Two New Cytotoxic Compounds from a Deep-Sea Penicillum citreonigrum XT20-134
by

Xi-Xiang Tang
1, 
Shun-Zhi Liu
2,
Xia Yan
3,
Bo-Wen Tang
2,
Mei-Juan Fang
2,
Xiu-Min Wang
2,
Zhen Wu
2,* and
Ying-Kun Qiu
2,* 1
Key Laboratory of Marine Biogenetic Resources, Third Institute of Oceanography State Oceanic Administration, Xiamen 361005, China
2
Fujian Provincial Key Laboratory of Innovative Drug Target Research, School of Pharmaceutical Sciences, Xiamen University, South Xiang-An Road, Xiamen 361102, China
3
Li Dak Sum Yip Yio Chin Kenneth Li Marine Biopharmaceutical Research Center, Ningbo University, Ningbo 315832, China
*
Authors to whom correspondence should be addressed.
Mar. Drugs 2019, 17(9), 509; https://doi.org/10.3390/md17090509
Submission received: 28 July 2019
/
Revised: 25 August 2019
/
Accepted: 27 August 2019
/
Published: 29 August 2019
(This article belongs to the Special Issue Deep-Sea Natural Products II)
Abstract
:Penicillum citreonigrum XT20-134 (MCCC 3A00956) is a fungus with cytotoxic activity, derived from deep-sea sediment. Five new compounds, adeninylpyrenocine (1), 2-hydroxyl-3-pyrenocine-thio propanoic acid (2), ozazino-cyclo-(2,3-dihydroxyl-trp-tyr) (3), 5,5-dichloro-1-(3,5-dimethoxyphenyl)-1,4-dihydroxypentan-2-one (4), and 2,3,4-trihydroxybutyl cinnamate (5), together with 19 known compounds (6–24), were isolated from an ethyl acetate (EtOAc) extract of its fermentation. The structures of the new compounds were comprehensively characterized by high-resolution electrospray ionization-mass spectrometry (HR-ESI-MS), 1D and 2D nuclear magnetic resonance (NMR). All isolates were evaluated for their cytotoxic activities. The heteroatom-containing new compounds 2 and 4 showed potent cytotoxicity to the human hepatoma tumor cell Bel7402 with IC50 values of 7.63 ± 1.46, 13.14 ± 1.41 μM and the human fibrosarcoma tumor cell HT1080 with IC50 values of 10.22 ± 1.32, 16.53 ± 1.67 μM, respectively.
1. Introduction
Considering the extreme environment of high salinity, darkness, high pressure, and high/low temperature [1], the discovery of new lead compounds from deep-sea microorganisms has become a hot topic in natural products research. Deep-sea fungi are attracting great interest because of their relatively large genome size, which may produce more second metabolites compared with bacteria. Cancer is the leading lethal disease in the world and deep-sea microorganism-originated compounds are thought to be the new anti-tumor drugs repository [2,3]. Recently, the new antitumor compounds diketopiperazine, cytochalasan alkaloids, chromone-derived polyketides, etc., have been isolated from deep-sea fungi [4,5,6].
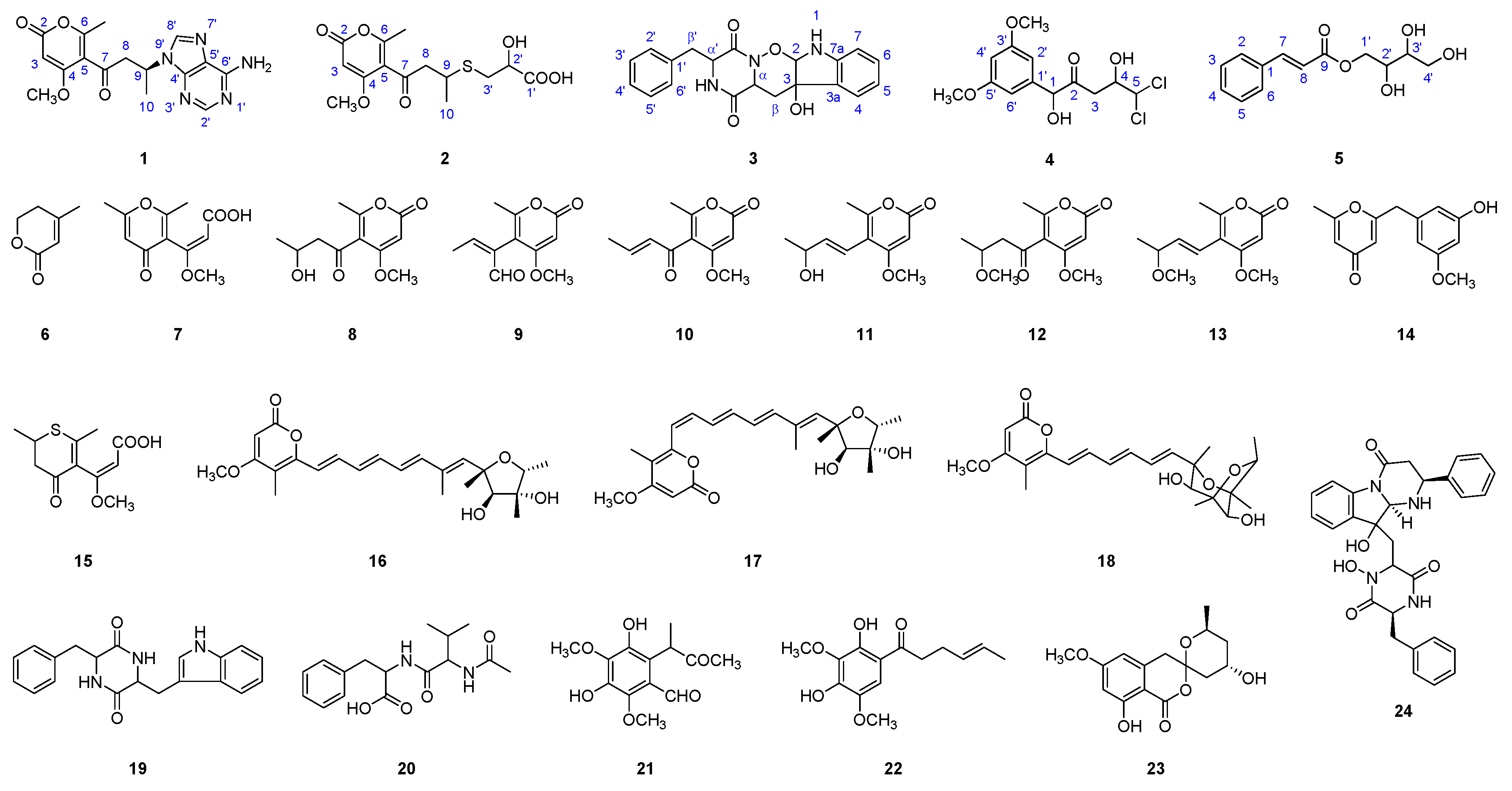
During the previous studies, several new cytotoxic compounds were characterized from deep-sea microbial resources [7,8,9,10,11]. In the present study, Penicillum citreonigrum XT20-134 (MCCC 3A00956), a fungal strain that originated from the deep-sea sediment in the southeast Indian Ocean, was found to possess cytotoxic activity. From its ethyl acetate (EtOAc) extract, five new compounds, adeninylpyrenocine (1), 2-hydroxyl-3-pyrenocine-thio propanoic acid (2), ozazino-cyclo-(2,3-dihydroxyl-trp-tyr) (3), 5,5-dichloro-1-(3,5-dimethoxyphenyl)-1,4-dihydroxypentan-2-one (4), and 2,3,4-trihydroxybutyl cinnamate (5) were isolated together with 19 known compounds (6–24) (Figure 1). The structures of the new compounds were comprehensively characterized by high-resolution electrospray ionization-mass spectrometry (HR-ESI-MS), 1D and 2D nuclear magnetic resonance (NMR). Their cytotoxic activities were studied. The new heteroatom-containing compounds 2 and 4 showed potent cytotoxicity to the human hepatoma tumor cell Bel7402 with IC50 values of 7.63 ± 1.46 and 13.14 ± 1.41 μM, and the human fibrosarcoma tumor cell HT1080 with IC50 values of 10.22 ± 1.32 and 16.53 ± 1.67 μM, respectively.
