Synthesis of Pseudellone Analogs and Characterization as Novel T-type Calcium Channel Blockers



Division of Chemistry and Structural Biology, Institute for Molecular Bioscience, the University of Queensland, Brisbane, Qld 4072, Australia
*
Authors to whom correspondence should be addressed.
Mar. Drugs 2018, 16(12), 475; https://doi.org/10.3390/md16120475
Submission received: 8 November 2018
/
Revised: 21 November 2018
/
Accepted: 26 November 2018
/
Published: 28 November 2018
(This article belongs to the Special Issue Synthesis of Marine Natural Products and Analogues)
Abstract
:T-type calcium channel (CaV3.x) blockers are receiving increasing attention as potential therapeutics for the treatment of pathophysiological disorders and diseases, including absence epilepsy, Parkinson’s disease (PD), hypertension, cardiovascular diseases, cancers, and pain. However, few clinically approved CaV3.x blockers are available, and selective pharmacological tools are needed to further unravel the roles of individual CaV3.x subtypes. In this work, through an efficient synthetic route to the marine fungal product pseudellone C, we obtained bisindole alkaloid analogs of pseudellone C with a modified tryptophan moiety and identified two CaV3.2 (2, IC50 = 18.24 µM; 3, IC50 = 6.59 µM) and CaV3.3 (2, IC50 = 7.71 µM; 3, IC50 = 3.81 µM) selective blockers using a FLIPR cell-based assay measuring CaV3.x window currents. Further characterization by whole-cell patch-clamp revealed a preferential block of CaV3.1 activated current (2, IC50 = 5.60 µM; 3, IC50 = 9.91 µM), suggesting their state-dependent block is subtype specific.
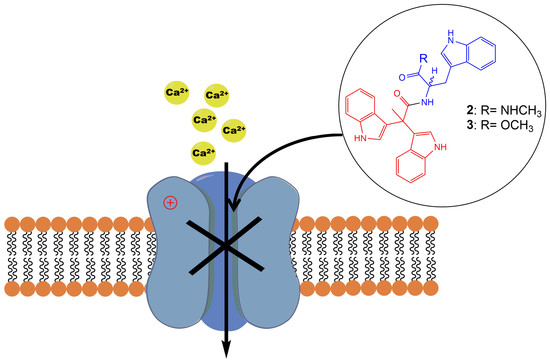
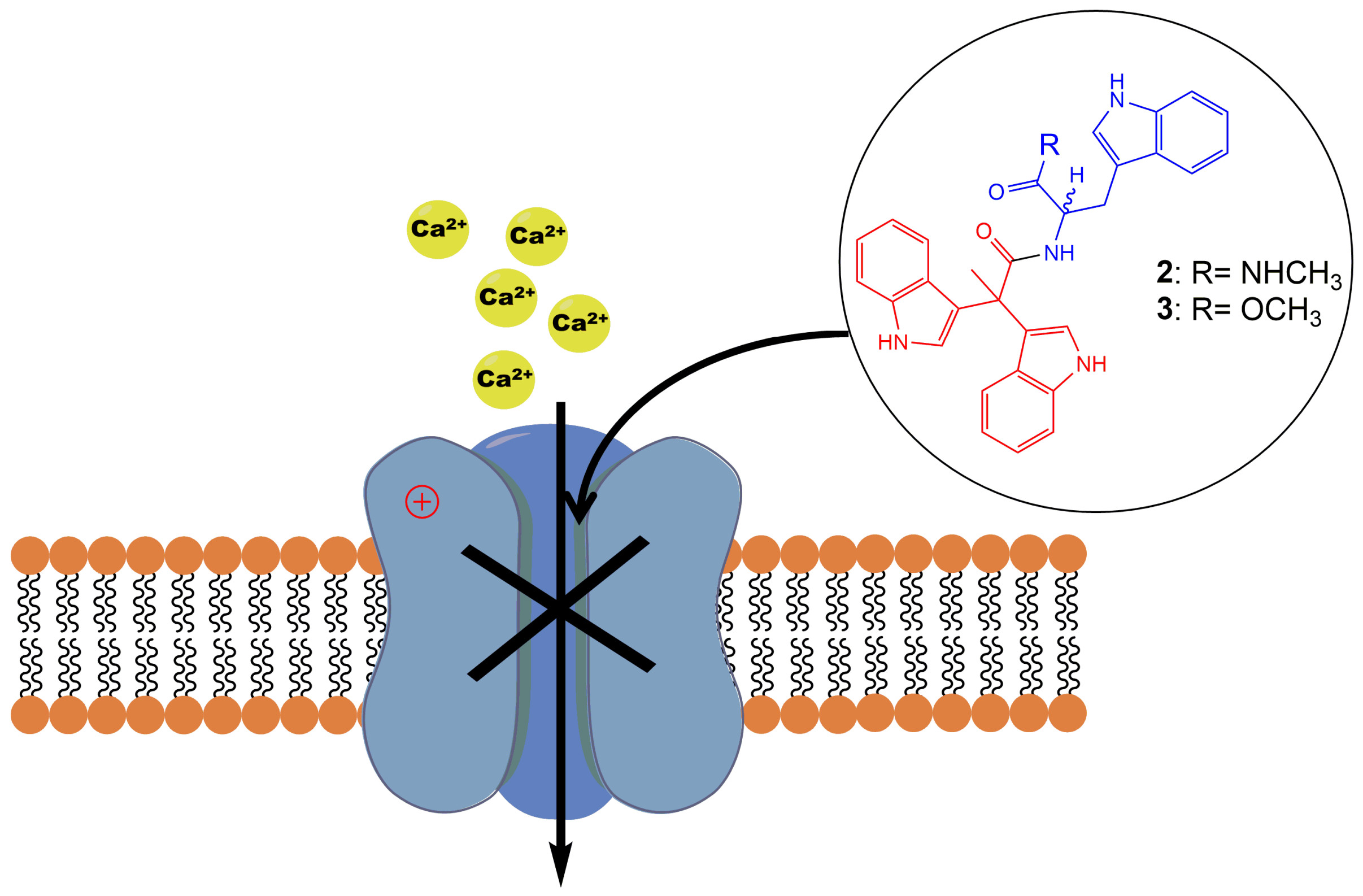
1. Introduction
Alkaloids produced by marine animals are mostly potent cytotoxins evolved for defense [1]. Marine indole alkaloids have been widely explored for their therapeutic potentials, providing potential new drug leads for the treatment of a wide range of diseases including cancer, neurological disorders, and parasitic infections [2,3]. However, the underlying pharmacological targets and mechanisms of the bioactive indole alkaloids remain largely undefined. Triptans, a family of tryptamine-based drugs used for the abortive treatment of migraine headaches, have been well established and characterized as selective agonists of 5-HT1 B and 5-HT1 D serotonin receptors [4]. As a result, pharmacological studies on indole alkaloids mainly focus on the discovery of novel serotonin receptor agonists [5]. Compounds with an indole moiety have been explored for their activities on voltage-gated ion channels previously, and a series of tremorgenic indole alkaloids have been reported to potently block potassium channels in smooth muscle [6]. In a more recent study, a novel aryl indole compound was identified as a potent and selective blocker of N-type CaV2.2, and showed robust in vivo efficacy in inflammatory and neuropathic rat pain models [7].
In 2015, Liu et al. isolated a tryptophan-derived bisindole alkaloid pseudellone C (1) from the marine fungus Pseudallescheria ellipsoidea along with two other epipolythiopiperazine alkaloids [8]. Following this discovery, in 2017 Sathieshkumar et al. reported the total synthesis of pseudellone C (44% overall yield) [9], although the biological activity of these natural products remains unstudied. Here, we describe an efficient total synthesis of pseudellone C and several new bisindole alkaloid analogs. To investigate their pharmacological potential, we explored their activity in voltage-gated calcium channels (VGCCs) using FLIPR cell-based assays, and further characterized two potent low voltage-activated (LVA) T-type calcium channel (CaV3.x) blockers by whole-cell patch-clamp using an automated electrophysiology platform, QPatch 16 X. For the first time, we identified two “pseudellone” bisindole alkaloids as potent and selective CaV3.x blockers, demonstrating their potential as leads for the development of new analgesic and antiepileptic agents.
