4. Materials and Methods
All anaerobic and moisture-sensitive manipulations were carried out with standard Schlenk techniques under argon. Solvents were dried and distilled by standard procedures. 1H- NMR and 13C-NMR spectra were recorded in CDCl3 on a Bruker Ascend-400 400 MHz or Bruker Ascend-500 500 MHz at room temperature. Chemical shifts (δ) are reported in ppm and are referenced to chloroform (δ 7.26 ppm for 1H, δ 77.16 ppm for 13C). Data for NMR spectra are reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, quint. = quintet, sext. = sextet, m = multiplet, br = broad signal, J = coupling constant in Hz. HRMS were recorded on an Agilent 6530-Q-TOF mass spectrometer equipped with an Agilent 1260- HPLC. Optical rotations were measured on a PerkinElmer 241 MC polarimeter.
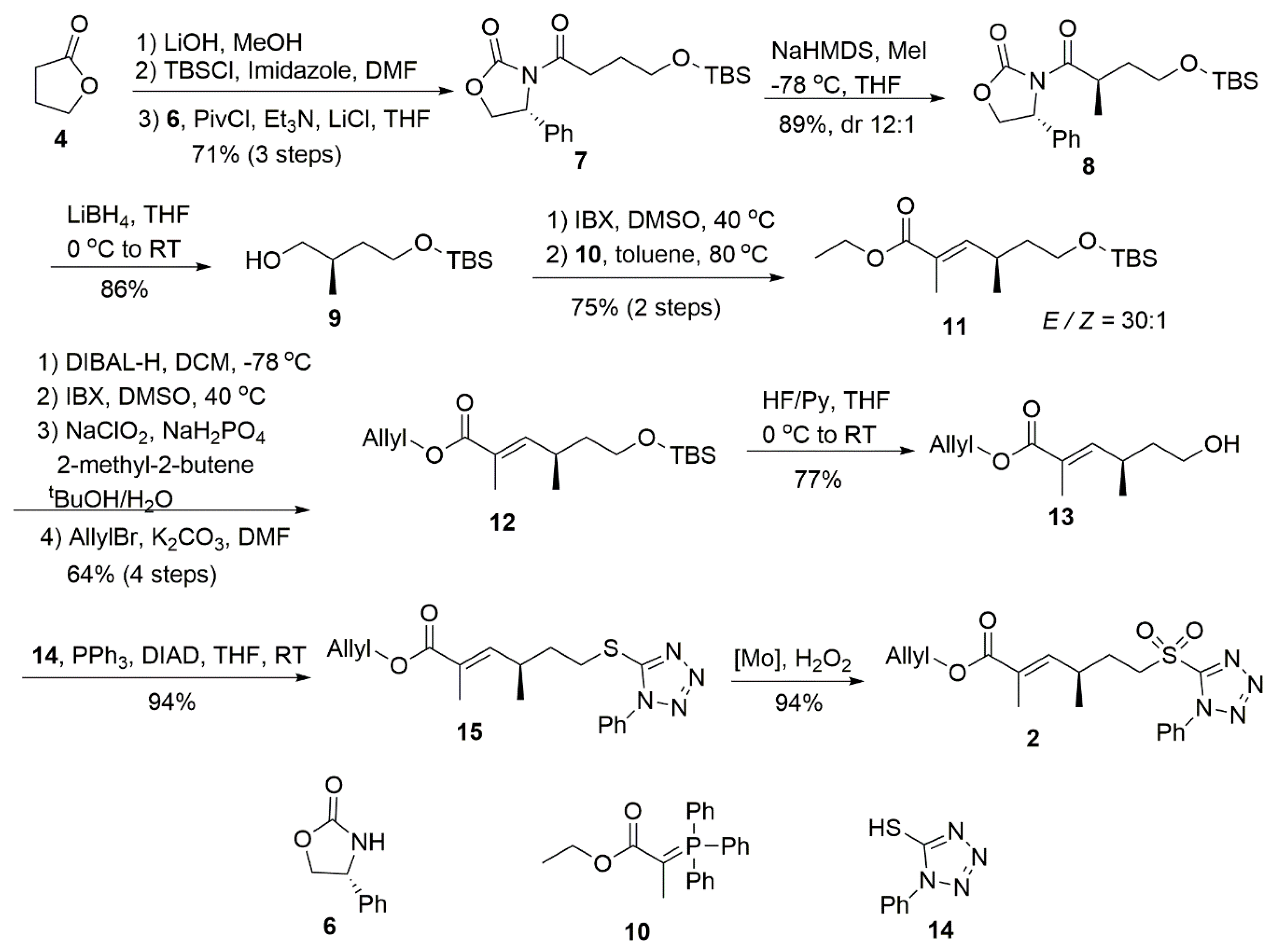
4.1. (R)-3-(4-((Tert-Butyldimethylsilyl)Oxy)Butanoyl)-4-Phenyloxazolidin-2-One (7)
To a solution of the lactone (5 g, 58 mmol) in MeOH (50 mL) at room temperature was added LiOH (2.44 g, 58 mmol) and stirred overnight. The solvent was removed from the reaction and the residue was dissolved in dimethylformamide (50 mL) at 0 °C. Imidazole (8 g, 116 mmol) was added to this solution, which was followed by TBSCl (8.7 g, 58 mmol) in two portions over 15 min. The reaction mixture was warmed to room temperature overnight with stirring and then diluted with 1 M HCl and extracted with EtOAc (3 × 100 mL). The combined organic layers were washed with brine and dried over Na2SO4. The organic layer was removed under vacuum and the crude acid was used without further purification.