2. Results
2.1. Structural Identification of New Compounds
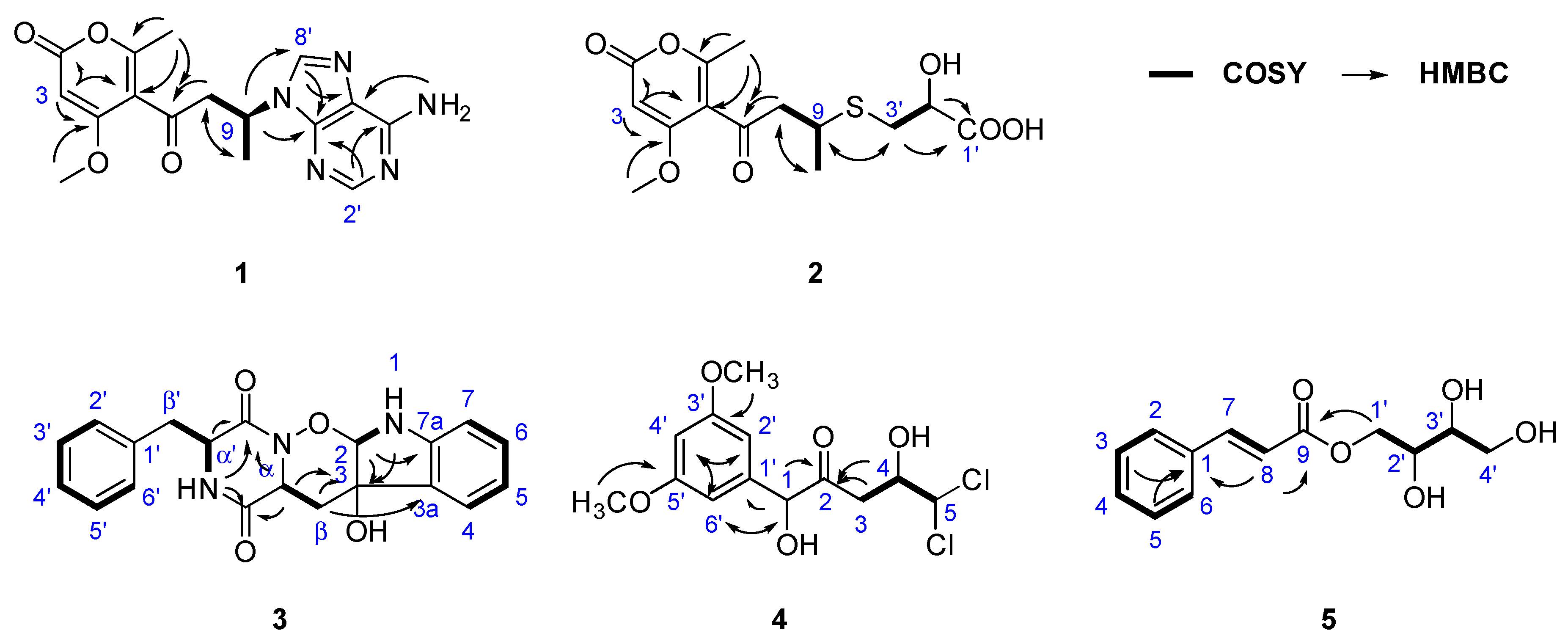
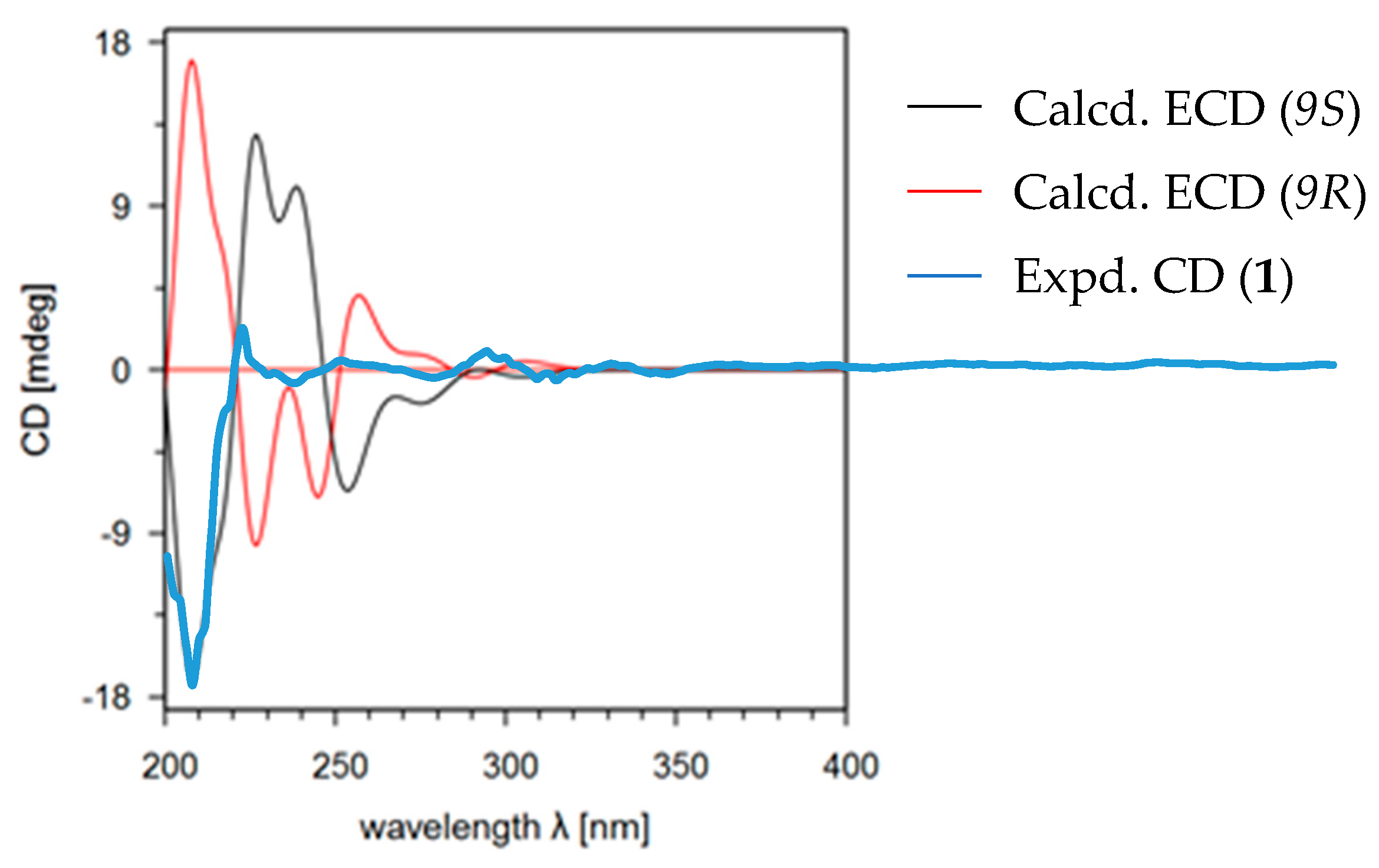
Adeninylpyrenocine (1) was isolated as a white amorphous powder. The infrared (IR) spectrum of 1 indicated the presence of conjugated ketone and conjugated lactone carbonyl signals at 1695 and 1642 cm−1, respectively. Its molecular formula of C16H17N5O4 was established by HR-ESI-MS at 344.1357 [M + H]+ (calcd. for 344.1353, C16H18N5O4). The unsaturation degree of 11 indicated the presence of heteroaromatic rings. With the aid of heteronuclear single quantum coherence (HSQC) spectra, three singlet signals in the low field of 1H NMR at δH 8.21 (1H, s, H-8’), 8.11 (1H, s, H-2’), and 5.62 (1H, s, H-3) were attributed to olefinic protons. Their corresponding carbon signals were found in 13C NMR at δC 140.3, 152.6, and 87.8, respectively. The signal at δH 7.16 (2H, br.s, 6’-NH2) should be assigned to the primary amino group due to the absence of carbon correlation in the HSQC spectrum. In the sp3 region of the 1H NMR, two singlet signals, belonging to a methoxyl at δH 3.74 (3H, s, 4-OCH3) and a methyl at δH 1.85 (3H, s, 6-CH3), could be found. In addition, a CH2–CH–CH3 fragment could be established, based on the ABXC3 spin system at δH [3.78 (1H dd, J = 17.6, 7.9 Hz) and 3.35 (1H dd, J = 17.6, 6.0 Hz), H-8], δH 5.06 (1H, m, H-9), and the doublet methyl signal and 1.53 (3H, d, J = 6.0 Hz, H-10). Considering the five nitrogen atoms in the molecular formula and the carbon signals at δC 156.4, 152.6, 149.5, 140.3, and 119.5, an adenine moiety is present in 1. With the help of distortionless enhancement by polarization transfer (DEPT-135) along 13C NMR, the four quaternary carbons at δC 156.4, 152.6, 149.5, 140.3, and the methine at δC 87.8 were attributed to an α-pyrone structure unit. In the 1H–1H homonuclear chemical shift correlation spectroscopy (COSY) spectrum, only the correlations in the CH2CH–CH3 were presented. In the 1H detected heteronuclear multiple bond correlation (HMBC) spectrum, key correlations were found to reveal the total structure. The correlations between H-9 (δH 5.06) and C-4’ (δC 149.5) and C-8’ (δC 140.3) indicated that the adenine moiety was connected to C-9 at N-9’. The conjugated ketone signal at δC 198.8 (C-7) in 13C NMR, is considered to link to C-8, due to the HMBC correlation between H-8 and C-7. The other two methyl groups were also assigned, as shown in Figure 2. The theoretical electronic circular dichroism (ECD) spectra of 9R-1 and 9S-1 were further calculated and compared with the experimental ones to determine the absolute configurations. As shown in Figure 3, the experimental ECD spectrum was similar to the calculated ECD spectrum of 9S-1 and the absolute configuration of 1 was determined as 9S.