2. Results
2.1. Synthesis of Pseudellone C and Its Bisindole Alkaloid Analogs
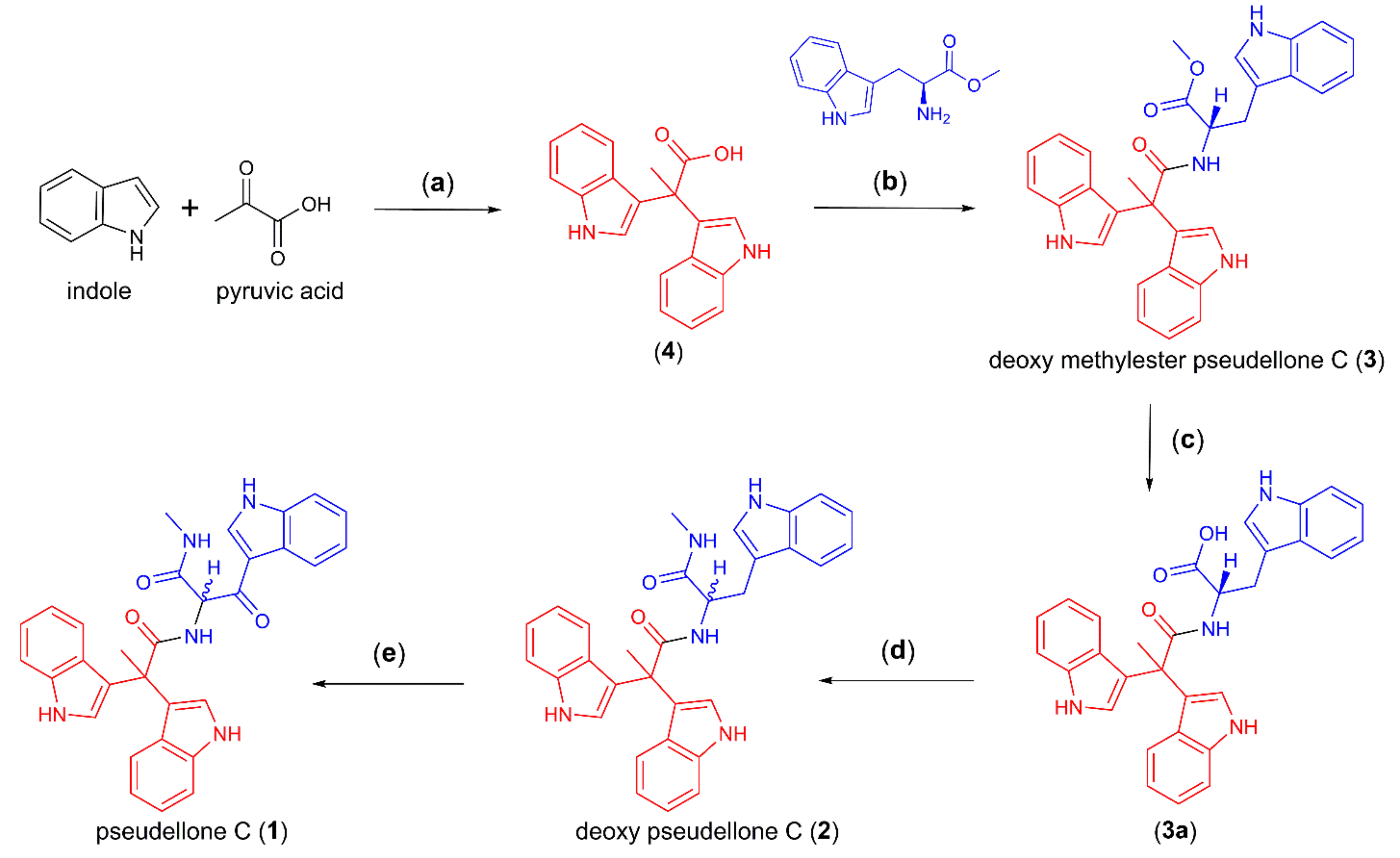
The synthesis of 2,2-bis (3,3′-indolyl) propionic acid (4) (85% yield) was achieved by a Friedel–Crafts condensation of indole and pyruvic acid [10]. As tryptophan is well known as a precursor for a wide range of pharmacologically important indole alkaloids [11,12], we employed a one-pot synthesis of deoxy-pseudellone C (2) by first coupling 4 with tryptophan methyl ester to yield 3 (38%) [9], followed by LiOH-mediated hydrolysis to yield the free acid 3a (85% yield), then DCC-initiated amide coupling of 3a with methylamine to yield deoxy-pseudellone C (3a) (37% yield, albeit with racemization about the single chiral center), and finally, DDQ oxidation of 2 to yield pseudellone C (1) (50% yield) (Scheme 1).
2.2. Evaluation of VGCC Activities of the Synthetic Compounds Using FLIPR Cell-Based Assays
Pseudellone C (1), along with its bisindole alkaloid analogs 2, 3, 3a, and its bisindole substrate 4, were evaluated for activity on VGCCs using FLIPR cell-based assays (see Table 1).
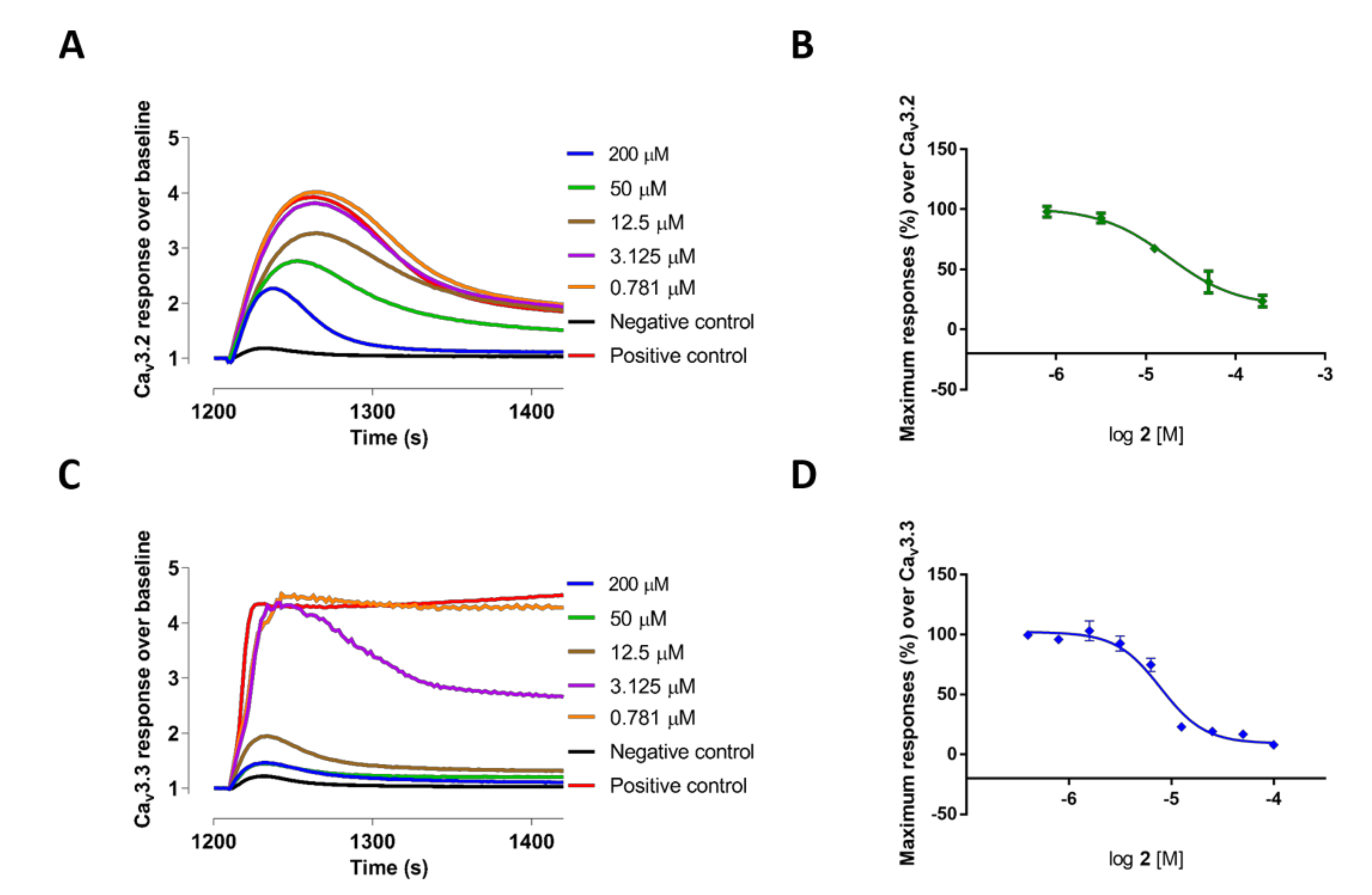
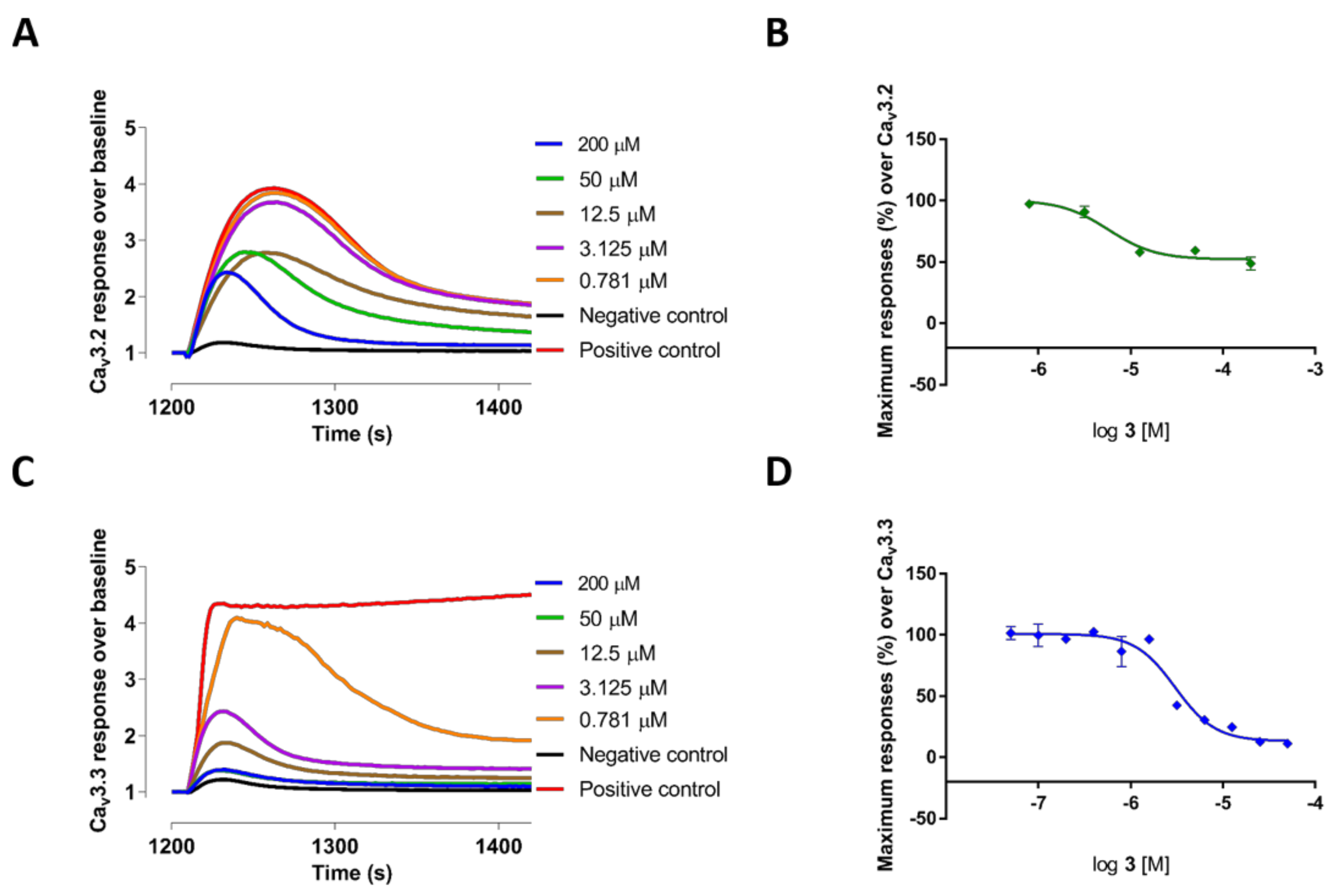
The 2,2-bis (3,3′-indolyl) propionic acid 4 and the bisindole alkaloid 3a showed poor inhibition of all the Ca2+ responses. In contrast, methyl-l-tryptophan-analog 3 showed good potency and selectivity for CaV3.3 currents measured in T-type window current assays with an IC50 value of 3.81 ± 1.08 µM (n = 3), which was >8-fold better than its potency at high voltage-activated (HVA) CaVs: 3 also potently blocked CaV3.2 responses with an IC50 value of 6.59 ± 0.66 µM (n = 3). Comparatively, deoxy pseudellone C (2), which had tryptophan moiety stabilized with a methyl amide group, had a 2-fold reduced potency for CaV3.3 window currents with an IC50 value of 7.71 ± 0.23 µM (n = 3) and a ~3-fold reduced potency for CaV3.2 window currents with an IC50 value of 18.24 ± 0.49 µM (n = 3), compared to 3. The natural product pseudellone C (1), which was generated by oxidation of the methylene group of the tryptophan moiety of deoxy pseudellone C (2), had >3-fold further reduced potency for CaV3.3 compared to 2. The fluorescent Ca2+ responses before and after addition of compounds 2 and 3, and their representative concentration response curves, are presented in Figure 1 (2) and Figure 2 (3), respectively.
