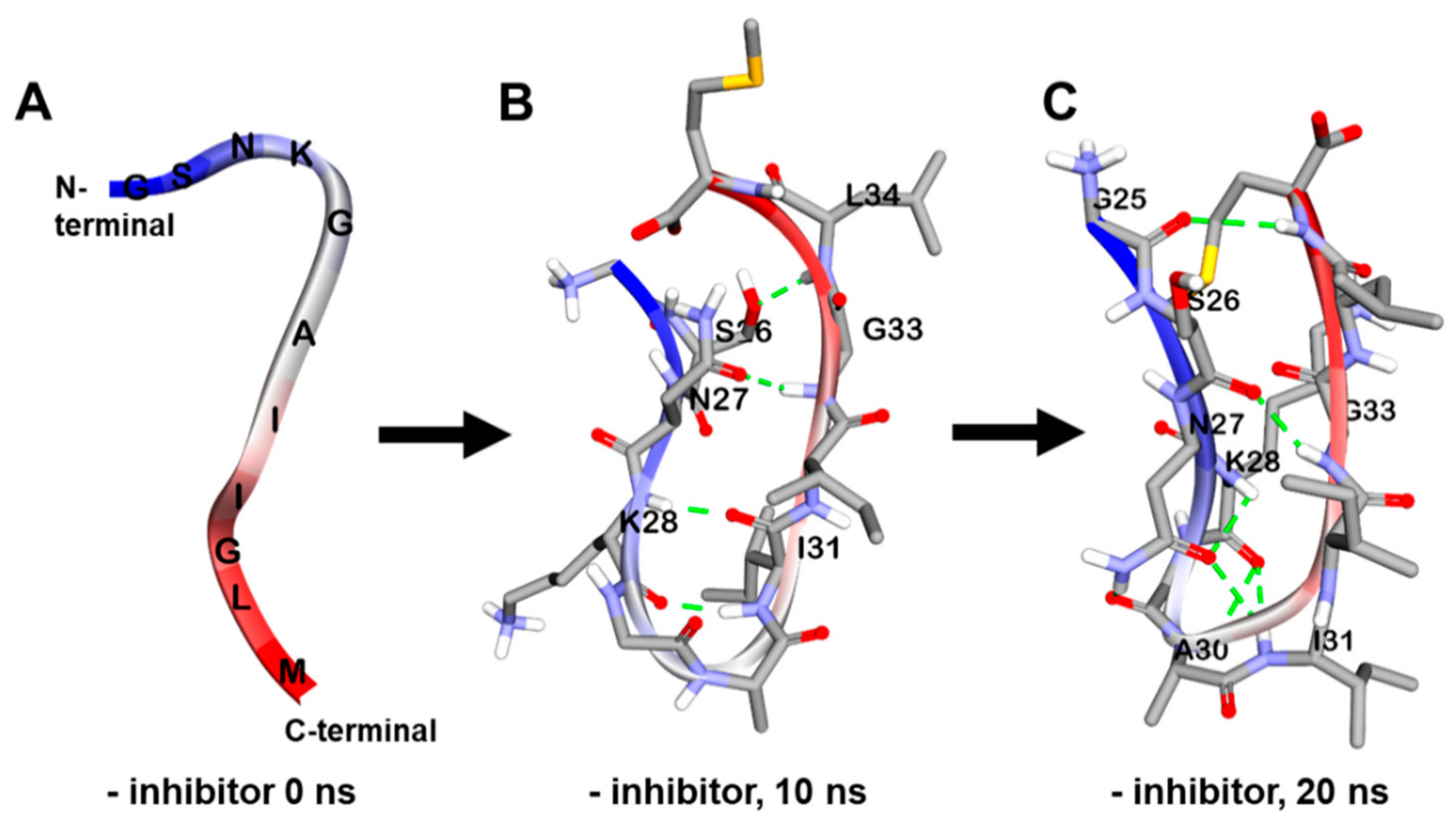
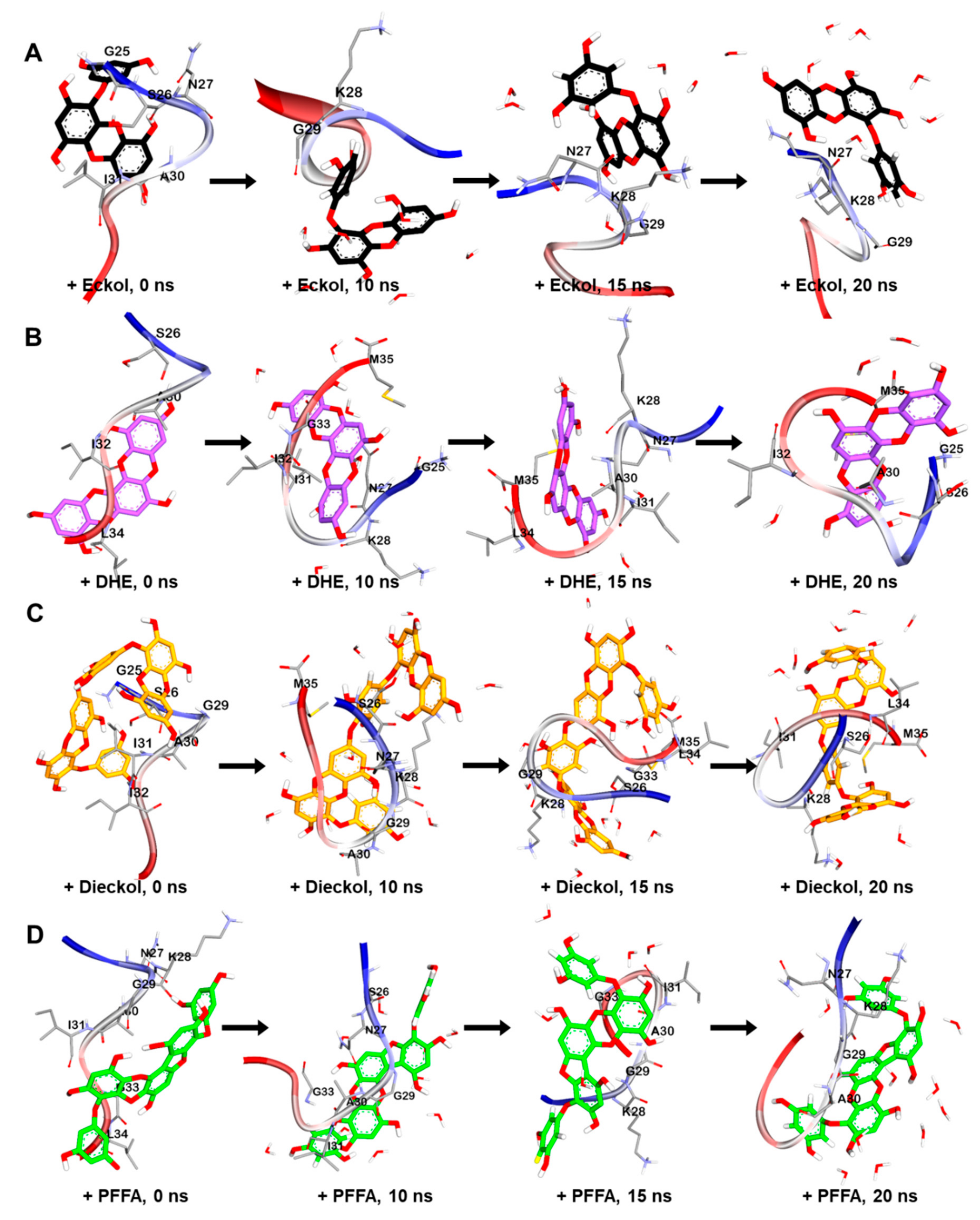
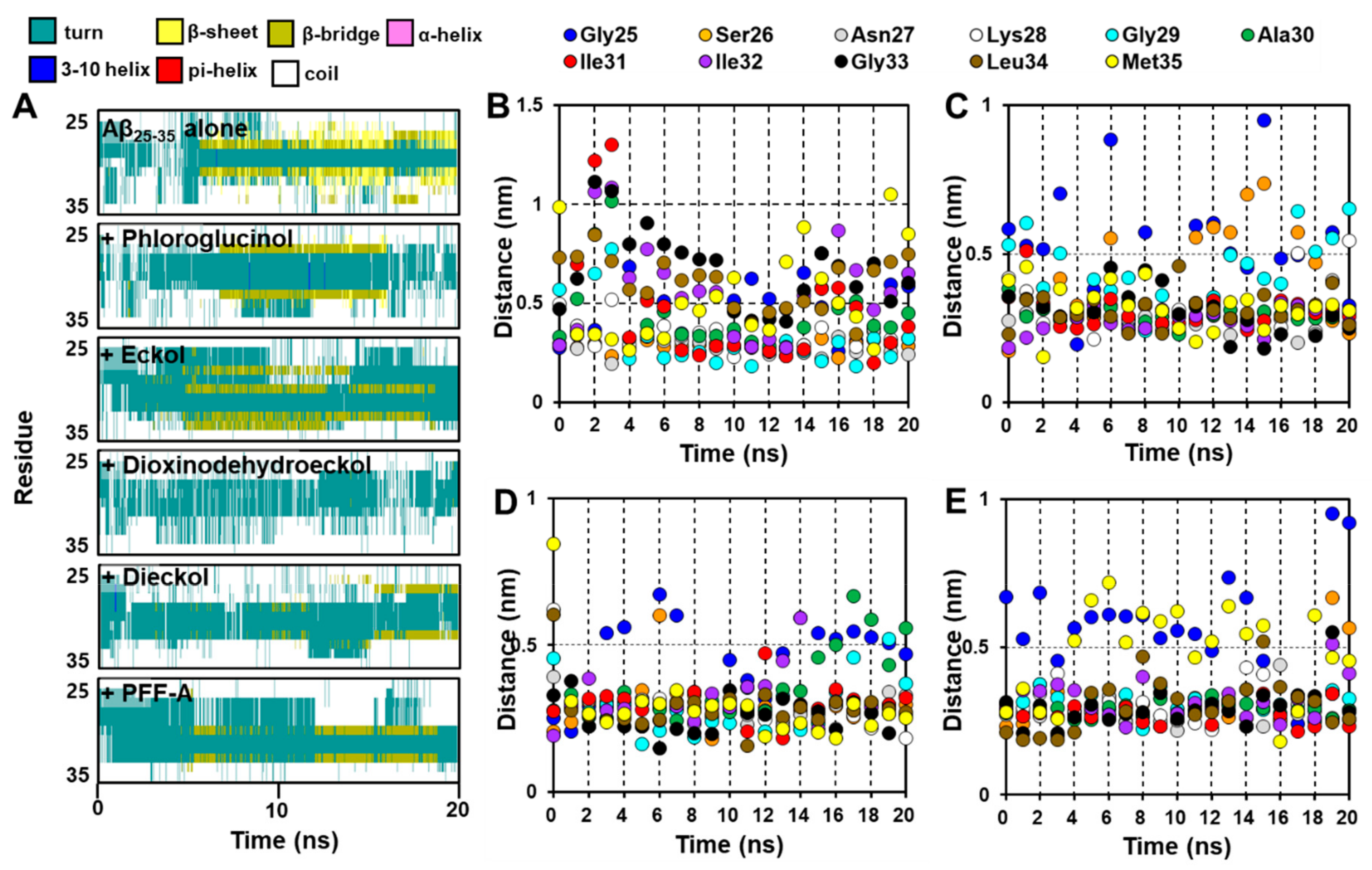
2.5. Dynamic Simulation of Phlorotannins Inhibiting Aβ25-35 Self-Aggregation
2.6. Docking Simulation for PFFA on Bovine Insulin
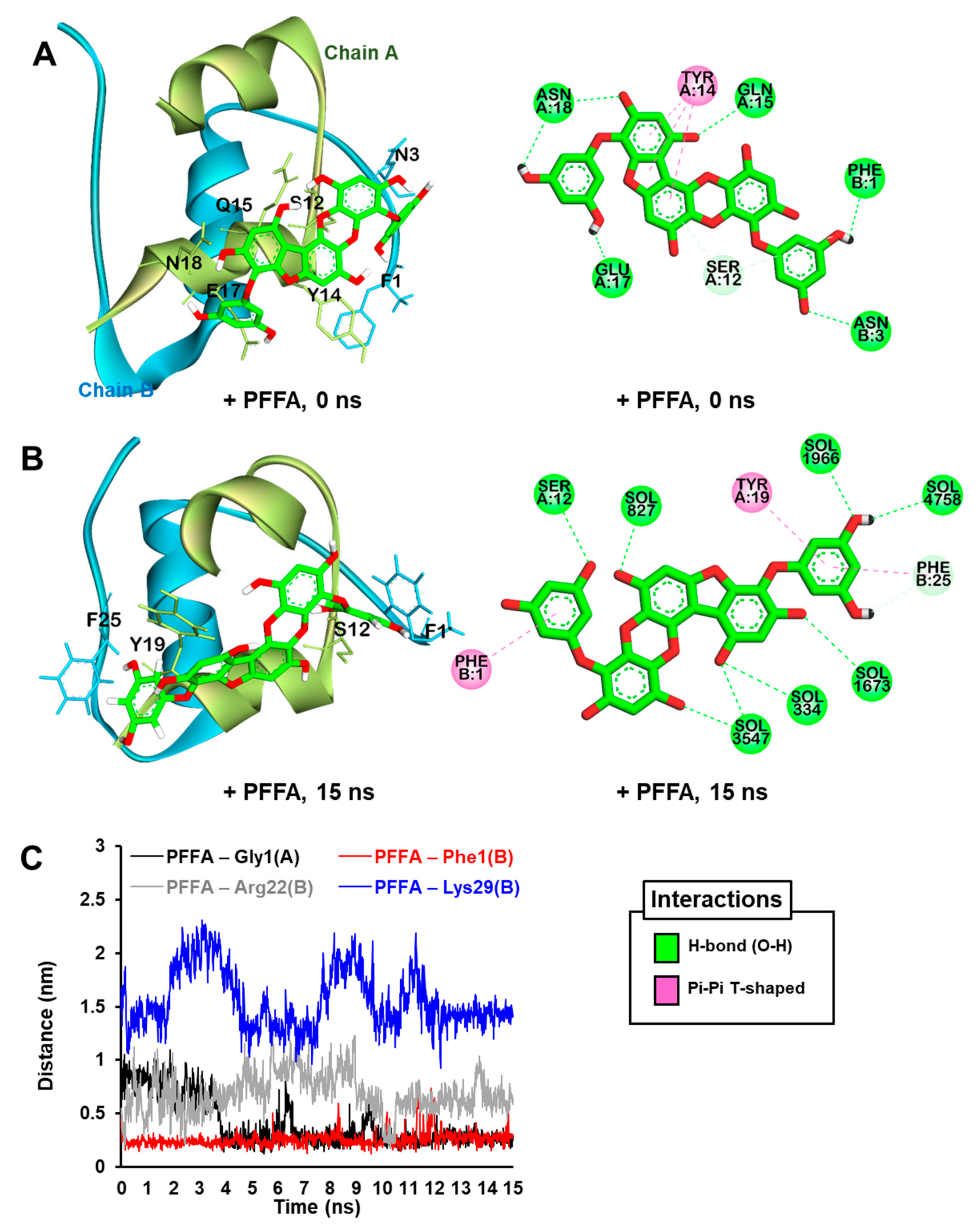
2.7. Dynamic Simulation of PFFA on Bovine Insulin
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Preparation of Phlorotannins
4.3. Assay for Aβ25-35 Self-Aggregation
4.4. Assay for Non-Enzymatic Insulin Glycation
4.5. Preparation of Rat Brain Homogenates
4.6. Lipid Peroxidation Assay
4.7. Molecular Docking Simulation
4.8. Molecular Dynamic simulation
4.9. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Cuanalo-Contreras, K.; Mukherjee, A.; Soto, C. Role of protein misfolding and proteostasis deficiency in protein misfolding diseases and aging. Int. J. Cell Biol. 2013, 2013, 638083. [Google Scholar] [CrossRef]
- Owen, M.C.; Gnutt, D.; Gao, M.; Wärmländer, S.K.; Jarvet, J.; Gräslund, A.; Winter, R.; Ebbinghaus, S.; Strodel, B. Effects of in vivo conditions on amyloid aggregation. Chem. Soc. Rev. 2019, 48, 3946–3996. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Kato, K.; Yanagisawa, K. Ganglioside-mediated assembly of amyloid β-protein: Roles in Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 2018, 156, 413–434. [Google Scholar] [PubMed]
- Ilie, I.M.; Caflisch, A. Simulation studies of amyloidogenic polypeptides and their aggregates. Chem. Rev. 2019, 119, 6956–6993. [Google Scholar] [CrossRef] [PubMed]
- Nasica-Labouze, J.; Nguyen, P.H.; Sterpone, F.; Berthoumieu, O.; Buchete, N.V.; Coté, S.; Simone, A.D.; Doig, A.J.; Faller, P.; Garcia, A.; et al. Amyloid β protein and Alzheimer’s disease: When computer simulations complement experimental studies. Chem. Rev. 2015, 115, 3518–3563. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.K.; Klimov, D.K. De novo aggregation of Alzheimer’s Aβ25-35 peptides in a lipid bilayer. Sci. Rep. 2019, 9, 7161. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Privitera, L.; Leznik, E.; Fà, M.; Staniszewski, A.; Palmeri, A.; Arancio, O. Picomolar amyloid-β positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 2008, 28, 14537–14545. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837. [Google Scholar] [CrossRef]