We isolated 2-hydroxyl-3-pyrenocine-thio propanoic acid (2, a pair of epimers with a ratio of 1:2) as a light yellow powder. In the IR spectrum of 2, unconjugated carbonyl and α-pyrone ketone signals emerged at 1714 and 1626 cm−1, respectively. Its molecular formula of C14H18O7S, which gave six degrees of unsaturation, was established by the positive HR-ESI-MS ion peaks at m/z 331.0845 [M + H]+, 353.0670 [M + Na]+ and negative ion peaks at m/z 329.0706 [M − H]−, respectively. The presence of a sulfur atom was supported by the isotope quasi-molecular ion peaks at m/z 333.0808 [M(34S) + H]+, 355.0627 [M(34S) + Na]+ and 331.0662 [M(34S) – H]−, respectively. In the 1H NMR spectrum of 2, an α-pyrone olefinic proton, a methyl and a methoxyl signal were found at δH 5.69 (1H, s, H-3), 1.23 (1H, d, J = 6.8 Hz, H-10) and 3.87 (3H, s, 4-OCH3), respectively. In the 1H–1H COSY spectrum, the correlations between δH 5.69 (H-10), δH 3.30 (H-9) and δH 2.99 & 2.90 (H-8) revealed the presence of a CH2CH–CH3 structure unit. In the 13C NMR, signals belong to the pyrenocine moiety were similar to those in 1, except for the C-9 signal at δC 36.3, indicating that they differed in the substituent at C-9 (Table 1). The adeninyl signals were absent in the NMR data of 2. Except for the adeninyl signals, the 1H-NMR of 2 presented other ABX system signals at δH 4.09 (1H, m, H-2’) and δH [2.83 (1H, dd, J = 13.5, 5.1 Hz) and 2.72 (1H, dd, J = 13.5, 9.5 Hz), H-3’]; and three additional carbon signals were at δC 174.5 (C-1’), 71.1 (C-2’) and 34.7 (C-3’). With the help of 1H–1H COSY and HMBC spectra, they were attributed to a –CH2–CH(OH)–COOH structure fragment. The HMBC correlations between H-3’ (δH 2.83, 2.71) and C-9 (δC 36.3), and between H-9 (δH 3.30) and C-3’ (δC 34.7) revealed that the two structure units connected at C-9 and C-3’. The 1H-NMR and 13C-NMR signals at positions C-1’, 2’, 3’ and C-8, 9, whose peak intensities were halved and appeared in pairs, indicated that compound 2 is a pair of epimers (Figure 2). Considering the similar structure unit in 1, the absolute configuration of C-9 is S, while C-2’ contains the pair of R and S configurations.