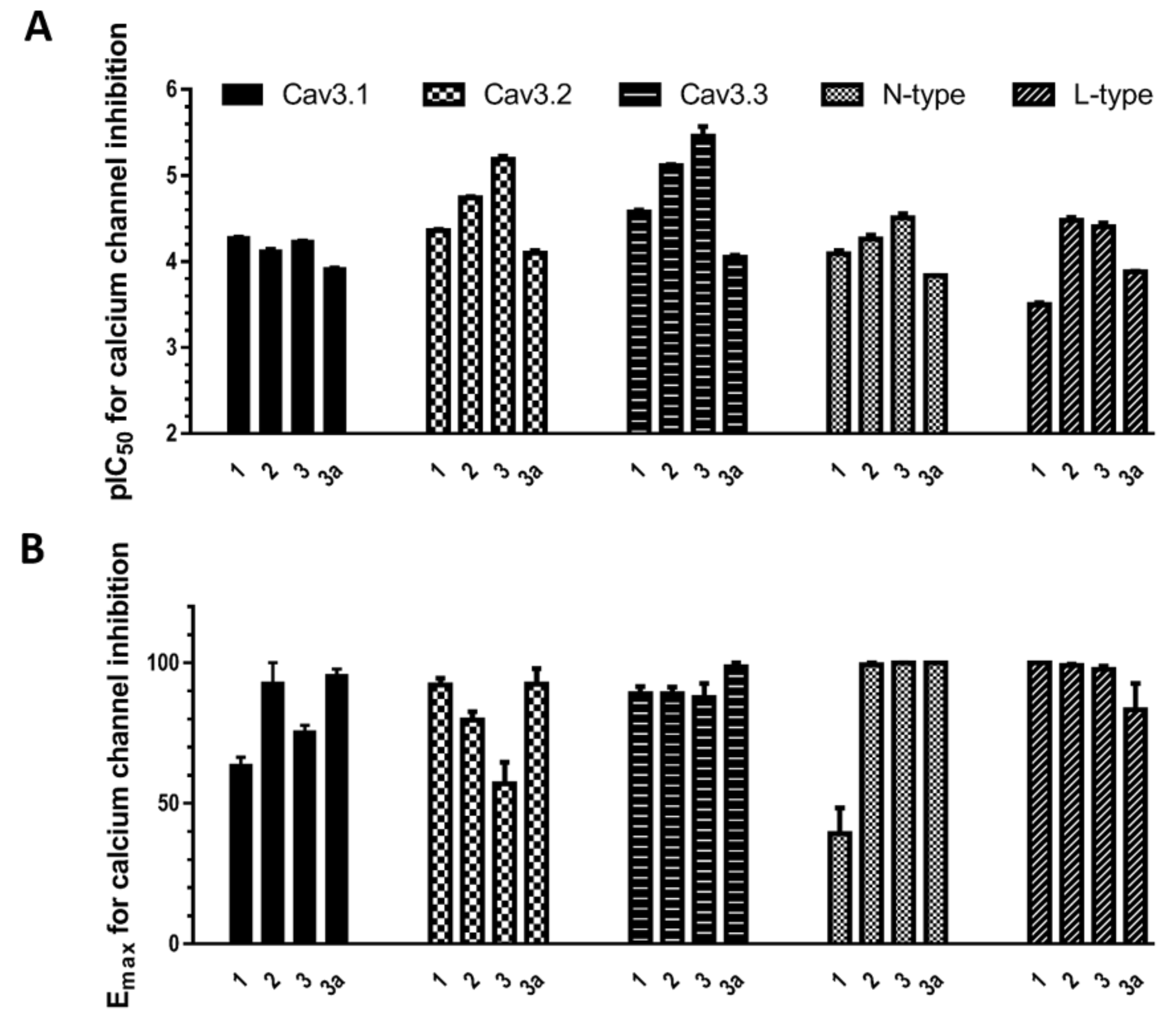
Interestingly, as indicated in Figure 2A,B and Figure 3, compound 3 showed potent partial blocking of the CaV3.2 window current. Also, pseudellone C is a weak calcium channel blocker that showed partial blocking of CaV3.1 and CaV2.2. Partial blocking of CaV3.x has been indicated to be useful for the treatment of epileptic behaviors by reducing synchronized oscillations of thalamocortical neurons [13,14]. Currently, compound 3 analogs are being designed for potential in rodent models of absence epilepsy and pain.
2.3. Electrophysiological Characterization of the Selective CaV3.x Blockers in QPatch Assays
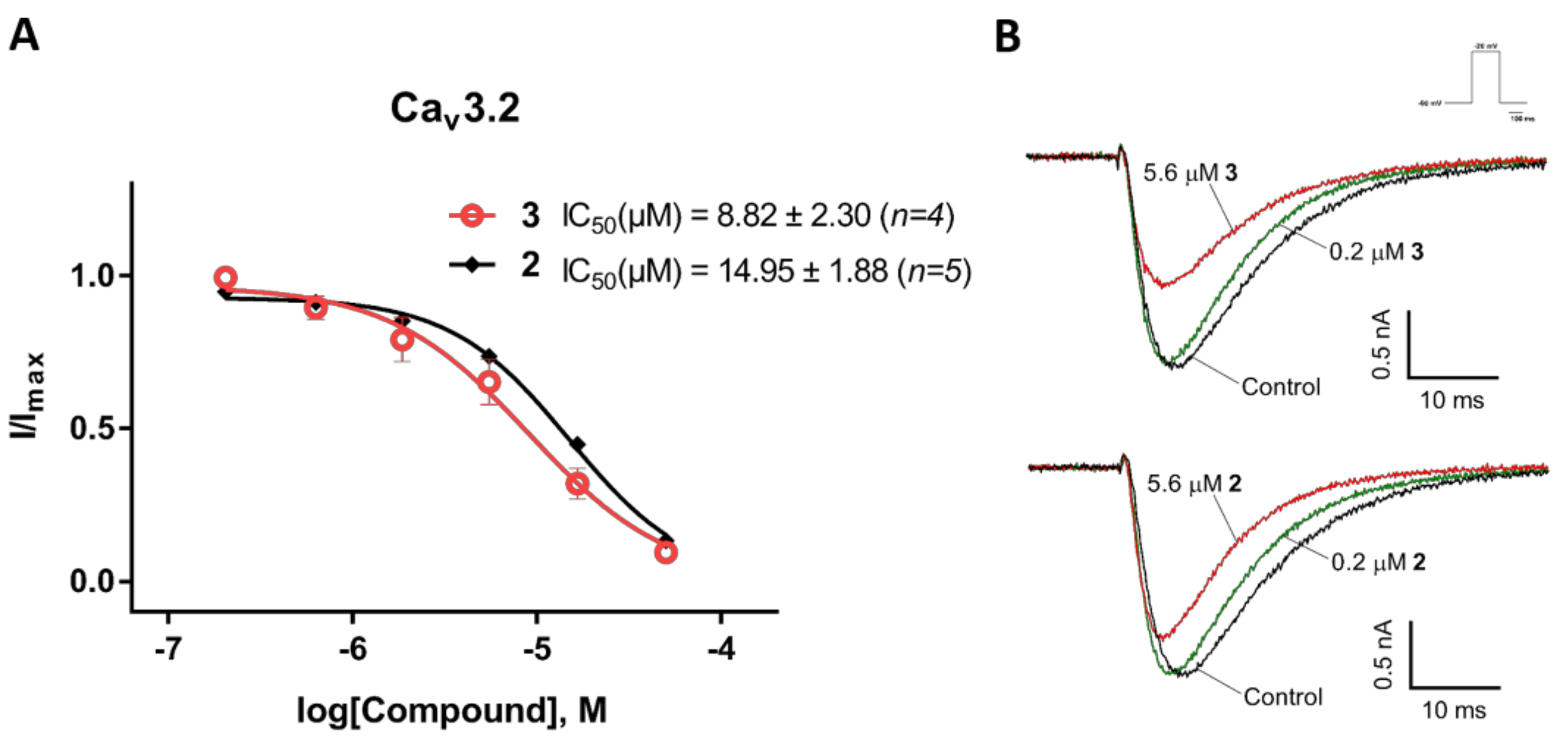
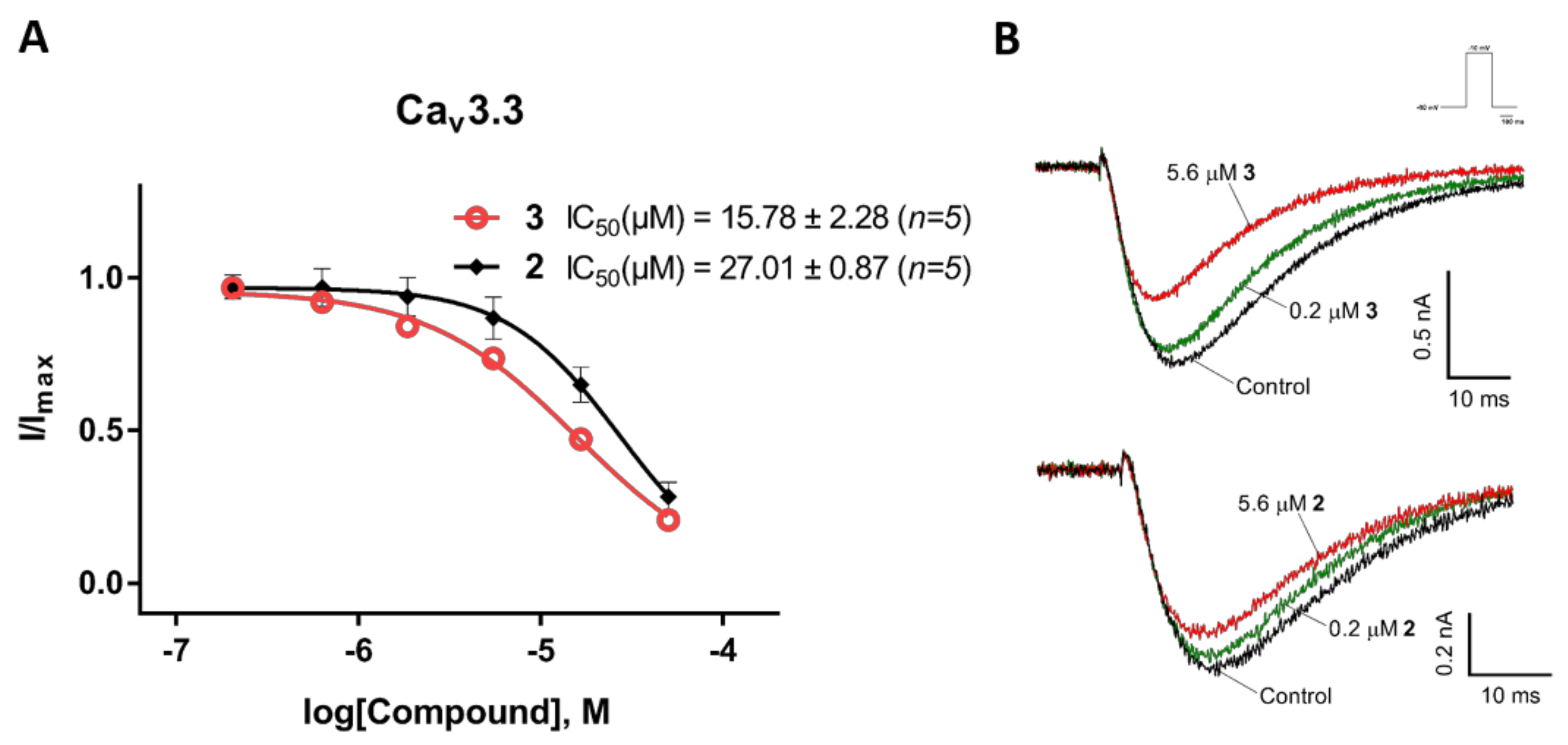
We also examined the effects of compounds 2 and 3 on the CaV3.x by whole-cell patch-clamp using the automated electrophysiology platform QPatch 16 X (Figure 4, Figure 5 and Figure 6). Surprisingly, both compounds showed preferential blocking of CaV3.1 currents (2: IC50 = 5.60 ± 0.26 µM (n = 5); 3: IC50 = 9.91 ± 3.00 µM (n = 6)). Unlike the full inhibition by 2 and 3 of CaV3.2 and CaV3.3 whole-cell currents (Emax value ≥ 95), 2 and 3 only showed partial block of the CaV3.1 current, with an Emax value of 70.31 ± 2.13 (n = 6) and 82.84 ± 2.55 (n = 5), respectively. Although 2 and 3 did not show comparably good potency for the CaV3.3 peak current compared to their effects on the CaV3.3 window current, low concentrations (200 nM) of 2 and 3 both showed weak inhibition (<10%) of the CaV3.3 peak current.
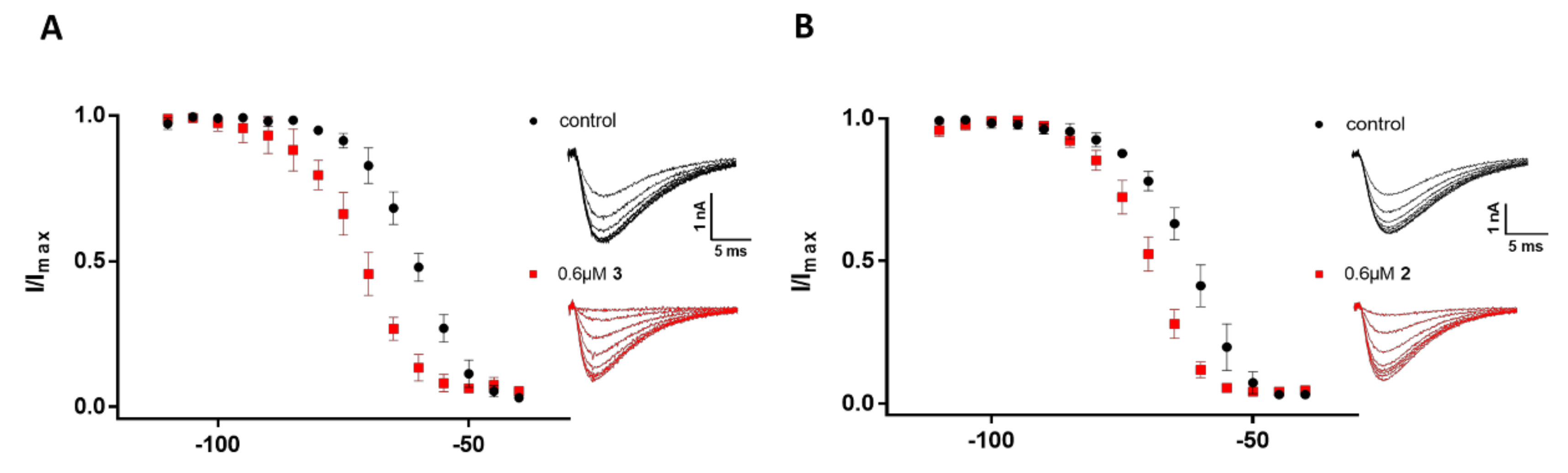
It is noteworthy that both 2 and 3 had a significant voltage-dependent effect on steady-state inactivation of CaV3.2, shifting half-maximal inactivation V50 to hyperpolarized potentials by ~6 mV and 11 mV, respectively (Figure 7). On average, control V50 (−65.40 ± 1.91 mV, n = 9) was shifted to −76.36 ± 1.56 mV (n = 5, p < 0.01) in the presence of compound 3, whereas in the presence of 2 the V50 was shifted to −72.40 ± 1.83 mV (n = 4, p < 0.05). In contrast, the voltage-dependence of steady-state inactivation for CaV3.1 and CaV3.3 was not significantly shifted in the presence of 2 and 3.