- Boyd, A.C.; Abdel-Wahab, Y.H.; McKillop, A.M.; McNulty, H.; Barnett, C.R.; O’Harte, F.P.; Flatt, P.R. Impaired ability of glycated insulin to regulate plasma glucose and stimulate glucose transport and metabolism in mouse abdominal muscle. Biochim. Biophys. Acta 2000, 1523, 128–134. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Borriello, M.; Carafa, V.; Altucci, L.; Vitiello, M.; Balestrieri, M.L.; Ricci, G.; Irace, G.; Sirangelo, I. D-ribose-glycation of insulin prevents amyloid aggregation and produces cytotoxic adducts. Biochim. Biophys. Acta 2016, 1862, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Fukami, K.; Yamagishi, S.; Ueda, S.; Okuda, S. Role of AGEs in diabetic nephropathy. Curr. Pharm. Des. 2008, 14, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W. AGEs and diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4867–4874. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, E.; Friess, U. Oxidative stress, AGE, and atherosclerosis. Kidney Int. Suppl. 2007, 106, S17–S26. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Chang, L.; Yan, L.J.; Zhao, B. Role of RAGE in alzheimer’s disease. Cell. Mol. Neurobiol. 2016, 36, 483–495. [Google Scholar] [CrossRef]
- Kim, A.R.; Shin, T.S.; Lee, M.S.; Park, J.Y.; Park, K.E.; Yoon, N.Y.; Kim, J.S.; Choi, J.S.; Jang, B.C.; Byun, D.S.; et al. Isolation and identification of phlorotannins from Ecklonia stolonifera with antioxidant and anti-inflammatory properties. J. Agric. Food Chem. 2009, 57, 3483–3489. [Google Scholar] [CrossRef]
- Artan, M.; Li, Y.; Karadeniz, F.; Lee, S.H.; Kim, M.M.; Kim, S.K. Anti-HIV-1 activity of phloroglucinol derivative, 6, 6′-bieckol, from Ecklonia cava. Bioorg. Med. Chem. 2008, 16, 7921–7926. [Google Scholar] [CrossRef]
- Kim, E.K.; Tang, Y.; Kim, Y.S.; Hwang, J.W.; Choi, E.J.; Lee, J.H.; Lee, S.H.; Jeon, Y.J.; Park, P.J. First evidence that Ecklonia cava-derived dieckol attenuates MCF-7 human breast carcinoma cell migration. Mar. Drugs 2015, 13, 1785–1797. [Google Scholar] [CrossRef]