Ozazino-cyclo-(2,3-dihydroxyl-trp-tyr) (3) was isolated as a white powder. The HR-ESI-MS cationized ion peaks revealed the presence of three nitrogen atoms. In the IR spectrum of 3, amide carbonyl signal was found at 1669 cm−1. The ultravoilet (UV) maximum absorption wavelengths at λmax (log ε) 240 (1.92) nm and 299 (1.55) nm, belong to the amide carbonyl and aromatic rings, respectively. In the 1H NMR spectrum of 3, a set of mono-substituted benzene ring signals could be found at δH 6.98 (2H, br. d, J = 7.2 Hz, H-2’, 6’), 6.86 (2H, br. t, J = 7.4 Hz, H-3’, 5’) and 6.75 (1H, m, H-4’), respectively. Another ortho-substituted benzene ring was elucidated by the signals at δH 6.90 (1H, br. d, J = 7.2 Hz, H-4), 6.56 (1H, br. t, J = 7.0 Hz, H-5), 7.00 (1H, br. t, J = 7.5 Hz, H-6), and 6.56 (1H, br. d, J = 7.7 Hz, H-7). In addition, two ABX spin system signals were present at δH 4.19 (1H, dd, J = 5.0, 2.8 Hz, H-α′), [2.87 (1H, dd, J = 13.9, 2.8 Hz) and 2.74 (1H, dd, J = 13.8, 5.0 Hz), H-β′], and at δH 4.17 (1H, dd, J = 9.1, 4.8 Hz, H-α), [1.91 (1H, dd, J = 13.6, 4.8 Hz) and 1.03 (1H, dd, J = 13.5, 9.1 Hz) H-β], and were attributed to two –CH–CH2– structure units. All these fragments were confirmed by the 13C NMR and DEPT spectra. In addition, two amide carbonyls at δC 166.1 and 161.5, together with a quaternary carbon at δC 74.7 and a methylidyne δC 99.3, indicated that 3 is a compound comprising two amino acid units, one of which is phenylalanine and the other one is a tryptophane-like structure unit. Similar structures have been isolated from P. citroviride [12] and from Penicillium sp. [13]. In the HSQC spectrum of 3, the proton signals at δH 7.96 (α′-NH) and δH 6.57 (1-NH) were attributed to two exchangeable protons, for the absence of correlation with carbon. With the help of the 1H–1H COSY spectrum, the signals belonging to the phenylalanine unit were attributed (Figure 2). The COSY correlation between δH 6.57 (1-NH) and δH 5.10 (1H, d, J = 2.8 Hz, H-2), together with the HMBC correlation between H-2 and C-3 (δC 74.6), revealed that C-2 and C-3 of the tryptophane unit were oxygen-connected. The HMBC correlations between δH 7.96 (α′-NH), 4.17 (H-α), 4.19 (H-α’), and δC 166.1 (C=O), 161.5 (C=O’) indicated the cyclo-dipeptide structure. Comparing the NMR data of 3 with those of cyclo-(L-tryptophyl-L-phenylalanyl) (19), a known compound reported previously [14], the chemical shift of 1-NH was significantly deduced from δH 10.83 to δH 6.57, indicating that the double bond between C-2 and C-3 in the indole ring disappeared. Moreover, absence of the α-NH signal (δH 7.96 in 19), and the high-field shifting of C=O’ (δC 167.3 in 19 and δC 161.5 in 3) were observed. Considering that the degree of unsaturation in 3 is 13, a six-membered ring between α-N and 2-O, connected by an ozazino bond, is prefered. Thus, the structrue of 3 is elucidated as ozazino-cyclo-(2,3-dihydroxyl-trp-tyr).
We isolated 5,5-dichloro-1-(3,5-dimethoxyphenyl)-1,4-dihydroxypentan-2-one (4) as a white powder. The HR-ESI-MS cationized ion peaks indicated a molecular formula of C13H16Cl2O5. The presence of the two chlorine atoms was confirmed by the isotope ion peak relative high ratio of 9:6:1. In the IR spectrum of 4, an unconjuated ketone carbonyl signal was at 1714 cm−1. A symmetrical 1,3,5-trisubstituted benzene ring was deduced by the 1H NMR signal at δH 6.37 (2H, d, J = 2.2 Hz, H-2’, 6’) and δH 6.27 (1H, d, J = 2.2 Hz, H-4’). The methoxyl signal, identical at δH 3.57 (6H, s), was attributed to 3’, 5’-OCH3. A –CH2–CH–CH– structural unit could be revealed by the peak splitting and coupling constants of the proton signals at δH [2.66 (1H, dd, J = 17.4, 8.8 Hz) and 2.52 (1H, dd, J = 17.4, 2.8 Hz), H-3], 4.07 (1H, ddd, J = 8.8, 3.3, 2.8 Hz, H-4), and 5.96 (1H, d, J = 3.3 Hz, H-5). The fragments were confirmed by the 1H–1H COSY correlations of 4. The 13C NMR of 4 showed an unconjuated ketone carbonyl at δC 207.8. The correlation between δH 4.85 (1H, s, H-1) and δC 79.7 (C-1), found in the HSQC spectrum, revealed an oxygen-linked methylidyne. The HMBC correlations between δH 4.85 (H-1) and δC 141.7 (C-1’), 207.8 (C-2), and between δH 2.66 and 2.52 (H-3) and δC 207.8 (C-2), allowed the elucidation of the structure of 4 (Figure 2).
We isolated 2,3,4-trihydroxybutyl cinnamate (5) as a white powder. The molecular formula of C13H16O5 was revealed by the HR-ESI-MS cationized ion peaks at m/z 275.0882 [M + Na]+ (calcd. for 275.0890 C13H16O5Na) in positive mode, and m/z 251.0922 [M − H]− (calcd. for 251.0925, C13H14O5) in negative mode. A conjugated ester carbonyl IR signal was present at 1700 cm−1. A mono-substituted benzene ring was deduced by the 1H NMR signal at δH 7.72 (2H, dd, J = 6.3, 3.0 Hz, H-2, 6) and δH 7.74 (3H, overlapped, H-3,5 and H-4). A pair of trans-alkene proton signals were exhibited at δH 7.69 (1H, d, J = 16.0 Hz, H-7) and δH 6.64 (1H, d, J = 16.0 Hz, H-8). With the help of 1H–1H COSY spectrum, two ABX spin systems at δH [4.33 (1H, dd, J = 11.3, 2.7 Hz) and 4.11 (1H, dd, J = 11.3, 7.1 Hz), H-1′], 3.66 (1H, m, H-2′), and at δH [3.58 (1H, br. d, J = 9.2 Hz) and 3.42 (1H, br. d, J = 9.2 Hz), H-4′], 3.44 (1H, m, H-3′), were connected to a structure fragment as O–CH2–CH(O)–CH(O)–CH2–O (Figure 2). In the HMBC spectrum, correlations from H-1’ (δH 4.33 & 4.11), H-8 (δH 6.64) to C-9 (δC 166.9), and from H-3, 5 (δH 7.44), H-8 (δH 6.64) to C-1 (δC 134.5), allowed the elucidation of 2,3,4-trihydroxybutyl cinnamate. The chlorogenic acid contained in the potato medium may be the original precursor of this compound.
The structures of compounds 6–24 were elucidated by the comparison of their MS and NMR data with those reported in the literature, and they were identified as: 4-methyl-5,6-dihydropyren-2-one (6) [15], citreo-g-pyrone (7) [16], pyrenocine B (8) [17], pyrenocine D (9) [18], pyrenocine A (10) [19], pyrenocine I (11) [20], pyrenocine E (12) [18], pyrenocine J (13) [21], citreovirenone (14) [12], citroethiolactone (15) [16], citreoviridin A (16) [22], isocitreoviridin A (17) [22], aurovertin U (18) [14], cyclo (phe-trp) (19) [14], N-(N-acetyl-valyl)-phenylalanine (20) [23], 2′,3′-dihydrosorbicillin (22) [24], citreoviranol (23) [25], and haenamindole (24) [26]. Most of the compounds have been isolated from the genus of Penicillum. Compound 21, a known compound without reported NMR data, was identified by MS and 1D, 2D NMR spectra data as 3,5-dihydroxy-2,4-dimethyl-6-(3-oxobutan-2-yl)benzaldehyde.
2.2. Cytotoxicity Evaluation
All of the isolated compounds were evaluated for their cytotoxic effects on four types of tumor cells; Bel7402, HT1080, CNE2 and A549. Compounds 2 and 4 showed potent cytotoxicity to the cell lines Bel7402 and HT1080, while they showed no obvious effects on Cne2 and A549 (Table 2). All other compounds exhibited much lower cytotoxicity to the four tumor cell lines, with IC50 values larger than 100 μM.
3. Discussion
In this study, five new compounds, including two new heteroatom-containing compounds were isolated from the ethyl acetate extract of a deep-sea fungus P. citreonigrum XT20-134 (MCCC 3A00956). Chemically, the relative configuration of these compounds was confirmed by their NOESY spectra; the absolute configuration of compound 1 was revealed by comparison of its CD spectra with the calculated ECD. All of the compounds were evaluated for cytotoxic activity. The new heteroatom-containing compounds 2 and 4 showed potent cytotoxicity to the tumor cell lines Bel7402 and HT1080.