3. Discussion
In this study, we demonstrated that two pseudellone C-derived bisindole alkaloids, 2 and 3, are potent and selective CaV3.x current blockers that showed preferential inhibition of CaV3.3 window currents measured in FLIPR assays and CaV3.1 whole-cell currents measured in QPatch assays. The FLIPR window current assay applied to CaV3.x was permissive of a window current resulting from incomplete inactivation [15]. The specific CaV3.x window current made CaV3.x a privileged gate for the entry of extracellular Ca2+ during secretion, neurotransmission, and cell proliferation [16,17]. Therefore, the inhibition of window currents in CaV3.x should now be targeted for the development of analgesic, antiepileptic, and anticancer drugs. Novel CaV3.x blockers that showed prominent experimental analgesic and antiepileptic efficacy [14,15,18] such as TTA-A2, Z941, and Z944, all showed better potency for T-type window currents than the activated current generated from the channel-closed state [14,15].
As mentioned above, tryptophan is the precursor for a wide range of pharmacologically important indole alkaloids [11,12]. Tryptophan-derived indole alkaloids have been receiving wide attention in drug discovery for their promising therapeutic potentials, represented by the powerful chemotherapy drugs vinblastine and vincristine [19,20,21] and the antipsychotic and antihypertensive drug reserpine [22,23,24]. In short, the marine fungal natural product pseudellone C was produced via oxidation of the intermediate 2, which in turn was obtained from amidation of the carboxylic acid 3a. The acid 3a was generated from hydrolysis of 3, which was in turn obtained from amide coupling of 4 and l-tryptophan methyl ester. The resulting suite of compounds have been evaluated for activity on VGCCs using FLIPR cell-based assays, and 2 and 3 were further explored for effects on CaV3.x whole-cell currents in QPatch assays.