To a stirred solution of the acid in dry THF (200 mL) at 0 °C under argon, Et3N (20 mL, 145 mmol) and PivCl (7.1 mL, 58 mmol) were added sequentially. After 1 h stirring at 0 °C, LiCl (0.62 g, 14.5 mmol), followed by oxazolidinone 6 (9.4 g, 58 mmol), were added. The reaction was continued for 1 h at 0 °C and another 2 h at room temperature prior to quenching with a saturated NH4Cl solution (50 mL) and extracted with DCM (2 × 200 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuum. Purification by column chromatography (PE/EA = 9:1) afforded compound 7 (15 g, 71% for three steps) as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 7.47–7.21 (m, 5H), 5.43 (dd, J = 8.7, 3.6 Hz, 1H), 4.69 (td, J = 8.8, 1.0 Hz, 1H), 4.28 (ddd, J = 8.9, 3.6, 1.3 Hz, 1H), 3.64 (t, J = 6.3 Hz, 2H), 3.03 (qt, J = 17.7, 7.4 Hz, 2H), 1.92–1.77 (m, 2H), 0.90 (s, 9H), 0.04 (d, J = 1.3 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 172.62, 153.76, 139.27, 129.18, 128.68, 125.93, 70.00, 62.25, 61.91, 57.56, 32.08, 27.09, 25.96, 18.32, −5.34, −5.62. = −42.63, (c 3.38, CHCl3). HRMS (ESI+): calcd. for C19H29NO4Si [M + H]+, 364.1939; found 364.1940.
4.2. (R)-3-((R)-4-((Tert-Butyldimethylsilyl)oxy)-2-Methylbutanoyl)-4-Phenyloxazolidin-2-one (8)
To a stirred solution of compound 7 (15 g, 41.2 mmol) in dry THF (100 mL) at −78 °C under argon, NaHMDS (2.0 M solution in THF, 24.7 mL, 49.4 mmol) was added dropwise. After 1 h, MeI (7.7 mL, 123.5 mmol) was added and the mixture was stirred overnight at the same temperature. The reaction mixture was quenched with a saturated NH4Cl solution (100 mL) and warmed up to room temperature before being extracted with DCM (2 × 100 mL). The combined organic extracts were washed with water and brine, dried over Na2SO4, and concentrated in vacuum. Purification by column chromatography (PE/EA = 13:1) gave pure 8 (13.8 g, 89%) as a white solid.
1H NMR (400 MHz, CDCl3) δ 7.45–7.23 (m, 5H), 5.44 (dd, J = 8.7, 3.7 Hz, 1H), 4.67 (t, J = 8.8 Hz, 1H), 4.25 (dd, J = 8.9, 3.7 Hz, 1H), 4.02–3.85 (m, 1H), 3.73–3.55 (m, 2H), 2.00 (td, J = 13.9, 6.4 Hz, 1H), 1.59 (dq, J = 12.2, 6.1 Hz, 1H), 1.15 (d, J = 7.0 Hz, 3H), 0.91 (s, 9H), 0.06 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 176.36, 153.35, 139.47, 129.28, 128.70, 125.75, 69.80, 61.02, 57.74, 35.72, 34.71, 26.00, 18.39, 17.83, −5.33. = −79.14, (c 2.34, CHCl3). HRMS (ESI+): calcd. for C20H31NO4Si [M + H]+, 378.2095, found 378.2095.
4.3. (R)-4-((Tert-Butyldimethylsilyl)Oxy)-2-Methylbutan-1-Ol (9)
To an ice-cold solution of compound
8 (13.8 g, 36.6 mmol) in THF (100 mL) moist with a catalytic amount of water, LiBH
4 (1.2 g, 54.9 mmol) was added portion wise under argon. After 12 h of stirring at room temperature, the reaction was quenched cautiously with a saturated NH
4Cl solution (50 mL) and then distilled under a reduced pressure followed by extraction with DCM. The combined organic solution was dried over Na
2SO
4 and concentrated in vacuum. Purification by column chromatography (PE/EA = 9:1) provided pure compound
9 (6.86 g, 86%) as a colorless oil [
11].
1H NMR (400 MHz, CDCl3) δ 3.81–3.69 (m, 1H), 3.68–3.60 (m, 1H), 3.46 (s, 1H), 3.43–3.33 (m, 1H), 3.19 (s, 1H), 1.86–1.70 (m, 1H), 1.59–1.46 (m, 2H), 0.89 (m, 12H), 0.05 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 68.21, 61.82, 37.53, 34.46, 25.98, 18.34, 17.46, −5.33, −5.36. = −10.98, (c 1.82, CHCl3). HRMS (ESI+): calcd. for C11H26O2Si [M + H]+, 219.1775, found 219.1771.