Marine microorganisms can utilize chloride ions or sulfate ions, the two most abundant anions in seawater, and produce heteroatom-containing compounds, with diverse chemical structures and various bioactivities [27]. The bioactive halogenated, mainly referring to chlorinated, natural products from microorganisms mainly manifest with cytotoxic and antibacterial activity, indicating that halogenated compounds produced from microorganisms, due to adaptability or defense from the extreme environment, have cytotoxic and antibacterial activity [28]. In this study, compound 4 that contained two choline atoms was also found to have potent cytotoxicity. The most abundant source of sulfur-containing natural products is also marine organisms. Sulfur can appear in a multitude of combinations and oxidation states: thiol, sulfide, disulfide, sulfoxide, sulfonate, etc. The diversity of sulfur-containing chemical structures leads to their various bioactivities [29]. Among them, psammaplin A, has been found to have a broad bioactive spectrum, especially in terms of antimicrobial and antiproliferative activities [30]. With a sulfur atom, compound 2 exhibited potent cytotoxic activity. Similar compounds without sulfur (1, 8, and 10) did not shown cytotoxic activity. However, compound 16 show no activity, indicating that the type of sulfur bond is important to the activity.
4. Materials and Methods
4.1. General Experimental Procedures
An electrospray ionization source (ESI)-equipped Q-Exactive mass spectrometer (Thermo Fisher Scientific Corporation, Waltham, MA, USA) was used to analyze the HR-ESI-MS data. A Shimadzu UV-260 spectrometer (Shimadzu Corporation, Tokyo, Japan) and a Perkin-Elmer 683 infrared spectrometer (PerkinElmer, Inc., Waltham, MA, USA) were used to obtain the UV and IR spectra, respectively. A JASCO P-200 polarimeter (JASCO Corporation, Tokyo, Japan) with a 5 cm cell was applied to measure the optical rotation value. The NMR spectra with TMS as the internal standard were taken on a Brucker Avance III 600 FT NMR spectrometer (Bruker Corporation, Billerica, MA, USA).
4.2. Eletronic Circular Dichroism (ECD) Calculations
The theoretical electronic circular dichroism (ECD) spectra of the isolated compounds were calculated on the basis of the relative configurations determined by their NOESY spectra and J values in 1H NMR. Conformational analyses and density functional theory (DFT) calculations were used to generate and optimize the conformers with energy. The ECD calculations were performed as previously described [31,32].
4.3. Fungal Strain and Fermentation
The strain Penicillium sp. XT20-134 was isolated from southeast Indian Ocean sediments at 2910 m by the tablet pour method. The internal transcribed spaces (ITS) region was amplified and sequenced using the general primers ITS1 and ITS4. The ITS region of the fungi is a 573 bp DNA sequence (GenBank Accession Number: KY 978587) that showed 99% identity to P. citreonigrum. The strain was deposited at the China Center for Type Culture Collection (CCTCC) as accession number M2017125 and Marine Culture Collection of China (MCCC) as accession number MCCC 3A00956. The fungus grew well on the rice medium in artificial seawater. Carbohydrate fermentation was conducted by subculturing the fungus in rice medium in artificial seawater and incubating at 28 °C for 30 days in a standing position.
4.4. Extraction and Isolation
The rice medium (10 kg) of P. citreonigrum XT20-134 was extracted with ethyl acetate (20 L) trice and concentrated under reduced pressure at 40 °C to yield 19.2 g of the residue.
The EtOAc extract (18 g) was fractionated over a column packed with silica gel (360 g, Yantai Chemical Industry Research Institute, Yantai, China), eluted with petroleum ether-ethyl acetate (v/v) (5:1, 1.0 L) and CHCl3–CH3OH (v/v) (5;1, 1.0 L) and CH3OH (1.0 L), to afford PE eluent (0.5 g), CM eluent (12.3 g), and methanol eluent (3.4 g). The PE eluent was purified by a silica gel (30 g) column and eluted with petroleum ether-ethyl acetate (v/v) (10:1, 5:1, and 3:1, each 200 L) to give compound 22 (13 mg). The CM eluent was separated over a Cosmosil reversed-phase C18 (300 g, 75 μm, Nakalai Tesque Co. Ltd., Kyoto, Japan) column and eluted with CH3OH/H2O (10%–100%, each 1.5 L) to provide 12 fractions (Fr. 1–Fr. 12). Compound 24 (83 mg) was obtained from Fr. 11 after recrystallization. Other fractions were purified over a preparative Cosmosil ODS column (250 mm × 20.0 mm i.d., 5 μm, Cosmosil, Nakalai Tesque Co. Ltd., Kyoto, Japan), and isocratically eluted with a mobile phase system of acetonitrile–H2O in different ratios. Eluting with 15% acetonitrile, preparative HPLC separation on Fr. 1, Fr. 2, and Fr. 3 resulted in the isolation of: compound 6 (6 mg) from Fr. 1, compounds 7 (8 mg), 8 (57 mg), 9 (5 mg) from Fr. 2, and compounds 1 (50 mg), 10 (90 mg), 11 (15 mg), 12 (8 mg) from Fr. 3, respectively. Preparative HPLC purification of Fr. 5, eluted with acetonitrile–H2O (30:70, v/v), led to the isolation of compound 2 (15 mg) and compound 15 (150 mg). Fr. 4, Fr. 6, and Fr. 7 were separated with 30% acetonitrile. As a result, compounds 5 (5 mg), 13 (4 mg), and 14 (5 mg) were isolated from Fr. 4; compound 19 (6 mg) from Fr. 6, and compounds 20 (13 mg) and 21 (20 mg) from Fr. 7, respectively. Fr. 8 was isolated with 35% acetonitrile to obtain compound 4 (4 mg). Separation of Fr. 9 and Fr. 10, eluted with 40% acetonitrile, obtained compound 3 (8 mg) and compound 23 (13 mg), respectively. The other three compounds, 16 (15 mg), 17 (18 mg) and 18 (13 mg), were obtained from Fr. 12, by eluting with 45% acetonitrile.
4.5. Structrural Elucidation of the New Compounds 1–5
Adeninylpyrenocine (1): white amorphous powder; [α − 14° (c = 0.1, CH3OH); IR (KBr) (νmax): 3441, 1695, 1642, 1402 and 1256 cm−1; UV (MeOH) λmax (log ε): 204 (2.49) and 261 (2.22) nm. HR-ESI-MS: m/z 344.1357 [M + H]+ (calcd. for 344.1353 C16H18N5O4) and 366.1172 [M + Na]+ (calcd. for 366.1173 C16H17N5O4Na) in positive mode, and m/z 342.1220 [M − H]− (calcd. for 342.1208, C16H16N5O4) in negative mode. 1H NMR (600 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6) spectra data are listed in Table 1, and Figures S1–S9 in supporting materials.
2-Hydroxyl-3-pyrenocine-thio propanoic acid (2): light yellow powder; [α + 26° (c = 0.1, CH3OH); IR (KBr) (νmax): 3435, 2927, 1714, 1626, 1453, 1400, 1260, and 1096 cm−1; UV (MeOH) λmax (log ε): 203 (2.10) nm and 260 (2.52) nm. HR-ESI-MS: m/z 331.0845 [M + H]+ (calcd. for 331.0859 C14H19O7S) and 353.0670 [M + Na]+ (calcd. for 353.0665 C14H18O7SNa) in positive mode, and m/z 329.0706 [M − H]− (calcd. for 329.0700, C14H17O7S) in negative mode. 1H NMR (600 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6) spectra data are listed in Table 1, and Figures S10–S17 in supporting materials.