- Manandhar, B.; Wagle, A.; Seong, S.H.; Paudel, P.; Kim, H.-R.; Jung, H.A.; Choi, J.S. Phlorotannins with potential anti-tyrosinase and antioxidant activity isolated from the marine seaweed Ecklonia stolonifera. Antioxidants 2019, 8, 240. [Google Scholar] [CrossRef]
- Kim, H.; Kong, C.S.; Lee, J.I.; Kim, H.; Baek, S.; Seo, Y. Evaluation of inhibitory effect of phlorotannins from Ecklonia cava on triglyceride accumulation in adipocyte. J. Agric. Food Chem. 2013, 61, 8541–8547. [Google Scholar] [CrossRef]
- Kang, M.C.; Wijesinghe, W.A.J.P.; Lee, S.H.; Kang, S.M.; Ko, S.C.; Yang, X.; Kang, N.; Jeon, B.T.; Kim, J.; Lee, D.H.; et al. Dieckol isolated from brown seaweed Ecklonia cava attenuates type ІІ diabetes in db/db mouse model. Food Chem. Toxicol. 2013, 53, 294–298. [Google Scholar] [CrossRef]
- Jung, H.A.; Roy, A.; Jung, J.H.; Choi, J.S. Evaluation of the inhibitory effects of eckol and dieckol isolated from edible brown alga Eisenia bicyclis on human monoamine oxidases A and B. Arch. Pharm. Res. 2017, 40, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Paudel, P.; Seong, S.H.; Wu, S.; Park, S.; Jung, H.A.; Choi, J.S. Eckol as a potential therapeutic against neurodegenerative diseases targeting dopamine D3/D4 receptors. Mar. Drugs 2019, 17, 108. [Google Scholar] [CrossRef] [PubMed]
- Seong, S.H.; Paudel, P.; Choi, J.W.; Ahn, D.H.; Nam, T.J.; Jung, H.A.; Choi, J.S. Probing multi-target action of phlorotannins as new monoamine oxidase inhibitors and dopaminergic receptor modulators with the potential for treatment of neuronal disorders. Mar. Drugs 2019, 17, 377. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.A.; Oh, S.H.; Choi, J.S. Molecular docking studies of phlorotannins from Eisenia bicyclis with BACE1 inhibitory activity. Bioorg. Med. Chem. Lett. 2010, 20, 3211–3215. [Google Scholar] [CrossRef] [PubMed]
- Yoon, N.Y.; Chung, H.Y.; Kim, H.R.; Choi, J.E. Acetyl-and butyrylcholinesterase inhibitory activities of sterols and phlorotannins from Ecklonia stolonifera. Fish. Sci. 2008, 74, 200–207. [Google Scholar] [CrossRef]