4.4. Ethyl (R,E)-6-((Tert-Butyldimethylsilyl)Oxy)-2,4-Dimethylhex-2-Enoate (11)
To a stirred solution of
9 (6.86 g, 31.5 mmol) in DMSO (50 mL) IBX (10.6 g, 37.8 mmol) was added. After 1 h stirring at 40 °C, the reaction was quenched with water (50 mL) and extracted with ether (2 × 100 mL). The combined organic layers were washed with brine, dried over Na
2SO
4, and removed under vacuum. The residue was refluxed with 10 (22 g, 63 mmol) in toluene (100 mL) at 80 °C for 3 h and the solvent was removed under vacuum. Purification by column chromatography (PE/EA = 40:1) provided pure compound
11 (7 g, 75%) as a colorless oil [
22].
1H NMR (400 MHz, CDCl3) δ 6.53 (dd, J = 10.1, 1.4 Hz, 1H), 4.17 (q, J = 7.1 Hz, 2H), 3.70–3.41 (m, 2H), 2.80–2.45 (m, 1H), 1.83 (d, J = 1.4 Hz, 3H), 1.66–1.44 (m, 2H), 1.28 (t, J = 7.1 Hz, 3H), 1.00 (d, J = 6.7 Hz, 3H), 0.87 (s, 9H), 0.01 (d, J = 2.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 168.57, 147.56, 126.83, 61.02, 60.52, 39.83, 29.78, 26.04, 20.06, 18.37, 14.42, 12.59, −5.23, −5.25. = −2.167, (c 0.1, CHCl3). HRMS (ESI+): calcd. for C16H32O3Si [M + H]+, 301.2193, found 301.2194.
4.5. Allyl (R,E)-6-((Tert-Butyldimethylsilyl)oxy)-2,4-Dimethylhex-2-Enoate (12)
To a stirred solution of compound 11 (7 g, 23.6 mmol) in dry DCM (60 mL) at −78 °C under argon, DIBAL-H (1.5 M solution in toluene, 18.0 mL, 27.0 mmol) was added dropwise. After 1 h, the reaction mixture was quenched with aqueous sodium-potassium tartrate solution (20 mL) and warmed up to room temperature before being extracted with DCM (2 × 100 mL). The combined organic extracts were washed with water and brine, dried over Na2SO4, and concentrated in vacuum. Purification by column chromatography (PE/EA = 8:1) gave alcohol (4.8 g, 79%) as a colorless oil.
To a stirred solution of above alcohol in DMSO (50 mL), IBX (6.2 g, 22 mmol) was added. After 1 h stirring at 40 °C, the reaction was quenched with water (50 mL) and extracted with ether (2 × 100 mL). The combined organic layers were washed with brine, dried over Na2SO4, and removed under vacuum. The crude anhydride was used without further purification.
To the solution of above crude aldehyde in tBuOH (40 mL), NaH2PO4 (11 g, 93 mmol) in H2O (40 mL), 2-methyl-2-butene (19 mL, 186 mmol), and NaClO2 (3.1 g, 28 mmol, >79.0% purity) were added. After stirring for 2 h at room temperature, the mixture was extracted with EA (50 mL × 3) and the combined organic layers were washed with brine, dried over Na2SO4, filtrated, and concentrated. The crude acid was taken into the next step without future purification.
To a stirred solution of above acid in dry DMF (50 mL) allylBr (3.2 g, 37.2 mmol) and K2CO3 (5.1 g, 37.2 mmol) were added separately. After being stirred for 12 h, the mixture was quenched with a saturated aqueous solution of NH4Cl and extracted with EA (100 mL × 3). The combined organic layers were washed with brine, dried over Na2SO4, filtrated, and concentrated. The residue was purified by column chromatography on silica gel (PE/EA = 40:1) to give 12 (4.7 g, 81%) for three steps as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 6.58 (dd, J = 10.1, 1.4 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.32 (ddd, J = 17.2, 3.0, 1.5 Hz, 1H), 5.23 (dd, J = 10.4, 1.3 Hz, 1H), 4.64 (dt, J = 5.6, 1.4 Hz, 2H), 3.66–3.42 (m, 2H), 2.82–2.58 (m, 1H), 1.86 (d, J = 1.4 Hz, 3H), 1.68–1.55 (m, 2H), 1.01 (d, J = 6.7 Hz, 3H), 0.88 (s, 10H), 0.02 (d, J = 2.2 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 168.16, 148.14, 132.76, 126.60, 117.84, 65.27, 61.00, 39.80, 29.82, 26.04, 20.02, 18.38, 12.62, −5.22. = −53.365, (c 0.52, CHCl3). HRMS (ESI+): calcd. for C17H32O3Si [M + H]+, 313.2193, found 313.2186.