Ozazino-cyclo-(2,3-dihydroxyl-trp-tyr) (3): white powder; [α + 43° (c = 0.1, CH3OH); IR (KBr) (νmax): 3437, 1669, 1623, 1460, 1396, and 1081 cm−1; UV (MeOH) λmax (log ε): 204 (2.55) nm, 240 (1.92) nm, and 299 (1.55) nm. HR-ESI-MS: m/z 366.1449 [M + H]+ (calcd. for 366.1448 C20H20N3O4) and 388.1265 [M + Na]+ (calcd. for 388.1268 C20H19N3ONa) in positive mode, and m/z 364.1303 [M − H]− (calcd. for 364.1303, C20H18N3O) in negative mode. 1H NMR (600 MHz, DMSO-d6) and 13C NMR (150 MHz, DMSO-d6) spectra data are listed in Table 3, and Figures S18–S25 in supporting materials.
5,5-Dichloro-1-(3,5-dimethoxyphenyl)-1,4-dihydroxypentan-2-one (4): white powder; [α − 23° (c = 0.1, CH3OH); IR (KBr) (νmax): 3447, 1608, 1396, and 1159 cm−1; UV (MeOH) λmax (log ε): 204 (2.36) nm and 275 (1.23) nm. HR-ESI-MS: m/z 323.0449 [M + H]+ (calcd. for 323.0448 C13H17O5Cl2) and 345.0261 [M + Na]+ (calcd. for 345.0267 C13H16O5Cl2Na) in positive mode, and m/z 321.0304 [M − H]− (calcd. for 321.0302, C13H15O5Cl2) in negative mode. 1H NMR (600 MHz, DMSO-d6) δH: 6.37 (2H, d, J = 2.2 Hz, H-2′, 6′), 6.27 (1H, t, J = 2.2 Hz, H-4′), 5.96 (1H, d, J = 3.3 Hz, H-5), 4.85 (1H, s, H-1), 4.07 (1H, ddd, J = 8.8, 3.3, 2.8 Hz, H-4), 3.57 (6H, s, H-3′, 5′-OCH3), [2.66 (1H, dd, J = 17.4, 8.8 Hz) & 2.52 (1H, dd, J = 17.4, 2.8 Hz), H-3]; 13C NMR (150 MHz, DMSO-d6) δC: 207.8 (C-2), 160.9 (C-3′, 5′), 141.7 (C-1′), 105.3 (C-2′, 6′), 100.0 (C-4′), 79.7 (C-1), 77.7 (C-5), 71.1 (C-4), 55.6 (C-3′, 5′-OCH3), 40.9 (C-3). The spectra are provided in Figures S26–S33 in supporting materials.
2,3,4-Trihydroxybutyl cinnamate (5): white powder; [α − 23° (c = 0.1, CH3OH); IR (KBr) (νmax): 3421, 1700, 1634, 1396, 1186 and 1085 cm−1; UV (MeOH) λmax (log ε): 216 (1.93) nm and 276 (1.99) nm. HR-ESI-MS: m/z 275.0882 [M + Na]+ (calcd. for 275.0890 C13H16O5Na) in positive mode, and m/z 251.0922 [M − H]− (calcd. for 251.0925, C13H14O5) in negative mode. 1H NMR (600 MHz, DMSO-d6) δH: 7.69 (1H, d, J = 16.0 Hz, H-7), 7.44 (1H, overlapped, H-4), 7.44 (2H, overlapped, H-3, 5), 7.72 (2H, dd, J = 6.3, 3.0 Hz, H-2, 6), 6.64 (1H, d, J = 16.0 Hz, H-8), 3.44 (1H, m, H-3′), 3.66 (1H, m, H-2′), [4.33 (1H, dd, J = 11.3, 2.7 Hz) and 4.11 (1H, dd, J = 11.3, 7.1 Hz), H-1′], [3.58 (1H, br. d, J = 9.2 Hz) and 3.42 (1H, br. d, J = 9.2 Hz), H-4′]; 13C NMR (150 MHz, DMSO-d6) δC: 166.9 (C-9), 144.8 (C-7), 134.5 (C-1), 130.9 (C-4), 129.4 (C-3, 5), 128.8 (C-2, 6), 118.8 (C-8), 72.8 (C-3′), 69.9 (C-2′), 67.0 (C-1′), 63.5 (C-4′). The spectra are provided in Figures S34–S42 in supporting materials.
4.6. Cytotoxicity Assay
Cytotoxicity assay was carried out according to the instructions of CCK-8 kit [7]. Briefly, compounds at different concentrations were added into the culture medium containing 105 /mL HT1080, Cne2 and Bel7402 cells and incubated for 24, 48, 72 h. Then, 10 μL of CCK-8 solution was added into each well of the 96-well plate, incubated for 2 h and then the absorbance was measured at 450 nm using a microplate reader (BIO-RAD, Hercules, California, USA). The inhibition rate = (A control − A treated)/A control × 100. The IC50 was the concentration at which it caused 50% inhibition of cell proliferation (50% reduction in the absorbance value in the treated cells, in respect to the control).
Supplementary Materials
The following are available online at https://www.mdpi.com/1660-3397/17/9/509/s1, Figures S1–S42: Supporting NMR, IR, and HR-ESI-MS spectra.
Author Contributions
X.-X.T. identified the fungus and performed the cytotoxicity activity assays. X.Y. and S.-Z.L. isolated the compounds. B.-W.T., M.-J.F., and X.-M.W. were responsible for the structural elucidation. Y.-K.Q. and Z.W. supervised the project.
Funding
The project was supported by the COMRA Project of China (DY135-B2-08), National Basic Research Program of China (973 Program) (No. 2015CB755901), Xiamen Ocean Economic Innovation and Development Demonstration Project (16PZP001SF16) and Xiamen Science and Technology Program (3502Z20182029), the Natural Science Foundation of Fujian Province of China (No. 2018J01132), and the Fundamental Research Funds for the Central Universities (No. 20720180051). The project was supported by the Natural Science Foundation of Fujian Province of China (No. 2018J01131).