Both compounds 2 and 3 inhibited CaV3.3 currents in the FLIPR window current assay, but to our surprise, they did not show comparable potency for CaV3.3 whole-cell currents in QPatch assays, where affinity is determined by interactions with the resting state of the channel. In contrast, 2 and 3 both showed >7-fold better potency for CaV3.1-activated currents than CaV3.1 window currents, whereas for CaV3.2, these two compounds did not show any preference for CaV3.2 window currents or CaV3.2-activated currents. Moreover, by measuring the voltage dependence of steady-state inactivation of CaV3.x, the addition of both 2 and 3 significantly shifted the half-maximal inactivation V50 of CaV3.2 to more hyperpolarized potentials, whereas they did not significantly alter CaV3.1 and CaV3.3 channel inactivation. In brief, these two new CaV3.x blockers preferentially blocked either the window or activated current depending on the subtype and exclusively modulated channel inactivation kinetics for CaV3.2, which may help in differentiating subtype-specific properties in further pharmacological studies on T-type CaV3.x.
In summary, through a five-step total synthesis of marine fungal product pseudellone C, we achieved several bisindole alkaloid analogs of pseudellone C, two of which later revealed promising and selective inhibition of T-type CaV3.x currents. For the first time, naturally sourced bisindole alkaloids were characterized as CaV3.x blockers. Moreover, the exquisite differential inhibition of compounds 2 and 3 for the three T-type subtypes makes them useful pharmacological tools to probe the roles of individual CaV3.x subtypes, and they could be subjected to further structural modification for the development of analgesic, antiepileptic, and anticancer drug candidates through targeting CaV3.x.
4. Materials and Methods
4.1. General Procedure for Chemical Synthesis
All reagents were used as purchased from sigma Aldrich without further purification. Anhydrous solvents were used. Flash column chromatography was performed using ethylacetate (EtOAc) and petroleum ether as solvent. Preparative HPLC was used for the purification of the compounds using water and acetonitrile containing 0.01% TFA as solvent. 1H and 13C NMR spectra were recorded on Bruker (600 MHz) spectrometers. Data for 1H NMR spectra are reported as chemical shift (ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublets, dt = doublet of triplets, br s = broad singlet), and coupling constant (J in Hz). Electrospray ionization mass spectrometry (ESIMS) experiments were carried out on a LC/MSD (quadrupole) instrument in both positive and negative modes. High-resolution ESIMS spectra were obtained on a micrOTOF mass spectrometer by direct injection in MeCN at 3 μL/min using sodium formate clusters as an internal calibrant.
4.1.1. Synthesis of 2,2-di(1H-indol-3-yl)propanoic Acid (4)
To a solution of indole (2.34 g, 20 mmol) in anhydrous CH3CN (20 mL), we added pyruvic acid (1.32 g, 30 mmol, 1.5 equiv.) and FeCl3 (5 mol % 162 mg). The mixture was stirred under nitrogen at room temperature for 12 h until TLC revealed the disappearance of the starting material. The solvent was removed in vacuo, and the residue was purified by flash column chromatography (30% EtOAc/petroleum ether) to give 4 (2.58 g, 85%) as a reddish solid. 1H NMR (600 MHz, DMSO-d6): δ 10.85 (s, 2H), 7.33 (d, J = 8.09, 1H), 7.31 (d, J = 8.02, 1H), 7.03 (s, 1H), 7.02 (s, 1H), 7.00 (t, J = 8.09, 2H), 6.81 (t, J = 8.02, 2H), 1.97 (s, 3H). 13 C NMR (150 MHz, DMSO-d6): δ 176.1, 136.7, 125.9, 123.0, 120.7, 120.5, 118.1, 117.9, 111.4, 29.7, 25.9. HRMS (ESI) calcd for C19 H16 N2 O2 [M + Na]+ 327.1104 found m/z = 327.1096.
4.1.2. Synthesis of l-Tryptophan Methyl Ester
To a solution of l-tryptophan (0.204 g, 1 mmol) in anhydrous MeOH (5 mL), we added thionyl chloride (0.236 g, 2 equiv.) at 0 °C in dropwise and stirred for 12 h at room temperature until TLC revealed the disappearance of the starting material. The solvent was removed in vacuo and the residue was purified by flash column chromatography (45% ethyl acetate in petroleum ether) to give 5 (0.163 g, 75%) as a white solid. 1H NMR (600 MHz, DMSO-d6): δ 11.14 (s, 3H), 8.66 (br s, 2H), 7.51 (d, J = 8.12, 1H), 7.36 (d, J = 8.12, 1H), 7.25 (s, 1H), 7.08 (t, J = 7.42, 1H), 7.00 (t, J = 7.19, 1H), 4.19 (t, J = 5.84, 1H), 3.63 (s, 3H) 3.36–3.27 (m, 2H). 13C NMR (150 MHz, DMSO-d6): δ 169.7, 136.1, 126.8, 124.9, 121.1, 118.6, 117.9, 111.5, 106.3, 52.6, 52.5, 26.0. HRMS (ESI) calcd for C12 H15 N2 O2 [M + H]+ 219.1128 found m/z = 219.1137.
4.1.3. Synthesis of Methyl (2,2-di(1H-indol-3-yl)propanoyl)-l-tryptophanate (3)
A solution of 4 (0.304 g, 1 mmol) in DMF (3 mL) with DCC (200 mg, 1.5 mmol) and HOBT (135 mg, 1 mmol) was stirred at room temperature. After 15 min, L-methyl ester tryptophan (0.218 g, 1 mmol) and TEA (404 mg, 4 mmol) were added, and the reaction mixture was stirred at 60 °C for 1 h. After the completion of the reaction, ice-cold water was used to quench the reaction and the mixture was extracted with ethyl acetate (3 × 15). The extract was concentrated in vacuo, and the residue was purified via flash column chromatography (40% EtOAc in petroleum ether) to yield 3 as an off-white solid (38%). Here, [α]D30 = −21 (c = 0.01, methanol). 1H NMR (600 MHz, Acetone-d6, ppm): δ 10.11 (s, 1H), 10.06 (s, 1H), 9.76 (s, 1H), 7.41 (d, J = 8.15, 1H), 7.39 (d, J = 8.15, 1H), 7.32 (d, J = 8.15, 1H), 7.26 (d, J = 9.16, 1H), 7.26 (s, 1H), 7.24 (d, J = 8.03, 1H), 7.22 (d, J = 8.03, 1H), 7.05–7.00 (m, 4H), 6.93 (t, J = 7.06, 1H), 6.81 (t, J = 8.03, 1H), 6.79 (t, J = 7.13, 1H), 6.59 (d, J = 7.13, 1H), 6.10 (s, 1H), 4.77–4.74 (m, 1H), 3.57 (s, 3H), 3.10–2.96 (m, 2H), 2.85 (s, 3H). 13C NMR (150 MHz, Acetone-d6): δ 175.2, 173.0, 138.3, 137.4, 128.1, 127.1, 124.8, 124.6, 124.1, 122.1, 122.0, 121.9, 121.9, 121.5, 119.6, 119.5, 119.3, 118.8, 112.4, 112.3, 112.1, 109.8, 53.8, 52.1, 47.85, 28.0, 26.1. HRMS (ESI) calcd for C31 H28 N4 O3 [M + Na]+ 527.2054 found: m/z = 527.2054.
4.1.4. Synthesis of (2,2-di(1H-indol-3-yl)propanoyl)-l-tryptophan (3a)
A solution of 3 (0.035 g, 0.07 mol) in 1:1 (MeOH/H2O) and LiOH (3.22 mg, 2 equiv.) was stirred for 3 h at pH 3 to yield a white solid that was filtered, dried, and purified by flash column chromatography to yield 3a (29 mg, 85%) as a white solid. Here, [α]D30 = −14 (c = 0.01, methanol). 1H NMR (600 MHz, Acetone-d6): δ 10.13 (s, 1H), 10.07 (s, 1H), 9.77 (s, 1H), 7.41–7.38 (m, 2H), 7.33–7.30 (m, 2H), 7.29 (d, J = 2.48, 1H), 7.27 (d, J = 8.19, 1H), 7.20 (d, J = 8.04, 1H), 7.03–7.00 (m, 3H), 6.98 (d, J = 2.58, 1H), 6.92–6.90 (m, 1H), 6.82–6.79 (m, 1H), 6.77–6.74 (m, 1H), 6.62 (d, J = 7.05, 1H), 6.13 (d, J = 2.3, 1H), 4.70–4.74 (m, 1H), 3.12–3.03 (m, 2H), 2.06 (s, 3H). 13 C NMR (150 MHz, Acetone-d6): δ 175.5, 173.4, 138.3, 138.2, 137.4, 128.2, 127.1, 127.0, 124.8, 124.6, 124.1, 122.0, 121.98, 121.93, 121.8, 121.4, 119.7, 119.5, 119.4, 119.2, 119.0, 112.3, 112.4, 112.3, 112.0, 110.0, 53.6, 47.8, 28.0, 26.1. HRMS (ESI) calcd for C30 H26 N4 O3 [M + Na]+ 513.1897 found m/z = 513.1886.