- Kang, I.J.; Jeon, Y.E.; Yin, X.F.; Nam, J.S.; You, S.G.; Hong, M.S.; Jang, B.G.; Kim, M.J. Butanol extract of Ecklonia cava prevents production and aggregation of beta-amyloid, and reduces beta-amyloid mediated neuronal death. Food Chem. Toxicol. 2011, 49, 2252–2259. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Yang, H.; Jeon, Y.J.; Lee, C.J.; Jin, Y.H.; Baek, N.I.; Kim, D.; Kang, S.M.; Yoon, M.; Yong, H.; et al. Phlorotannins of the edible brown seaweed Ecklonia cava Kjellman induce sleep via positive allosteric modulation of gamma-aminobutyric acid type A–benzodiazepine receptor: A novel neurological activity of seaweed polyphenols. Food Chem. 2012, 132, 1133–1142. [Google Scholar] [CrossRef]
- Iannuzzi, C.; Borriello, M.; Irace, G.; Cammarota, M.; Di Maro, A.; Sirangelo, I. Vanillin affects amyloid aggregation and non-enzymatic glycation in human insulin. Sci. Rep. 2017, 7, 15086. [Google Scholar] [CrossRef]
- Asgary, S.; Naderi, G.A.; Sarraf Zadegan, N.; Vakili, R. The inhibitory effects of pure flavonoids on in vitro protein glycosylation. J. Herb. Pharmacother. 2002, 2, 47–55. [Google Scholar] [CrossRef]
- Braughler, J.M.; Duncan, L.A.; Chase, R.L. The involvement of iron in lipid peroxidation. Importance of ferric to ferrous ratios in initiation. J. Biol. Chem. 1986, 261, 10282–10289. [Google Scholar]
- Pike, C.J.; Burdick, D.; Walencewicz, A.J.; Glabe, C.G.; Cotman, C.W. Neurodegeneration induced by beta-amyloid peptides in vitro: The role of peptide assembly state. J. Neurosci. 1993, 13, 1676–1687. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zheng, J.; Huang, C.; Zhao, J.; Lin, J.; Zhou, X.; Naman, C.B.; Wang, N.; Gerwick, W.H.; Wang, Q.; et al. Eckmaxol, a phlorotannin extracted from Ecklonia maxima, produces anti-β-amyloid oligomer neuroprotective effects possibly via directly acting on glycogen synthase kinase 3β. ACS Chem. Neurosci. 