Conflicts of Interest
The authors have no conflict of interest to declare. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Chunxiao, S.; Shah, M.; Zhenzhen, Z.; Yanyan, F.; Yimin, C.; Qian, C.; Qianqun, G.; Tianjiao, Z.; Guojian, Z.; Dehai, L. Secondary Metabolites from Deep-sea Derived Microorganisms. Curr. Med. Chem. 2019, 26, 1. [Google Scholar]
- Deshmukh, S.K.; Prakash, V.; Ranjan, N. Marine Fungi: A Source of Potential Anticancer Compounds. Front. Microbiol. 2018, 8, 2536. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Xue, Y.R.; Liu, C.H. A Brief Review of Bioactive Metabolites Derived from Deep-Sea Fungi. Mar. Drugs 2015, 13, 4594–4616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.; Sun, Z.H.; Liu, Z.; Chen, Y.C.; Liu, H.X.; Li, H.H.; Zhang, W.M. Dichotocejpins A-C: New Diketopiperazines from a Deep-Sea-Derived Fungus Dichotomomyces cejpii FS110. Mar. Drugs 2016, 14, 164. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, Z.M.; Chen, Y.C.; Tan, H.B.; Li, S.N.; Li, H.H.; Gao, X.X.; Liu, H.X.; Zhang, W.M. Chromone-Derived Polyketides from the Deep-Sea Fungus Diaporthe phaseolorum FS431. Mar. Drugs 2019, 17, 182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.H.; Min, X.T.; Huang, J.J.; Zhong, Y.; Wu, Y.H.; Li, X.X.; Deng, Y.Y.; Jiang, Z.D.; Shao, Z.Z.; Zhang, L.H.; et al. Cytoglobosins H and I, New Antiproliferative Cytochalasans from Deep-Sea-Derived Fungus Chaetomium globosum. Mar. Drugs 2016, 14, 233. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Tang, X.X.; Qin, D.; Yi, Z.W.; Fang, M.J.; Wu, Z.; Qiu, Y.K. Biosynthetic Functional Gene Analysis of Bis-Indole Metabolites from 25D7, a Clone Derived from a Deep-Sea Sediment Metagenomic Library. Mar. Drugs 2016, 14, 107. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Tang, X.X.; Chen, L.; Yi, Z.W.; Fang, M.J.; Wu, Z.; Qiu, Y.K. Two New Cytotoxic Indole Alkaloids from a Deep-Sea Sediment Derived Metagenomic Clone. Mar. Drugs 2014, 12, 2156–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, P.; Tang, X.X.; Yi, Z.W.; Qiu, Y.K.; Wu, Z. Two new compounds from marine-derived fungus Penicillium sp F11. J. Asian Nat. Prod. Res. 2012, 14, 197–203. [Google Scholar] [CrossRef]
- Liu, D.; Lin, H.; Proksch, P.; Tang, X.X.; Shao, Z.Z.; Lin, W.H. Microbacterins A and B, New Peptaibols from the Deep Sea Actinomycete Microbacterium sediminis sp. nov. YLB-01(T). Org. Lett. 2015, 17, 1220–1223. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, X.X.; Shao, Z.Z.; Ren, J.W.; Liu, D.; Proksch, P.; Lin, W.H. Indole-based alkaloids from deep-sea bacterium Shewanella piezotolerans with antitumor activities. J. Antibiot. 2014, 67, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Shizuri, Y.; Nagahama, M.; Yamamura, S.; Kawai, K.; Kawai, N.; Furukawa, H. Isolation and Structures of Citreovirenone and Citreovirone. Chem. Lett. 1986, 15, 1129–1132. [Google Scholar] [CrossRef]
- Yang, M.H.; Li, T.X.; Wang, Y.; Liu, R.H.; Luo, J.; Kong, L.Y. Antimicrobial metabolites from the plant endophytic fungus Penicillium sp. Fitoterapia 2017, 116, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Tani, K.; Kojima, A.; Sotoma, G.; Okada, K.; Shimada, A. Cyclo-(l-tryptophyl-l-phenylalanyl), a plant growth regulator produced by the fungus Penicillium. Phytochemistry 1996, 41, 665–669. [Google Scholar] [CrossRef]
- Shintani, R.; Hayashi, T. Rhodium-Catalyzed Asymmetric 1,4-Addition of Sodium Tetraarylborates to β, β-Disubstituted α, β-Unsaturated Esters. Org. Lett. 2011, 13, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Nakada, T.; Sudo, S.; Kosemura, S.; Yamamura, S. Two new metabolites of hybrid strains KO 0201 and 0211 derived from Penicillium citreo-viride B. IFO 6200 and 4692. Tetrahedron Lett. 1999, 40, 6831–6834. [Google Scholar] [CrossRef]
- Sato, H.; Konoma, K.; Sakamura, S. Phytotoxins Produced by Onion Pink Root Fungus, Pyrenochaeta terrestris. J. Agric. Chem. Soc. Jpn. 1979, 43, 2409–2411. [Google Scholar]
- Amagata, T.; Minoura, K.; Numata, A. Cytotoxic metabolites produced by a fungal strain from a Sargassum alga. J. Antibiot. 1998, 51, 432–434. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.L.; Zhang, H.P. Isolation of Secondary Metabolites of 9F Series Marine Fungi and Their Bioactivities against Pyricularia oryzae. Nat. Prod. Res. Dev. 2005, 17, 677–680. [Google Scholar]
- Hashida, J.; Niitsuma, M.; Iwatsuki, M.; Mori, M.; Ishiyama, A.; Namatame, M.; Nishihara-Tsukashima, A.; Nonaka, K.; Ui, H.; Masuma, R.; et al. Pyrenocine I, a new pyrenocine analog produced by Paecilomyces sp. FKI-3573. J. Antibiot. 2010, 63, 559–561. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Yang, S.X.; Li, L.; Wang, Y. Chemical Constituents and Their Toxic Activity from the Endophytic Fungus Phomopsissp. KY-12,Isolated from Pleioblastus amarus. Nat. Prod. Res. Dev. 2014, 26, 1389–1392. [Google Scholar]
- Da Rocha, M.W.; Resck, I.S.; Caldas, E.D. Purification and full characterisation of citreoviridin produced by Penicillium citreonigrum in yeast extract sucrose (YES) medium. Food Addit. Contam. A 2015, 32, 584–595. [Google Scholar] [CrossRef]
- Sarubbi, E.; Seneci, P.F.; Angelastro, M.R.; Peet, N.P.; Denaro, M.; Islam, K. Peptide Aldehydes as Inhibitors of Hiv Protease. FEBS Lett. 1993, 319, 253–256. [Google Scholar] [CrossRef]
- Teng, X.; Zhuang, Y.; Wang, Y.; Liu, P.; Xu, Z.; Zhu, W. Secondary metabolites from Penicillium sp.gxwz406 symbiotic with the gorgonian Echinogorgia flora. Chin. J. Mar. Drugs 2010, 29, 11–15. [Google Scholar]
- Shizuri, Y.; Shigemori, H.; Sato, R.; Yamamura, S.; Kawai, K.; Furukawa, H. 4 New Metabolites Produced by Penicillium-Citreo-Viride B on Addition of Nabr. Chem. Lett. 1988, 17, 1419–1422. [Google Scholar] [CrossRef]
- Matsunaga, K.; Shizuri, Y.; Yamamura, S.; Kawai, K.; Furukawa, H. Isolation and Structure of Citreoindole, a New Metabolite of Hybrid Strain Ko-0052 Derived from Penicillium-Citreo-Viride B—Ifo-6200 and B—Ifo-4692. Tetrahedron Lett. 1991, 32, 6883–6884. [Google Scholar] [CrossRef]
- Jimenez, C. Marine sulfur-containing natural products. Stud. Nat. Prod. Chem. 2001, 25, 811–917. [Google Scholar]
- Wang, X.; Wang, P.; Zhu, T.; Zhu, W. Advances in studies of bioactive halo-natural products from microorganisms. Zhongguo Haiyang Yaowu 2011, 30, 40–51. [Google Scholar]
- Marakalala, M.B.; Mmutlane, E.M.; Kinfe, H.H. β-hydroxy sulfides and their syntheses. Beilstein J. Org. Chem. 2018, 14, 1668–1692. [Google Scholar] [CrossRef] [PubMed]
- Jing, Q.; Hu, X.; Ma, Y.; Mu, J.; Li, Z.; Bai, J.; Hua, H.; Li, D.; Liu, W.; Xu, F. Marine-Derived Natural Lead Compound Disulfide-Linked Dimer Psammaplin A: Biological Activity and Structural Modification. Mar. Drugs 2019, 17, 384. [Google Scholar] [CrossRef]
- Tang, X.X.; Yan, X.; Fu, W.H.; Yi, L.Q.; Tang, B.W.; Yu, L.B.; Fang, M.J.; Wu, Z.; Qiu, Y.K. New beta-Lactone with Tea Pathogenic Fungus Inhibitory Effect from Marine-Derived Fungus MCCC3A00957. J. Agric. Food Chem. 2019, 67, 2877–2885. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.H.; Ou-Yang, H.; Yan, X.; Tang, B.W.; Fang, M.J.; Wu, Z.; Chen, J.W.; Qiu, Y.K. Open-Ring Butenolides from a Marine-Derived Anti-Neuroinflammatory Fungus Aspergillus terreus Y10. Mar. Drugs 2018, 16, 428. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structures of compounds 1–24 isolated from an extract of Penicillum citreonigrum XT20-134.