4.1.5. Synthesis of (S)-N-(3-(1H-indol-3-yl)-1-(methylamino)-1-oxopropan-2-yl)-2,2-di(1H-indol-3-yl)propanamide (2)
A solution of 3a (0.15 g, 0.29 mmol) in DMF (3 mL) with DCC (200 mg, 1.5 mmol) and HOBT (135 mg, 1 mmol) was stirred at room temperature. After 15 min methyl amine (3 equiv., ethanolic solution) and TEA (404 mg, 4 mmol) was added, and the reaction mixture was stirred at 60 °C for 1 h. After the completion of the reaction, cold water was used to quench the reaction and the mixture was extracted with EtoAc (3 × 15 mL), and the combined EtOAc concentrated in vacuo with the residue purified by HPLC (phenomenex Luna C18 250 × 21 mm, 10 µm, 20 mL/min elution, 10% to 100% MeCN/H2 O with 0.01% TFA as modifier) yielded 2 (54 mg, 37%) as a white solid. Here, [α]D30 = 0.0 (c = 0.10, methanol) and [α]D30 = 0.0 (c = 0.10, methanol). 1H NMR (600 MHz, Acetone-d6): δ 10.17 (s, 1H), 10.07 (s, 1H), 9.89 (s, 1H), 7.45 (d, J = 8.19, 1H), 7.39 (d, J = 8.21, 1H), 7.36–7.33 (m, 2H), 7.19 (d, J = 2.45, 1H), 7.10–7.05 (m, 3H), 7.01 (t, J = 7.60, 1H), 6.93 (t, J = 7.60, 1H), 6.89–6.88 (m, 2H), 6.72 (d, J = 7.44, 1H), 6.68 (t, J = 7.45, 1H), 6.34 (s, 1H), 6.26–6.19 (m, 1H), 4.68 (q, J = 6.52, 1H), 3.33 (s, 1H), 3.13–2.96 (m, 2H) 2.31 (d, J = 4.50, 3H), 2.04 (s, 3H). 13C NMR (150, Acetone-d6): δ 175.4, 172.5, 138.3, 137.5, 128.3, 126.9, 126.6, 124.9, 124.3, 121.1, 122.0, 121.9, 121.5, 119.53, 119.52, 119.4, 119.2, 112.6, 112.5, 112.0, 54.7, 47.8, 28.5, 26.1, 25.9. HRMS (ESI) calcd for C31 H29 N5 O2 [M + Na]+ 526.2213 found m/z = 526.2228.
4.1.6. Synthesis of Pseudellone C (1)
A solution of 2 (35 mg, 0.07 mol) in 9:1 MeCN/H2O (1 mL) was treated with DDQ (32 mg, 0.14 mmol) in 6 portions over intervals of 10 min, and the resulting mixture was stirred at room temperature for 3 h, after which the residue was dissolved in EtOAc, washed with saturated NaCl (5 mL) and water (10 mL), dried over anhydrous Na2 SO4, and concentrated in vacuo to give residue, which was purified by HPLC (phenomenex Luna C18 250 × 21 mm, 10 µm, 20 mL/min elution, 10% to 100% MeCN/H2 O with 0.01% TFA as modifier) to afford pseudellone C (14.4 mg, 97%) as a white solid. Here, [α]D30 = 0.0 (c = 0.10, methanol) and 1H NMR (600 MHz, Acetone-d6): δ 11.17 (s, 1H), 10.25 (s, 1H), 10.21 (s, 1H), 8.49 (d, J = 3.24, 1H), 8.14 (d, J = 7.44, 1H), 7.59, (d, J = 6.07, 1H), 7.50 (dt, J = 1.03, 8.07, 1H), 7.47 (m, 2H), 7.41 (d, J = 8.15, 1H), 7.38 (d, J = 8.14, 1H), 7.29 (d, J = 2.48, 1H), 7.26 (d, J = 2.47, 1H), 7.23–7.20 (m, 1H), 7.19–7.15 (m, 1H), 7.10–7.07 (m, 1H), 7.05–7.01, (m, 1H), 6.96–6.91 (m, 1H), 6.83–6.81 (m, 1H), 6.45 (d, J = 5.09, 1H), 5.69 (d, J = 5.97, 1H), 2.45 (d, J = 4.87, 3H), 2.14 (s, 3H). 13C NMR (150 MHz, Acetone-d6): δ 187.0, 175.4, 168.0, 138.4, 138.3, 137.6, 136.7, 127.0, 126.9, 126.8, 125.1, 124.9, 124.5, 124.3, 124.2, 123.1, 122.6, 122.2, 122.1, 121.6, 121.4, 119.7, 119.6, 119.5, 112.9, 112.7, 112.5, 62.4, 47.9, 26.2, 26.1. HRMS (ESI) calcd for C31 H27 N5 O3 [M + Na]+ 540.2006 found m/z = 540.2026.
4.2. Cell Culture and Transient Expression
The human embryonic kidney 293 (HEK 293) cell lines (from Emmanuel Bourinet, Montpellier, France) stably expressing CaV3.2 or CaV3.3 were cultured under 5% carbon dioxide at 37 °C in Dulbecco’s Modified Eagle Medium (DMEM) Glutamax (Gibco, Life Technologies, Carlsbad, CA, USA) supplemented with 10% (v/v) fetal bovine serum (FBS), 100 U/mL penicillin, 100 μg/mL streptomycin (Gibco, Life Technologies), and 750 μg/mL geneticin (G418) (Gibco, Life Technologies). The Chinese Hamster Ovary (CHO) cell lines (Emmanuel Bourinet, Montpellier, France) expressing CaV3.1 were cultured under 5% carbon dioxide at 37 °C in Alpha Minimum Essential Media (MEM) Glutamax (Gibco, Life Technologies), supplemented with 10% (v/v) fetal bovine serum (FBS) and 300 μg/mL geneticin (G418) (Gibco, Life Technologies). The human neuroblastoma SH-SY5 Y cells (Victor Diaz, Goettingen, Germany) were cultured under 5% carbon dioxide at 37 °C in RPMI 1640 antibiotic-free medium (Invitrogen, Carlsbad, CA, USA), supplemented with 15% FBS and 2 mM GlutaMAX™ (Invitrogen). D-PBS (Gibco, Life Technologies) was used to wash the cells, and 0.25% Trypsin-EDTA (Gibco, Life Technologies) was used to detach cells from the flask surface. They were split in a ratio of 1:5 (ideally 1000 cells/cm2) when they reached 70%–80% confluence (every 2–3 days). Transiently transfected CaV3.1HEK293 T cells were used in FLIPR Assays. HEK 293 T cells were cultured under 5% carbon dioxide at 37 °C in DMEM supplemented with 10% (v/v) FBS. D-PBS was used to wash the cells, and 0.25% Trypsin-EDTA was used to detach cells from the flask surface. The cells were split and seeded at 6 million cells per T175 flask, to reach 70%–80% confluence after 24 h. The next day, 12 μg DNA of human CaV3.1 was incubated in 900 μL serum-free DMEM with 36 μL FuGENE HD transfection reagent (Promega Corporation, Madison, WI, US) (1:3 DNA/Fugene ratio) for 20 min, and then the mixture was added into the cell flask slowly, drop by drop. After the transfection, the cells were cultured under 5% carbon dioxide at 37 °C for 24 h and then moved to a 28 °C incubator.
4.3. T-Type Calcium Channel Window Current FLIPR Assays
HEK 293 cells stably expressing CaV3.2 or CaV3.3 were seeded into 384-well black wall clear bottom plates (Corning, Lowell, MA, USA) at a density of 30,000 cells per well. Transiently transfected CaV3.1HEK293 T cells were seeded into 384-well black wall clear bottom plates at a density of 60,000 cells per well. Once the cells reached 90%–95% confluence after 24 h, the media were removed from the wells and replaced with 20 μL of 10% calcium 4 dye (Molecular Devices, Sunnyvale, CA, USA) in HBSS-HEPES (containing 5 mM KCl, 10 mM HEPES, 140 mM NaCl, 10 mM glucose, and 0.5 mM CaCl2, pH 7.4) with 0.1% bovine serum albumin (BSA). The cells were incubated for 30 min at 37 °C in the presence of 5% carbon dioxide. Each well on the reagent plates for the first addition contained 15 μL different concentrations of compounds dissolved in HBSS-HEPES containing 0.1% BSA and <0.1% DMSO and was incubated for 20 min after loading. Positive and negative controls contained 15 μL of HBSS-HEPES (0.1% BSA) alone. The plates were placed in the FLIPRTETRA (Molecular Devices, Sunnyvale, CA, USA) programmed to measure maximum fluorescence intensity following a second addition of the agonist 5 mM CaCl2. The data acquisition parameters were adjusted as follows: Baseline fluorescence of 1500–2000 arbitrary fluorescence units (AFU), emission wavelength of 515–575 nm, and excitation wavelength of 470–495 nm. HBSS-HEPES (0.1% BSA) was used in the second addition as a negative control. One fluorescence reading was taken before the second addition. Then the fluorescence readings were recorded every two seconds for 300 s. Raw fluorescence readings in the form of relative light units were converted to response over baseline using ScreenWorks® (Molecular Devices, version 3.2.0.14) software. Using this approach, pimozide (CaV3.2, IC50 = 4.2 ± 1.06 μM (n = 3); CaV3.3, IC50 = 7.71 ± 0.70 μM (n = 3)) and mibefradil (CaV3.2, IC50 = 1.02 ± 0.14 μM (n = 3); CaV3.3, 2.0 ± 0.2 μM (n = 3)) inhibited CaV3.x calcium influx with IC50 s consistent with literature values [25].
4.4. HVA Calcium Channel FLIPR Assays
SH-SY5Y cells were seeded into 384-well black wall clear bottom plates at a density of 15,000 cells per well, resulting in 90%–95% confluence after 24 h. The media were then removed from the wells and replaced with 20 μL of 10% calcium 4 dye (Molecular Devices) in physiological salt solution (PSS) (containing 5.9 mM KCl, 1.4 mM MgCl2, 10 mM HEPES, 1.2 mM NaH2 PO4, 5 mM NaHCO3, 140 mM NaCl, 11.5 mM glucose, and 1.8 mM CaCl2, pH 7.4) with 0.1% BSA. As reported [26], for N-type calcium channel FLIPR assays the cells were pre-incubated with 10 µM nifedipine added in the dye to ensure full inhibition of L-type calcium responses. For L-type calcium channel FLIPR assays, the cells were pre-incubated with 1 µM CVID added in the dye to ensure full inhibition of N-type calcium responses. Positive control on the first reagent plate contained 15 μL of PSS (0.1% BSA), whereas PSS (0.1% BSA) containing 1 µM CVID and 10 µM nifedipine (final concentration) was used as a negative control. The fluorescence readings were recorded and converted as described previously [26], and agonist containing 90 mM KCl + 5 mM CaCl2 was used in the second addition.