2018, 9, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.R.; Moon, H.E.; Kim, H.R.; Jung, H.A.; Choi, J.S. Neuroprotective effect of edible brown alga Eisenia bicyclis on amyloid beta peptide-induced toxicity in PC12 cells. Arch. Pharm. Res. 2012, 35, 1989–1998. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Youn, K.; Kim, D.H.; Ahn, M.R.; Yoon, E.; Kim, O.Y.; Jun, M. Anti-neuroinflammatory property of phlorotannins from Ecklonia cava on Aβ25-35-induced damage in PC12 cells. Mar. Drugs 2019, 17, 7. [Google Scholar] [CrossRef]
- Jarosz-Griffiths, H.H.; Noble, E.; Rushworth, J.V.; Hooper, N.M. Amyloid-β receptors: The good, the bad, and the prion protein. J. Biol. Chem. 2016, 291, 3174–3183. [Google Scholar] [CrossRef]
- Larini, L.; Shea, J.E. Role of β-hairpin formation in aggregation: The self-assembly of the amyloid-β (25-35) peptide. Biophys. J. 2012, 103, 576–586. [Google Scholar] [CrossRef]
- Millucci, L.; Ghezzi, L.; Bernardini, G.; Santucci, A. Conformations and biological activities of amyloid beta peptide 25-35. Curr. Protein Pept. Sci. 2010, 11, 54–67. [Google Scholar] [CrossRef]
- Pike, C.J.; Walencewicz-Wasserman, A.J.; Kosmoski, J.; Cribbs, D.H.; Glabe, C.G.; Cotman, C.W. Structure-activity analyses of β-amyloid peptides: Contributions of the β25–35 region to aggregation and neurotoxicity. J. Neurochem. 1995, 64, 253–265. [Google Scholar] [CrossRef]
- Reggiani, A.M.; Simoni, E.; Caporaso, R.; Meunier, J.; Keller, E.; Maurice, T.; Minarini, A.; Rosini, M.; Cavalli, A. In vivo characterization of ARN14140, a memantine/galantamine-based multi-target compound for Alzheimer’s disease. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Wei, G.; Shea, J.E. Effects of solvent on the structure of the Alzheimer amyloid-β (25-35) peptide. Biophys. J. 2006, 91, 1638–1648. [Google Scholar] [CrossRef]
- Man, V.H.; He, X.; Derreumaux, P.; Ji, B.; Xie, X.Q.; Nguyen, P.H.; Wang, J. Effects of all-atom molecular mechanics force fields on amyloid peptide assembly: The case of Aβ16−22 dimer. J. Chem. Theory Comput. 2019, 15, 1440–1452. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Derreumaux, P. Understanding amyloid fibril nucleation and aβ oligomer/drug interactions from computer simulations. ACC Chem. Res. 2014, 47, 603–611. [Google Scholar] [CrossRef]