4.6. Allyl (R,E)-6-Hydroxy-2,4-Dimethylhex-2-Enoate (13)
To a stirred solution of 12 (4.7 g, 15 mmol) in dry THF (10 mL), the HF/Py complex (2 mL) was added at 0 °C. After being stirred for 1 h, the mixture was quenched with a saturated aqueous solution of NaHCO3 and extracted with DCM (20 mL × 3). The combined organic layers were washed with 1 M HCl brine, dried over Na2SO4, filtrated, and concentrated. The residue was purified by column chromatography on silica gel (PE/EA = 6:1) to give 13 (2.3 g, 77%) as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 6.59 (dd, J = 10.1, 1.4 Hz, 1H), 5.97 (ddt, J = 17.1, 10.6, 5.6 Hz, 1H), 5.34 (dq, J = 17.2, 1.5 Hz, 1H), 5.24 (ddd, J = 10.5, 2.6, 1.3 Hz, 1H), 4.65 (dt, J = 5.6, 1.4 Hz, 2H), 3.75–3.44 (m, 2H), 2.78–2.63 (m, 1H), 1.88 (d, J = 1.4 Hz, 3H), 1.75–1.62 (m, 1H), 1.62–1.50 (m, 2H), 1.05 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.07, 147.61, 132.63, 126.88, 117.97, 65.34, 60.94, 39.55, 29.97, 20.09, 12.62. = −55.72, (c 0.79, CHCl3). HRMS (ESI+): calcd. for C11H18O3 [M + H]+, 199.1329, found 199.1328.
4.7. Allyl (R,E)-2,4-Dimethyl-6-((1-Phenyl-1H-Tetrazol-5-yl)Thio)Hex-2-Enoate (15)
To a stirred solution of 13 (2.3 g, 11.6 mmol) in anhydrous dry THF (50 mL) at 0 °C under argon, PPh3 (6.1 g, 23.2 mmol), 14 (2 g, 11.6 mmol), and DIAD (4.6 mL, 23.2 mmol) were added sequentially. The reaction was continued further for 1 h at room temperature prior to quenching with the saturated NH4Cl solution (50 mL) and extracted with DCM (2 × 100 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuum. Purification by column chromatography (PE/EA = 15:1) afforded compound 15 (3.9 g, 94%) as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 7.67–7.46 (m, 5H), 6.55 (dd, J = 10.1, 1.4 Hz, 1H), 5.94 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.42–5.29 (m, 1H), 5.22 (dd, J = 10.4, 1.3 Hz, 1H), 4.62 (dt, J = 5.6, 1.4 Hz, 2H), 3.41–3.13 (m, 2H), 2.77–2.60 (m, 1H), 2.06–1.90 (m, 1H), 1.89–1.74 (m, 4H), 1.05 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.69, 154.18, 146.10, 133.74, 132.49, 130.22, 129.89, 127.82, 123.90, 118.07, 65.40, 35.95, 32.63, 31.37, 19.89, 12.76. = −58.87, (c 1.47, CHCl3). HRMS (ESI+): calcd. for C18H22N4O2S [M + H]+, 359.1536, found 359.1532.
4.8. Allyl (R,E)-2,4-Dimethyl-6-((1-Phenyl-1H-Tetrazol-5-yl)sulfonyl)Hex-2-Enoate (2)
To a stirred solution of 15 (3.9 g, 10.1 mmol) in ethanol (100 mL) at 0 °C, (NH4)6Mo7O24 (2.9 g, 2.2 mmol) and H2O2 (20 mL) were added sequentially. The reaction was continued further for 12 h at room temperature prior to quenching with saturated NH4Cl solution (50 mL) and extracted with DCM (2 × 100 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuum. Purification by column chromatography (PE/EA = 8:1) afforded compound 2 (3.6 g, 94%) as a white solid.
1H NMR (400 MHz, CDCl3) δ 7.76–7.51 (m, 5H), 6.53 (dd, J = 10.1, 1.4 Hz, 1H), 5.97 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.48–5.32 (m, 1H), 5.25 (dd, J = 10.4, 1.3 Hz, 1H), 4.65 (dt, J = 5.6, 1.3 Hz, 2H), 3.77–3.58 (m, 2H), 2.95–2.62 (m, 1H), 2.24–2.04 (m, 1H), 2.01–1.92 (m, 1H), 1.88 (d, J = 1.4 Hz, 3H), 1.11 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.47, 153.49, 144.44, 133.12, 132.42, 131.64, 129.88, 128.93, 125.17, 118.32, 65.61, 54.40, 32.31, 28.74, 19.98, 12.86. = −13.87, (c 0.79, CHCl3). HRMS (ESI+): calcd. for C18H22N4O2S2 [M + H]+, 391.1435, found 391.1435.