4.5. Whole-Cell Patch-Clamp Electrophysiology
Whole-cell patch-clamp experiments were performed on an automated electrophysiology platform QPatch 16 X (Sophion Bioscience A/S, Ballerup, Denmark) in single-hole configuration using 16-channel planar patch chip QPlates (Sophion Bioscience A/S). The extracellular recording solution contained, in mM: TEACl 157, MgCl2 0.5, CaCl2 5, and HEPES 10; pH 7.4 adjusted with TEAOH; and osmolarity 320 mOsm. The intracellular pipette solution contained, in mM: CsF 140, EGTA 1, HEPES 10, and NaCl 10; pH 7.2 adjusted with CsOH; and osmolarity 325 mOsm. Compounds were diluted in extracellular recording solution with 0.1% BSA at the concentrations stated (DMSO ≤ 0.1%), and the effects of compounds were compared to the control (extracellular solution with 0.1% BSA) parameters within the same cell. Compounds’ incubation time varied from two (for the highest concentration) to five (for the lowest concentration) minutes by applying the voltage protocol 10–30 times at 10 s intervals to ensure steady-state inhibition was achieved. The effects of compounds were obtained using 200 ms voltage steps to peak potential from a holding potential of −90 mV. The steady-state inactivation kinetics of CaV3.x currents were examined by applying steps lasting 1 s from −110 mV to the indicated voltages in 5-mV increments, followed by 60-ms test pulses to −20 mV. Data were fitted with a single Boltzmann distribution: I/Imax = {1 + exp[V − V50]/k}−1, where V50 is the half-availability voltage and k is the slope factor. Off-line data analysis was performed using QPatch Assay Software v5.6 (Sophion Bioscience A/S) and Excel 2013 (Microsoft Corporation, Redmond, WA, USA).
4.6. Data Analysis
Data were plotted and analyzed using GraphPad Prism v7.0 (GraphPad Software Inc., San Diego, CA, USA). A four-parameter logistic Hill equation with variable Hill coefficients was fitted to the data for concentration-response curves. Data are means ± SEM of n independent experiments. Statistical analysis was performed with a paired Student’s t-test with statistical significance at p < 0.05.
Author Contributions
D.W. and P.N. are joint first authors. Conceptualization, R.J.L. and R.J.C.; methodology, D.W., P.N., and L.R.; formal Analysis, D.W. and P.N.; chemical Investigation, P.N.; biological investigation, D.W.; data curation, D.W. and P.N.; writing–original draft preparation, D.W. and P.N.; writing–review and editing, R.J.L. and R.J.C.; supervision, R.J.L., L.R., and R.J.C; funding acquisition, R.J.L.
Funding
This research was funded by the NHMRC Program, grant number APP1072113, and the APC was funded by the NHMRC Principal Research Fellowship.
Acknowledgments
The authors thank Emmanuel Bourinet for donating stable cell lines for T-type calcium channels and Gerald W. Zamponi for donating plasmids for T-type calcium channels.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Fattorusso, E.; Taglialatela-Scafati, O. Modern Alkaloids: Structure, Isolation, Synthesis, and Biology; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Gul, W.; Hamann, M.T. Indole alkaloid marine natural products: An established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci. 2005, 78, 442–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netz, N.; Opatz, T. Marine indole alkaloids. Mar. Drugs 2015, 13, 4814–4914. [Google Scholar] [CrossRef] [PubMed]
- Sheftell, F.D.; Bigal, M.E.; Tepper, S.J.; Rapoport, A.M. Sumatriptan: A decade of use and experience in the treatment of migraine. Expert Rev. Neurother. 2004, 4, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Kochanowska-Karamyan, A.J.; Hamann, M.T. Marine indole alkaloids: Potential new drug leads for the control of depression and anxiety. Chem. Rev. 2010, 110, 4489–4497. [Google Scholar] [CrossRef] [PubMed]
- Knaus, H.-G.; McManus, O.B.; Lee, S.H.; Schmalhofer, W.A.; Garcia-Calvo, M.; Helms, L.M.; Sanchez, M.; Giangiacomo, K.; Reuben, J.P. Tremorgenic indole alkaloids potently inhibit smooth muscle high-conductance calcium-activated potassium channels. Biochemistry 1994, 33, 5819–5828. [Google Scholar] [CrossRef] [PubMed]
- Tyagarajan, S.; Chakravarty, P.K.; Park, M.; Zhou, B.; Herrington, J.B.; Ratliff, K.; Bugianesi, R.M.; Williams, B.; Haedo, R.J.; Swensen, A.M. A potent and selective indole N-type calcium channel (Ca v 2.2) blocker for the treatment of pain. Bioorg. Med. Chem. Lett. 2011, 21, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, H.-J.; Xu, M.-Y.; Ju, Y.-C.; Wang, L.-Y.; Xu, J.; Yang, D.-P.; Lan, W.-J. Pseudellones A–C, three alkaloids from the marine-derived fungus Pseudallescheria ellipsoidea F42–3. Org. Lett. 2015, 17, 5156–5159. [Google Scholar] [CrossRef] [PubMed]
- Sathieshkumar, P.P.; Latha, P.; Nagarajan, R. Total Synthesis of Pseudellone C. Eur. J. Org. Chem. 2017, 2017, 3161–3164. [Google Scholar] [CrossRef]
- Garbe, T.R.; Kobayashi, M.; Shimizu, N.; Takesue, N.; Ozawa, M.; Yukawa, H. Indolyl carboxylic acids by condensation of indoles with α-keto acids. J. Nat. Products 2000, 63, 596–598. [Google Scholar] [CrossRef]
- Talapatra, S.K.; Talapatra, B. Quinine. Cinchona Alkaloids (Tryptophan Derived Quinoline Alkaloids). In Chemistry of Plant Natural Products; Springer: Berlin, Germany, 2015; pp. 855–874. [Google Scholar]
- Ramani, S.; Patil, N.; Nimbalkar, S.; Jayabaskaran, C. Alkaloids derived from tryptophan: Terpenoid indole alkaloids. In Natural Products; Springer: Berlin, Germany, 2013; pp. 575–604. [Google Scholar]
- Crunelli, V.; Lightowler, S.; Pollard, C. AT-type Ca2+ current underlies low-threshold Ca2+ potentials in cells of the cat and rat lateral geniculate nucleus. J. Physiol. 1989, 413, 543–561. [Google Scholar] [CrossRef] [PubMed]
- Tringham, E.; Powell, K.L.; Cain, S.M.; Kuplast, K.; Mezeyova, J.; Weerapura, M.; Eduljee, C.; Jiang, X.; Smith, P.; Morrison, J.-L. T-type calcium channel blockers that attenuate thalamic burst firing and suppress absence seizures. Sci. Transl. Med. 2012, 4, 121ra119. [Google Scholar] [CrossRef] [PubMed]
- Uebele, V.N.; Nuss, C.E.; Fox, S.V.; Garson, S.L.; Cristescu, R.; Doran, S.M.; Kraus, R.L.; Santarelli, V.P.; Li, Y.; Barrow, J.C. Positive allosteric interaction of structurally diverse T-type calcium channel antagonists. Cell Biochem. Biophys. 2009, 55, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.S.; Macdonald, T.L. The pharmacology and regulation of T type calcium channels: New opportunities for unique therapeutics for cancer. Cell Calcium 2006, 40, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Rossier, M.F. T-type calcium channel: A privileged gate for calcium entry and control of adrenal steroidogenesis. Front. Endocrinol. 2016, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Francois, A.; Kerckhove, N.; Meleine, M.; Alloui, A.; Barrere, C.; Gelot, A.; Uebele, V.N.; Renger, J.J.; Eschalier, A.; Ardid, D. State-dependent properties of a new T-type calcium channel blocker enhance CaV3. 2 selectivity and support analgesic effects. Pain 2013, 154, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Noble, R.L. The discovery of the vinca alkaloids—Chemotherapeutic agents against cancer. Biochem. Cell Biol. 1990, 68, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-J.; Rahmani, R. Preclinical and clinical pharmacology of vinca alkaloids. Drugs 1992, 44, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Finnbladder, N.B.C.S.G.; de Tratamiento Oncologico, C.U.; EORTC Genito-Urinary Group; Australian Bladder Cancer Study Group; National Cancer Institute of Canada Clinical Trials Group. Neoadjuvant cisplatin, methotrexate, and vinblastine chemotherapy for muscle-invasive bladder cancer: A randomised controlled trial. Lancet 1999, 354, 533–540. [Google Scholar]
- Shore, P. Reserpine: A survey of its pharmacology. In Alterations of Chemical Equilibrium in the Nervous System; Springer: Berlin, Germany, 1971; pp. 349–356. [Google Scholar]
- Jerie, P. Milestones of cardiovascular therapy. IV. Reserpine. Casopis Lekaru Ceskych 2007, 146, 573–577. [Google Scholar] [PubMed]
- Preskorn, S.H. The evolution of antipsychotic drug therapy: Reserpine, chlorpromazine, and haloperidol. J. Psychiatr. Pract. 2007, 13, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Van Deusen, A.L.; Vitko, I.; Babu, D.A.; Davies, L.A.; Huynh, N.; Cheng, H.; Yang, N.; Barrett, P.Q.; Perez-Reyes, E. Validation of high throughput screening assays against three subtypes of Cav3 T-type channels using molecular and pharmacologic approaches. Assay Drug Dev. Technol. 2007, 5, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.R.; Vetter, I.; Ragnarsson, L.; Lewis, R.J. Expression and pharmacology of endogenous Cav channels in SH-SY5Y human neuroblastoma cells. PLoS ONE 2013, 8, e59293. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Synthesis of pseudellone C (1) and analogs 2, 3, 3a, and 4. (a) MeCN, FeCl3, r.t., 12 h, 85%; (b) DCC, benzotriazole, TEA, DMF, 60 °C, 1 h, 38%; (c) LiOH, MeOH/H2O (1:1), r.t., 5 h, 85%; (d) DCC, methylamine, benzotriazole, DMF, 60 °C, 1 h, 37%; (e) DDQ, MeOH/H2O (1:1), 50%.