- Doig, A.J.; Del Castillo-Frias, M.P.; Berthoumieu, O.; Tarus, B.; Nasica-Labouze, J.; Sterpone, F.; Nguyen, P.H.; Hooper, N.M.; Faller, P.; Derreumaux, P. Why is research on amyloid-β failing to give new drugs for Alzheimer’s disease. ACS Chem. Neurosci. 2017, 8, 1435–1437. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Lourenco, M.V.; Ferreira, S.T. How does brain insulin resistance develop in Alzheimer’s disease? Alzheimers Dement. 2014, 10, S26–S32. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Kuo, Y.M.; Huang, C.C.; Hsu, K.S. Insulin rescues amyloid β-induced impairment of hippocampal long-term potentiation. Neurobiol. Aging 2009, 30, 377–387. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Screening for type 2 diabetes. Diabetes Care 2002, 25, S21–S24. [Google Scholar] [CrossRef]
- Chen, X.; Su, T.; Chen, Y.; He, Y.; Liu, Y.; Xu, Y.; Wei, Y.; Li, J.; He, R. D-Ribose as a Contributor to Glycated Haemoglobin. EBioMedicine 2017, 25, 143–153. [Google Scholar] [CrossRef]
- O’Harte, F.P.; Hojrup, P.; Barnett, C.R.; Flatt, P.R. Identification of the site of glycation of human insulin. Peptides 1996, 17, 1323–1330. [Google Scholar] [CrossRef]
- Zoete, V.; Meuwly, M.; Karplus, M. Investigation of glucose binding sites on insulin. Proteins 2004, 55, 568–581. [Google Scholar] [CrossRef]
- Cini, M.; Fariello, R.Y.; Bianchettei, A.; Moretti, A. Studies on lipid peroxidation in the rat brain. Neurochem. Res. 1994, 19, 283–288. [Google Scholar] [CrossRef]
- Stocks, J.; Gutteridge, J.M.C.; Sharp, R.J.; Dormandy, T.L. Assay using brain homogenate for measuring the antioxidant activity of biological fluids. Clin. Sci. 1974, 47, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Wills, E.D. Mechanisms of lipid peroxide formation in animal tissues. Biochem. J. 1966, 99, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.H.; Yang, Z.; Yoon, B.; He, Y.; Uhm, S.; Shin, H.C.; Lee, B.H.; Yoo, Y.C.; Lee, K.B.; Han, S.Y.; et al. Blood-brain barrier-permeable fluorone-labeled dieckols acting as neuronal ER stress signaling inhibitors. Biomaterials 2015, 61, 52–60. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Liu, J.; Liang, J.; Liu, X.; Li, W.; Liu, Z.; Ding, Z.; Tuo, D. Towards improvements for penetrating the blood–brain barrier—recent progress from a material and pharmaceutical perspective. Cells 2018, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, J.; Kim, S.K.; Shim, M.S. Antimicrobial, antioxidant, and anticancer activities of biosynthesized silver nanoparticles using marine algae Ecklonia cava. Nanomaterials 2016, 6, 235. [Google Scholar] [CrossRef]
- Naldi, M.; Fiori, J.; Pistolozzi, M.; Drake, A.F.; Bertucci, C.; Wu, R.; Mlynarczyk, K.; Filipek, S.; De Simone, A.; Andrisano, V. Amyloid β-peptide 25-35 self-assembly and its inhibition: A model undecapeptide system to gain atomistic and secondary structure details of the Alzheimer’s disease process and treatment. ACS Chem. Neurosci. 2012, 3, 952–962. [Google Scholar] [CrossRef]
- Seong, S.H.; Ali, M.Y.; Jung, H.A.; Choi, J.S. Umbelliferone derivatives exert neuroprotective effects by inhibiting monoamine oxidase A, self-amyloidβ aggregation, and lipid peroxidation. Bioorg. Chem. 2019, 92, 10323. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated docking of flexible ligands: Applications of AutoDock. J. Mol. Recognit. 1996, 9, 1–5. [Google Scholar] [CrossRef]
- D’Ursi, A.M.; Armenante, M.R.; Guerrini, R.; Salvadori, S.; Sorrentino, G.; Picone, D. Solution structure of amyloid beta-peptide (25-35) in different media. J. Med. Chem. 2004, 12, 4231–4238. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Daura, X.; Mark, A.E.; Van Gunsteren, W.F. Parametrization of aliphatic CHn united atoms of GROMOS96 force field. J. Comput. Chem. 1998, 19, 535–547. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N_log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (μM) a | EC50 (μM) a | ||
|---|---|---|---|---|
| Aβ25-35 Aggregation | d-Ribose-Induced Insulin Glycation | d-Glucose-Induced Insulin Glycation | Lipid Peroxidation | |
| Phloroglucinol | >100 | >100 | >100 | >75 |