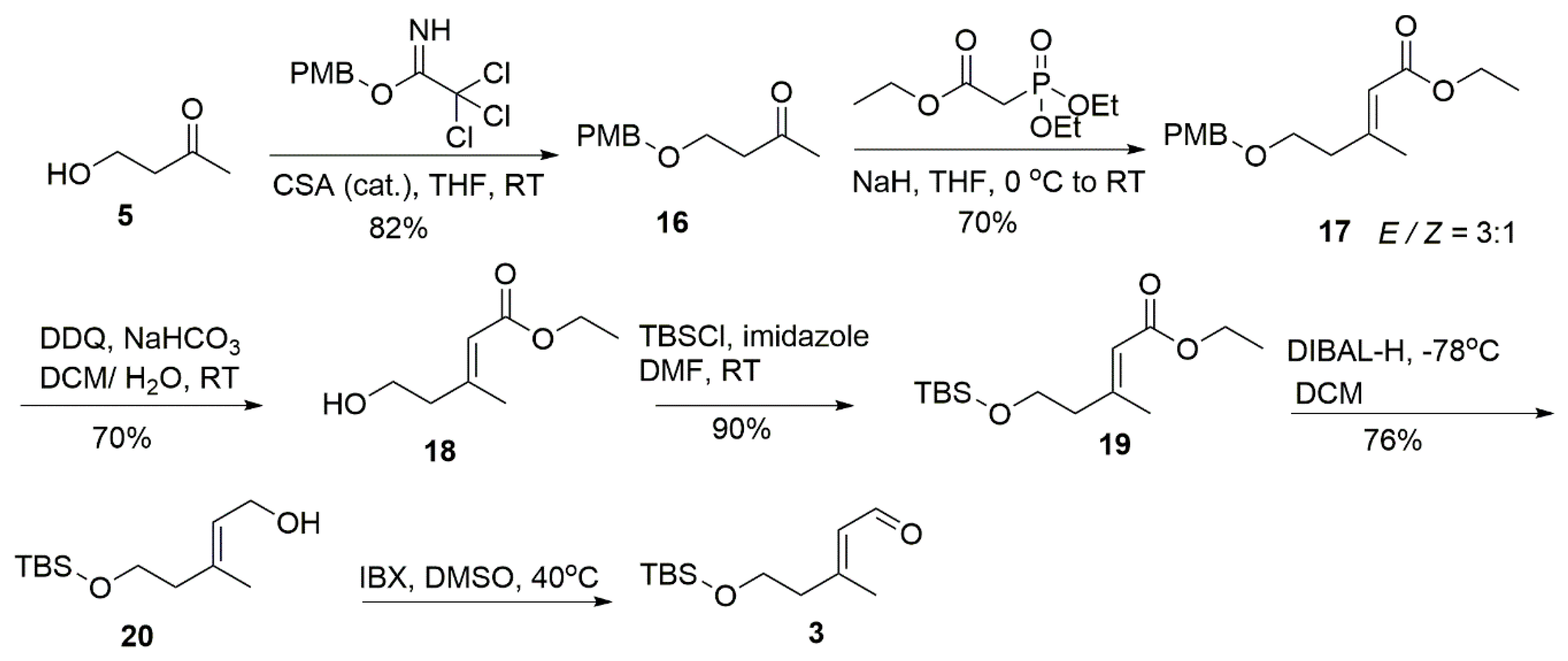
4.9. 4-((4-Methoxybenzyl)Oxy)Butan-2-One (16)
To a stirred solution of 4-hydroxy-2-butanone (1.1 mL ,12.8 mmol) in dry THF (20 mL) at 0 °C, 4-methoxybenzyl 2,2,2-trichloroacetimidate (2 mL, 10.7 mmol) and triphenylcarbenium tetrafluoroborate (cat.) were added sequentially. The reaction was continued further for 1 h at room temperature prior to quenching with the saturated NH
4Cl solution (50 mL) and extracted with DCM (2 × 100 mL). The combined organic layers were washed with brine, dried over Na
2SO
4, and concentrated in vacuum. Purification by column chromatography (PE/EA = 6:1) afforded compound
16 (1.8 g, 82%) as a colorless oil [
14].
1H NMR (400 MHz, CDCl3) δ 7.33–7.18 (m, 2H), 6.94–6.81 (m, 2H), 4.45 (s, 2H), 3.81 (s, 3H), 3.72 (t, J = 6.3 Hz, 2H), 2.71 (t, J = 6.3 Hz, 2H), 2.18 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 207.51, 159.27, 130.16, 129.42, 113.84, 72.91, 64.96, 55.31, 43.81, 30.48. HRMS (ESI+): calcd. for C12H16O3 [M + H]+, 209.1172, found 209.1170.
4.10. Ethyl (E)-5-((4-Methoxybenzyl)Oxy)-3-Methylpent-2-Enoate (17)
To a stirred solution of triethyl phosphonoacetate (3.5 mL, 17.5 mmol) in dry THF (50 mL) at 0 °C, NaH (0.7 g, 17.5 mmol) was added. After stirring at room temperature for 1 h, 16 (1.8 g, 8.8 mmol) in THF (5 mL) was added at 0 °C. The reaction was continued further for 12 h at room temperature prior to quenching with a saturated NH4Cl solution (50 mL) and extracted with DCM (2 × 100 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuum. Purification by column chromatography (PE/EA = 20:1) afforded compound 17 in the E/Z mixture (1.7 g, 70%) as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 7.27 (d, J = 8.6 Hz, 2H), 6.99–6.72 (m, 2H), 5.82–5.63 (m, 1H), 4.47 (d, J = 2.8 Hz, 2H), 4.23–4.08 (m, 2H), 3.82 (d, J = 2.1 Hz, 3H), 3.70–3.51 (m, 2H), 2.98 (t, J = 6.6 Hz, 1H), 2.45 (t, J = 6.4 Hz, 2H), 2.19 (d, J = 1.2 Hz, 2H), 1.96 (d, J = 1.3 Hz, 1H), 1.29 (td, J = 7.1, 5.4 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 166.73, 166.34, 159.33, 159.19, 157.81, 156.72, 130.70, 130.30, 129.38, 129.27, 117.46, 117.10, 113.90, 113.81, 72.76, 72.46, 68.74, 67.59, 59.60, 55.35, 40.89, 33.73, 26.16, 19.00, 14.42. HRMS (ESI+): calcd. for C16H22O4 [M + H]+, 279.1591; found 279.1589.