Scheme 1.
Synthesis of pseudellone C (1) and analogs 2, 3, 3a, and 4. (a) MeCN, FeCl3, r.t., 12 h, 85%; (b) DCC, benzotriazole, TEA, DMF, 60 °C, 1 h, 38%; (c) LiOH, MeOH/H2O (1:1), r.t., 5 h, 85%; (d) DCC, methylamine, benzotriazole, DMF, 60 °C, 1 h, 37%; (e) DDQ, MeOH/H2O (1:1), 50%.

Figure 1.
(A) Representative fluorescent traces of the CaV3.2 window current before and after addition of compound 2; (B) the concentration-response curve for compound 2 (IC50 = 18.16 μM, n = 4); (C) representative fluorescent traces of the CaV3.3 window current before and after addition of compound 2; and (D) the concentration response curve for compound 2 (IC50 value of 7.83 μM, n = 4).
Figure 1.
(A) Representative fluorescent traces of the CaV3.2 window current before and after addition of compound 2; (B) the concentration-response curve for compound 2 (IC50 = 18.16 μM, n = 4); (C) representative fluorescent traces of the CaV3.3 window current before and after addition of compound 2; and (D) the concentration response curve for compound 2 (IC50 value of 7.83 μM, n = 4).

Figure 2.
(A) Representative fluorescent traces of the CaV3.2 window current before and after addition of compound 3; (B) the concentration-response curve for compound 3 (IC50 = 5.77 μM, n = 4); (C) representative fluorescent traces of the CaV3.3 window current before and after addition of compound 3; and (D) the concentration response curve for compound 3 (IC50 value of 3.05 μM, n = 4).
Figure 2.
(A) Representative fluorescent traces of the CaV3.2 window current before and after addition of compound 3; (B) the concentration-response curve for compound 3 (IC50 = 5.77 μM, n = 4); (C) representative fluorescent traces of the CaV3.3 window current before and after addition of compound 3; and (D) the concentration response curve for compound 3 (IC50 value of 3.05 μM, n = 4).

Figure 3.
(A) pIC50 values for bisindole alkaloids 1, 2, 3, and 3a inhibition of T-, N- and L-type calcium channels; (B) Emax values for bisindole alkaloids 1, 2, 3, and 3a inhibition of calcium channels. Here, 1 produced 63.56% ± 2.80% (n = 3) inhibition for CaV3.1 and 39.26% ± 9.18% (n = 3) inhibition for N-type CaV2.2, and 3 produced 75.47% ± 2.29% (n = 3) inhibition for CaV3.1 and 56.91% ± 7.74% (n = 3) inhibition for CaV3.2.
Figure 3.
(A) pIC50 values for bisindole alkaloids 1, 2, 3, and 3a inhibition of T-, N- and L-type calcium channels; (B) Emax values for bisindole alkaloids 1, 2, 3, and 3a inhibition of calcium channels. Here, 1 produced 63.56% ± 2.80% (n = 3) inhibition for CaV3.1 and 39.26% ± 9.18% (n = 3) inhibition for N-type CaV2.2, and 3 produced 75.47% ± 2.29% (n = 3) inhibition for CaV3.1 and 56.91% ± 7.74% (n = 3) inhibition for CaV3.2.

Figure 4.
Inhibition of CaV3.1 current by compounds 2 and 3. (A) Concentration-response curves of compounds 2 and 3 on recombinant hCaV3.1 channels (n = 5–6). Data are means ± SEM. (B) Representative ICa during 200 ms depolarizations to Vmax (−20 mV) from a holding potential of −90 mV before and after perfusions of 0.2 µM and 5.6 µM of compounds 2 and 3, as indicated.
Figure 4.
Inhibition of CaV3.1 current by compounds 2 and 3. (A) Concentration-response curves of compounds 2 and 3 on recombinant hCaV3.1 channels (n = 5–6). Data are means ± SEM. (B) Representative ICa during 200 ms depolarizations to Vmax (−20 mV) from a holding potential of −90 mV before and after perfusions of 0.2 µM and 5.6 µM of compounds 2 and 3, as indicated.

Figure 5.
Inhibition of CaV3.2 current by compounds 2 and 3. (A) Concentration-response curves of compounds 2 and 3 on recombinant hCaV3.2 channels (n = 4–5). Data are means ± SEM. (B) Representative ICa during 200 ms depolarizations to Vmax (−20 mV) from a holding potential of −90 mV before and after perfusions of 0.2 µM and 5.6 µM of compounds 2 and 3, as indicated.
Figure 5.
Inhibition of CaV3.2 current by compounds 2 and 3. (A) Concentration-response curves of compounds 2 and 3 on recombinant hCaV3.2 channels (n = 4–5). Data are means ± SEM. (B) Representative ICa during 200 ms depolarizations to Vmax (−20 mV) from a holding potential of −90 mV before and after perfusions of 0.2 µM and 5.6 µM of compounds 2 and 3, as indicated.

Figure 6.
Inhibition of CaV3.3 current by compounds 2 and 3. (A) Concentration-response curves of compounds 2 and 3 on recombinant hCaV3.3 channels (n = 5). Data are means ± SEM. (B) Representative ICa during 200 ms depolarizations to Vmax (−10 mV) from a holding potential of −90 mV before and after perfusions of 0.2 µM and 5.6 µM of compounds 2 and 3, as indicated.
Figure 6.
Inhibition of CaV3.3 current by compounds 2 and 3. (A) Concentration-response curves of compounds 2 and 3 on recombinant hCaV3.3 channels (n = 5). Data are means ± SEM. (B) Representative ICa during 200 ms depolarizations to Vmax (−10 mV) from a holding potential of −90 mV before and after perfusions of 0.2 µM and 5.6 µM of compounds 2 and 3, as indicated.

Figure 7.
Comparison of the steady-state inactivation kinetics of CaV3.2 before (black) and after (red) the addition of 0.6 µM of 2 and 3. Steps lasting 1 s were given directly from −110 mV to the indicated voltages in 5-mV increments, followed by 60-ms test pulses to −20 mV. (A) Normalized I/Imax under conditioning steps from −110 mV to −40 mV (n = 5), along with representative elicited ICa under potentials from −95 mV to −60 mV before (black) and after (red) the addition of 0.6 µM of 3. Error bars represent the SEM. (B) Normalized I/Imax under conditioning steps from −110 mV to −40 mV (n = 4), along with representative elicited ICa under potentials from −95 mV to −60 mV before (black) and after (red) the addition of 0.6 µM of 2. Error bars represent the SEM.
Figure 7.
Comparison of the steady-state inactivation kinetics of CaV3.2 before (black) and after (red) the addition of 0.6 µM of 2 and 3. Steps lasting 1 s were given directly from −110 mV to the indicated voltages in 5-mV increments, followed by 60-ms test pulses to −20 mV. (A) Normalized I/Imax under conditioning steps from −110 mV to −40 mV (n = 5), along with representative elicited ICa under potentials from −95 mV to −60 mV before (black) and after (red) the addition of 0.6 µM of 3. Error bars represent the SEM. (B) Normalized I/Imax under conditioning steps from −110 mV to −40 mV (n = 4), along with representative elicited ICa under potentials from −95 mV to −60 mV before (black) and after (red) the addition of 0.6 µM of 2. Error bars represent the SEM.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Effects of pseudellone C-based bisindole compounds on low voltage-activated (LVA) and high voltage-activated (HVA) calcium channels.
Table 1.
Effects of pseudellone C-based bisindole compounds on low voltage-activated (LVA) and high voltage-activated (HVA) calcium channels.
| Compounds | LVA CaVs (IC50 (µM) N = 3) | HVA CaVs (IC50 (µM) N = 3) | |||
|---|---|---|---|---|---|
| CaV3.1 | CaV3.2 | CaV3.3 | CaV2.2 | CaV1 | |
| 1 | 53.49 ± 2.72 | 43.96 ± 1.60 | 26.59 ± 1.23 | 82.55 ± 7.60 | 322.00 ± 21.63 |
| 2 | 76.36 ± 5.39 | 18.24 ± 0.49 | 7.71 ± 0.23 | 55.29 ± 5.58 | 33.66 ± 2.62 |
| 3 | 58.57 ± 2.62 | 6.59 ± 0.66 | 3.81 ± 1.08 | 31.32 ± 3.40 | 39.76 ± 3.98 |
| 3a | 122.23 ± 5.96 | 80.65 ± 6.21 | 90.05 ± 4.99 | 146.70 ± 1.72 | 132.03 ± 1.63 |
| 4 | 72.28 ± 0.41 | 132.17 ± 6.13 | 119.17 ± 8.66 | 179.30 ± 19.68 | 264.30 ± 11.89 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, D.; Neupane, P.; Ragnarsson, L.; Capon, R.J.; Lewis, R.J. Synthesis of Pseudellone Analogs and Characterization as Novel T-type Calcium Channel Blockers. Mar. Drugs 2018, 16, 475. https://doi.org/10.3390/md16120475
AMA Style
Wang D, Neupane P, Ragnarsson L, Capon RJ, Lewis RJ. Synthesis of Pseudellone Analogs and Characterization as Novel T-type Calcium Channel Blockers. Marine Drugs. 2018; 16(12):475. https://doi.org/10.3390/md16120475
Chicago/Turabian StyleWang, Dan, Pratik Neupane, Lotten Ragnarsson, Robert J. Capon, and Richard J. Lewis. 2018. "Synthesis of Pseudellone Analogs and Characterization as Novel T-type Calcium Channel Blockers" Marine Drugs 16, no. 12: 475. https://doi.org/10.3390/md16120475
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.