| Eckol | 34.36 ± 1.11 | 258.54 ± 10.81 | >100 | 38.64 ± 1.16 |
| Dioxinodehydroeckol | 8.31 ± 0.23 | >100 | >100 | 12.43 ± 1.50 |
| Dieckol | 7.93 ± 0.16 | 63.67 ± 3.83 | >100 | 15.48 ± 2.14 |
| Phlorofucofuroeckol-A | 6.18 ± 0.18 | 29.50 ± 0.53 | 43.55 ± 2.38 | 10.96 ± 0.16 |
| Curcumin b | 10.73 ± 1.40 | ‒ | ‒ | ‒ |
| Vanillin c | ‒ | >500 | ‒ | ‒ |
| Rutin b | ‒ | 5.19 ± 1.35 | ||
| Trolox b | ‒ | ‒ | ‒ | 49.01 ± 3.50 |
| Ligands | Binding Energy (kcal/mol) | Hydrogen Bonding Interactions | Other Interactions |
|---|---|---|---|
| Target protein: human Aβ25-35 | |||
| Phloroglucinol | −3.19 | Gly25, Asn27, Ile32 | Ala30 (Pi-Alkyl), Ser26 (Pi-Lone pair) |
| Eckol | −4.73 | Gly25, Ser26, Asn27 | Ile31 (Pi-Alkyl), Ala30 (Pi-sigma) |
| Dioxinodehydroeckol | −4.94 | Ser26, Leu34 | Ala30 (Pi-sigma, Pi-Alkyl), Ala30 (Pi-Alkyl), Ile32 (Pi-Alkyl) |
| Dieckol | −3.51 | Gly25, Ile32, Ala30, Ser26, Gly29 | Ile31 (Pi-sigma), Ile31 (Pi-Alkyl), Ile32 (Pi-Alkyl) |
| PFFA | −5.33 | Gly29, Lys28, Asn27, Ile31, Leu34, Gly33 | Ala30 (Pi-Alkyl) |
| Target protein: bovine insulin | |||
| PFFA | −5.03 | Ser12 (A), Gln15 (A), Glu17 (A), Asn18 (A), Asn3 (B), Phe1 (B) | Tyr14 (Pi-Amide stacked) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seong, S.H.; Paudel, P.; Jung, H.A.; Choi, J.S. Identifying Phlorofucofuroeckol-A as a Dual Inhibitor of Amyloid-β25-35 Self-Aggregation and Insulin Glycation: Elucidation of the Molecular Mechanism of Action. Mar. Drugs 2019, 17, 600. https://doi.org/10.3390/md17110600
Seong SH, Paudel P, Jung HA, Choi JS. Identifying Phlorofucofuroeckol-A as a Dual Inhibitor of Amyloid-β25-35 Self-Aggregation and Insulin Glycation: Elucidation of the Molecular Mechanism of Action. Marine Drugs. 2019; 17(11):600. https://doi.org/10.3390/md17110600
Chicago/Turabian StyleSeong, Su Hui, Pradeep Paudel, Hyun Ah Jung, and Jae Sue Choi. 2019. "Identifying Phlorofucofuroeckol-A as a Dual Inhibitor of Amyloid-β25-35 Self-Aggregation and Insulin Glycation: Elucidation of the Molecular Mechanism of Action" Marine Drugs 17, no. 11: 600. https://doi.org/10.3390/md17110600