4.11. Ethyl (E)-5-Hydroxy-3-Methylpent-2-Enoate (18)
To a stirring solution of
17 (1.7 g, 6.1 mmol) in DCM (20 mL) and water (4 mL) DDQ (1.67 g, 7.3 mmol) was added at room temperature. After 1 h, the reaction mixture was quenched with saturated aqueous NaHCO
3 (10 mL) and 1 M aqueous NaHSO
3 (10 mL) and extracted with DCM (30 mL × 3). The combined organic layers were washed with brine, dried over Na
2SO
4, and concentrated. The crude product was purified by column chromatography (PE/EA = 3:1) to give the desired compound
18 as a colorless oil (672 mg, 70% yield) [
13].
1H NMR (400 MHz, CDCl3) δ 5.71 (d, J = 1.2 Hz, 1H), 4.12 (q, J = 7.1 Hz, 2H), 3.76 (t, J = 6.4 Hz, 2H), 2.47–2.30 (m, 2H), 2.16 (d, J = 1.2 Hz, 3H), 1.92 (s, 1H), 1.25 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 166.67, 156.12, 117.73, 60.22, 59.78, 43.82, 18.82, 14.37. HRMS (ESI+): calcd. for C8H14O3 [M + H]+, 159.1016, found 159.1014.
4.12. Ethyl (E)-5-((Tert-Butyldimethylsilyl)Oxy)-3-Methylpent-2-Enoate (19)
To a stirred solution of
18 (672 mg, 4.3 mmol) in anhydrous DMF (10 mL) at 0 °C, imidazole (7.7 g, 11.4 mmol) and TBSCl (1.1 g, 6.84 mmol) were added sequentially. The reaction was continued further for 12 h at room temperature prior to quenching with saturated NH
4Cl solution (20 mL) and extracted with EA (2 × 20 mL). The combined organic layers were washed with brine, dried over Na
2SO
4, and concentrated in vacuum. Purification by column chromatography (PE/EA = 40:1) created compound
19 (1.05 g, 90%) as a colorless oil [
13].
1H NMR (400 MHz, CDCl3) δ 5.66 (d, J = 1.1 Hz, 1H), 4.12 (q, J = 7.1 Hz, 2H), 2.32 (t, J = 6.6 Hz, 2H), 2.15 (d, J = 1.2 Hz, 3H), 1.24 (t, J = 7.1 Hz, 3H), 0.86 (s, 9H), 0.02 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 166.74, 156.92, 117.34, 61.41, 59.53, 44.12, 25.97, 19.21, 18.36, 14.42, −5.29. HRMS (ESI+): calcd. for C14H28O3Si [M + H]+, 273.1880, found 273.1880.
4.13. (E)-5-((Tert-Butyldimethylsilyl)Oxy)-3-Methylpent-2-En-1-Ol (20)
To a stirred solution of compound
19 (1.05 g, 3.87 mmol) in dry DCM (15 mL) at −78 °C under argon, DIBAL-H (1.5 M solution in toluene, 3.9 mL, 5.8 mmol) was added dropwise. After 1 h, the reaction mixture was quenched with the aqueous sodium-potassium tartrate solution (20 mL) and warmed up to room temperature before being extracted with DCM (2 × 20 mL). The combined organic extracts were washed with water and brine, dried over Na
2SO
4, and concentrated in vacuum. Purification by column chromatography (PE/EA = 7:1) gave
20 (680 mg, 76%) as a colorless oil [
23].
1H NMR (400 MHz, CDCl3) δ 5.45 (td, J = 6.9, 1.2 Hz, 1H), 4.16 (d, J = 6.9 Hz, 2H), 3.71 (t, J = 7.0 Hz, 2H), 2.25 (t, J = 7.0 Hz, 2H), 1.71 (s, 3H), 1.53 (s, 1H), 0.90 (s, 9H), 0.06 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 136.95, 125.38, 62.20, 59.42, 42.91, 26.05, 18.45, 16.80, −5.17. HRMS (ESI+): calcd. for C12H26O2Si ([M + H]+, 231.1775, found 231.1775.