Figure 1.
Structures of compounds 1–24 isolated from an extract of Penicillum citreonigrum XT20-134.

Figure 2.
Key 1H–1H COSY, HMBC correlations of compounds 1–5.

Figure 3.
Calculated (9R and 9S) and experimental electronic circular dichroism (ECD) spectra of compound 1.
Figure 3.
Calculated (9R and 9S) and experimental electronic circular dichroism (ECD) spectra of compound 1.


{kind=link}
{kind=link}
{kind=link}
Table 1.
1H NMR and 13C NMR (DMSO-d6) data of compounds 1 and 2.
| Positions | 1 | 2 | 2’ | |||
|---|---|---|---|---|---|---|
| δC | δH | δC | δH | δC | δH | |
| 2 | 162.0 | 162.2 | 162.2 | |||
| 3 | 87.8 | 5.62, s | 87.9 | 5.69, s | 87.9 | 5.69, s |
| 4 | 168.3 | 168.5 | 168.5 | |||
| 5 | 115.1 | 115.4 | 115.4 | |||
| 6 | 162.8 | 163.2 | 163.2 | |||
| 7 | 198.8 | 199.5 | 199.5 | |||
| 8 | 49.3 | 3.78, dd (17.6, 7.9) | 51.7 | 2.99, dd (17.1, 6.6) | 51.6 | 2.99, dd (17.1, 6.6) |
| 3.35, dd (17.6, 6.0) | 2.90, dd (17.1, 3.7) | 2.89, dd (17.1, 3.7) | ||||
| 9 | 47.7 | 5.06, m | 36.3 | 3.30, m | 36.2 | 3.30, m |
| 10 | 21.1 | 1.54, br. d (6.6) | 21.8 | 1.23, d (6.8) | 21.8 | 1.23, d (6.8) |
| 6-CH3 | 18.0 | 1.85, s | 18.5 | 2.18, d (2.6) | 18.5 | 2.18, d (2.6) |
| 4-OCH3 | 57.4 | 3.74, s | 57.5 | 3.87, s | 57.5 | 3.87, s |
| 2′ | 152.6 | 8.11, br. s | ||||
| 4′ | 149.5 | |||||
| 5′ | 119.5 | |||||
| 6′ | 156.4 | |||||
| 8′ | 140.3 | 8.21, br. s | ||||
| 6′-NH2 | 7.16, br. s | |||||
| 1′ | 174.5 | 174.5 | ||||
| 2′ | 71.1 | 4.09, m | 71.0 | 4.09, m | ||
| 3′ | 34.7 | 2.83, dd (13.5, 5.1) | 34.6 | 2.82, dd (13.5, 5.1) | ||
| 2.72, dd (13.6, 9.5) | 2.71, dd (13.6, 9.5) | |||||
Table 2.
Cytotoxic activities of compounds 2 and 4 (IC50, μM).
| Compd. | Bel7402 | HT1080 | Cne2 | A549 |
|---|---|---|---|---|
| 2 | 7.63 ± 1.46 | 10.22 ± 1.32 | 73.14 ± 5.32 | 87.08 ± 7.32 |
| 4 | 13.14 ± 1.41 | 16.53 ± 1.67 | 83.56 ± 6.49 | 92.47 ± 6/33 |
| paclitaxel | <1 | <1 | <1 | <1 |
| DMSO | None | None | None | None |
Table 3.
1H NMR and 13C NMR (DMSO-d6) data of compounds 3 and 19.
| Positions | 3 | 19 | ||
|---|---|---|---|---|
| δC | δH | δC | δH | |
| 2 | 99.3 | 5.10, d (2.8) | 121.4 | 6.88, br. s |
| 3 | 74.6 | 109.2 | ||
| 4 | 123.1 | 6.90, br. d (7.2) | 118.9 | 7.43, br. d (7.2) |
| 5 | 118.3 | 6.56, br. t (7.0) | 119.2 | 6.93, br. t (7.0) |
| 6 | 129.1 | 7.00, br. t (7.5) | 124.9 | 7.01, br. t (7.4) |
| 7 | 109.6 | 6.56, br. d (7.7) | 111.8 | 7.26, br. d (7.7) |
| 3a | 131.9 | 128.0 | ||
| 7a | 148.1 | 136.5 | ||
| 1′ | 136.0 | 137.0 | ||
| 2′, 6′ | 130.4 | 6.98, br. d (7.2) | 130.2 | 7.10, overlapped |
| 3′, 5′ | 128.2 | 6.86, br. t (7.4) | 128.5 | 6.64, br. t (7.4) |
| 4′ | 126.5 | 6.75, m | 126.9 | 7.12, overlapped |
| C=O | 166.1 | 166.7 | ||
| C=O′ | 161.5 | 167.3 | ||
| α | 54.6 | 4.17, dd (9.1, 4.8) | 55.8 | 3.92, m |
| α′ | 54.8 | 4.19, dd (5.0, 2.8) | 56.1 | 3.79, m |
| β′ | 37.7 | 2.87, dd (13.9, 2.8) | 39.5 | 2.40, overlapped |
| 2.74, dd (13.8, 5.0) | 1.78, dd (13.5, 7.0) | |||
| β | 36.0 | 1.91, dd (13.6, 4.8) | 30.2 | 2.74, dd (14.0, 4.4) |
| 1.03, dd (13.5, 9.1) | 2.44, overlapped | |||
| 1-NH | 6.57, br. s | 10.83, br. s | ||
| α-NH | 7.85, br. s | |||
| α′-NH | 7.96, br. s | 7.64, br. s | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tang, X.-X.; Liu, S.-Z.; Yan, X.; Tang, B.-W.; Fang, M.-J.; Wang, X.-M.; Wu, Z.; Qiu, Y.-K. Two New Cytotoxic Compounds from a Deep-Sea Penicillum citreonigrum XT20-134. Mar. Drugs 2019, 17, 509. https://doi.org/10.3390/md17090509
AMA Style
Tang X-X, Liu S-Z, Yan X, Tang B-W, Fang M-J, Wang X-M, Wu Z, Qiu Y-K. Two New Cytotoxic Compounds from a Deep-Sea Penicillum citreonigrum XT20-134. Marine Drugs. 2019; 17(9):509. https://doi.org/10.3390/md17090509
Chicago/Turabian StyleTang, Xi-Xiang, Shun-Zhi Liu, Xia Yan, Bo-Wen Tang, Mei-Juan Fang, Xiu-Min Wang, Zhen Wu, and Ying-Kun Qiu. 2019. "Two New Cytotoxic Compounds from a Deep-Sea Penicillum citreonigrum XT20-134" Marine Drugs 17, no. 9: 509. https://doi.org/10.3390/md17090509
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.