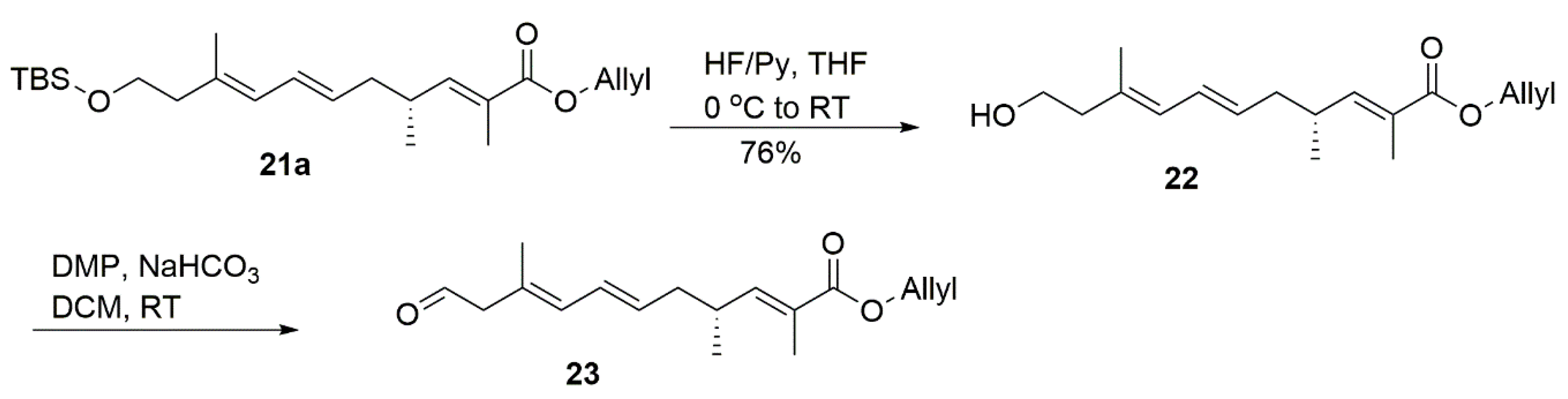
4.14. Allyl (R,2E,6E,8E)-11-((Tert-butyldimethylsilyl)Oxy)-2,4,9-Trimethylundeca-2,6,8-Trienoate (21a)
To a stirred solution of 20 (680 mg, 3.0 mmol) in DMSO (10 mL) IBX (990 mg, 3.5 mmol) was added. After 1 h of stirring at 40 °C, the reaction was quenched with water (10 mL) and extracted with ether (2 × 20 mL). The combined organic layers were washed with brine, dried over Na2SO4, and removed under vacuum. The crude aldehyde 3 was used without further purification.
To a stirred solution of crude 3 and compound 2 (1.3 g, 3.3 mmol) in dry THF (30 mL) at −78 °C under argon, KHMDS (1.0 M solution in THF, 27.0 mL, 27.0 mmol) was added dropwise. After 1 h, the reaction mixture was quenched with the saturated NH4Cl solution (20 mL) and warmed up to room temperature before being extracted with DCM (2 × 100 mL). The combined organic extracts were washed with water and brine, dried over Na2SO4, and concentrated in vacuum. Purification by column chromatography (PE/EA = 40:1) gave 21a (1.0 g, 86%, 2 steps) as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 6.61 (dd, J = 10.0, 1.4 Hz, 1H), 6.23 (dd, J = 15.0, 10.8 Hz, 1H), 5.97 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.79 (d, J = 10.8 Hz, 1H), 5.48 (dt, J = 14.8, 7.3 Hz, 1H), 5.33 (ddd, J = 17.2, 3.1, 1.5 Hz, 1H), 5.23 (dd, J = 10.4, 1.3 Hz, 1H), 4.64 (d, J = 5.5 Hz, 2H), 3.68 (t, J = 7.0 Hz, 2H), 2.64–2.47 (m, 1H), 2.24 (t, J = 7.0 Hz, 2H), 2.14 (t, J = 7.1 Hz, 2H), 1.85 (d, J = 1.3 Hz, 3H), 1.74 (s, 3H), 1.02 (d, J = 6.7 Hz, 3H), 0.88 (s, 9H), 0.04 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 168.13, 147.84, 134.13, 132.75, 129.61, 128.53, 126.49, 126.40, 117.83, 65.27, 62.39, 43.29, 40.02, 33.96, 26.09, 19.68, 18.47, 17.20, 12.73, −5.14. = −32.72, (c 0.38, CHCl3). HRMS (ESI+): calcd. for C23H40O3Si [M + H]+, 293.2819, found 293.2819.
4.15. Allyl (R,2E,6E,8E)-11-Hydroxy-2,4,9-Trimethylundeca-2,6,8-Trienoate (22)
To a stirred solution of 21a (1.0 g, 2.6 mmol) in dry THF (5 mL), HF/Py complex (1 mL) was added at 0 °C. After being stirred for 1 h, the mixture was quenched with a saturated aqueous solution of NaHCO3 and extracted with DCM (20 mL × 3). The combined organic layers were washed with 1 M HCl, brine, dried over Na2SO4, filtrated, and concentrated. The residue was purified by column chromatography on silica gel (PE/EA = 6:1) to give 22 (550 mg, 76%) as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 6.60 (dd, J = 10.0, 1.4 Hz, 1H), 6.24 (dd, J = 15.0, 10.8 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.85 (d, J = 10.8 Hz, 1H), 5.52 (dt, J = 14.8, 7.3 Hz, 1H), 5.32 (ddd, J = 17.2, 3.0, 1.5 Hz, 1H), 5.22 (dd, J = 10.4, 1.3 Hz, 1H), 4.63 (d, J = 5.6 Hz, 2H), 3.69 (t, J = 6.3 Hz, 2H), 2.65–2.51 (m, 1H), 2.29 (t, J = 6.3 Hz, 2H), 2.15 (t, J = 7.0 Hz, 2H), 1.84 (d, J = 1.4 Hz, 3H), 1.75 (s, 3H), 1.54 (s, 1H), 1.01 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.09, 147.69, 132.93, 132.70, 130.49, 128.14, 127.38, 126.53, 117.82, 65.26, 60.49, 42.95, 39.97, 33.84, 19.67, 16.50, 12.72. = −38.23, (c 0.96, CHCl3). HRMS (ESI+): calcd. for C17H26O3 [M + H]+, 279.1955, found 279.1954.
4.16. Allyl (2E,4R,6E,8E,11R)-11-Hydroxy-2,4,9-Trimethyl-13-Oxotetradeca-2,6,8-Trienoate (24a)
To the above alcohol 22 (200 mg, 0.72 mmol) in dry DCM (5 mL, 0 °C), DMP (610 mg, 1.44 mmol) and NaHCO3 (240 mg, 2.9 mmol) was added sequentially. After being stirred for 30 min, the mixture was carefully quenched with a solution of saturated aqueous NaHCO3 and Na2S2O3. The resulting mixture was extracted with DCM (20 mL × 3) and the combined organic layers were washed with brine, dried over Na2SO4, and concentrated. The crude aldehyde 23 was used without future purification.
To a stirred solution of (+)-IPCBCl (1.8 M in heptane, 1.08 mmol, 0.6 mL) in dry ether (3 mL) at −0 °C under argon, Et3N (0.2 mL, 1.44 mmol) and acetone (80 uL, 1.08 mmol) were added sequentially. After 1 h stirring at −0 °C, aldehyde 23 in ether (2 mL) was added at −78 °C. The reaction was continued further for 1 h at −78 °C and another 12 h at −20 °C prior to quenching with a mixture of PH 7 buffer (1 mL), methanol (1 mL), and H2O2 (1 mL). The mixture was warmed up to room temperature before being extracted with ether (2 × 10 mL). The combined organic extracts were washed with water and brine, dried over Na2SO4, and concentrated in vacuum. Purification by column chromatography (PE/EA = 4:1) created 24a (132 mg, 55%) as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 6.60 (dd, J = 10.0, 1.2 Hz, 1H), 6.22 (dd, J = 15.0, 10.8 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.83 (d, J = 10.8 Hz, 1H), 5.52 (dt, J = 14.8, 7.3 Hz, 1H), 5.32 (dd, J = 17.2, 1.4 Hz, 1H), 5.23 (dd, J = 10.4, 1.1 Hz, 1H), 4.64 (d, J = 5.5 Hz, 2H), 4.20 (tt, J = 11.6, 5.9 Hz, 1H), 2.79 (s, 1H), 2.66–2.47 (m, 3H), 2.24 (dd, J = 13.4, 7.5 Hz, 1H), 2.19–2.07 (m, 6H), 1.84 (d, J = 1.2 Hz, 3H), 1.75 (s, 3H), 1.02 (d, J = 6.7 Hz, 3H).13C NMR (101 MHz, CDCl3) δ 209.56, 168.10, 147.68, 132.82, 132.72, 130.79, 128.10, 127.99, 126.58, 117.85, 65.91, 65.29, 49.62, 47.13, 40.02, 33.86, 30.98, 19.72, 16.89, 12.75. = −33.92, (c 0.34, CHCl3). HRMS (ESI+): calcd. for C20H30O4 [M + H]+, 335.2217, found 335.2217.
4.17. Allyl (2E,4R,6E,8E,11S)-11-Hydroxy-2,4,9-Trimethyl-13-Oxotetradeca-2,6,8-Trienoate (24b)
To a stirred solution of (−)-IPCBCl (1.7 M in heptane, 1.08 mmol, 0.64 mL) in dry ether (3 mL) at 0 °C under argon, Et3N (0.2 mL, 1.44 mmol) and acetone (80 uL, 1.08 mmol) were added sequentially. After 1 h stirring at 0 °C, aldehyde 23 in ether (2 mL) was added at −78 °C. The reaction was continued further for 1 h at −78 °C and another 12 h at −20 °C prior to quenching with a mixture of PH 7 buffer (1 mL), methanol (1 mL), and H2O2 (1 mL). The mixture was warmed up to room temperature before being extracted with ether (2 × 10 mL). The combined organic extracts were washed with water and brine, dried over Na2SO4, and concentrated in vacuum. Purification by column chromatography (PE/EA = 4:1) provided 24b (142 mg, 59%) as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 6.60 (dd, J = 10.0, 1.2 Hz, 1H), 6.23 (dd, J = 15.0, 10.8 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.83 (d, J = 10.8 Hz, 1H), 5.53 (dt, J = 14.8, 7.3 Hz, 1H), 5.32 (dd, J = 17.2, 1.4 Hz, 1H), 5.23 (dd, J = 10.4, 1.1 Hz, 1H), 4.64 (d, J = 5.5 Hz, 2H), 4.29–4.12 (m, 1H), 2.66–2.51 (m, 3H), 2.23 (dd, J = 13.5, 7.6 Hz, 1H), 2.19–2.08 (m, 6H), 1.84 (d, J = 1.2 Hz, 3H), 1.75 (s, 3H), 1.02 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 209.53, 168.10, 147.67, 132.83, 132.72, 130.76, 128.12, 127.98, 126.57, 117.86, 65.92, 65.29, 49.66, 47.11, 39.97, 33.86, 30.98, 19.69, 16.91, 12.74. = −26.04, (c 0.37, CHCl3). HRMS (ESI+): calcd. for C20H30O4 [M + H]+, 335.2217, found 335.2216.
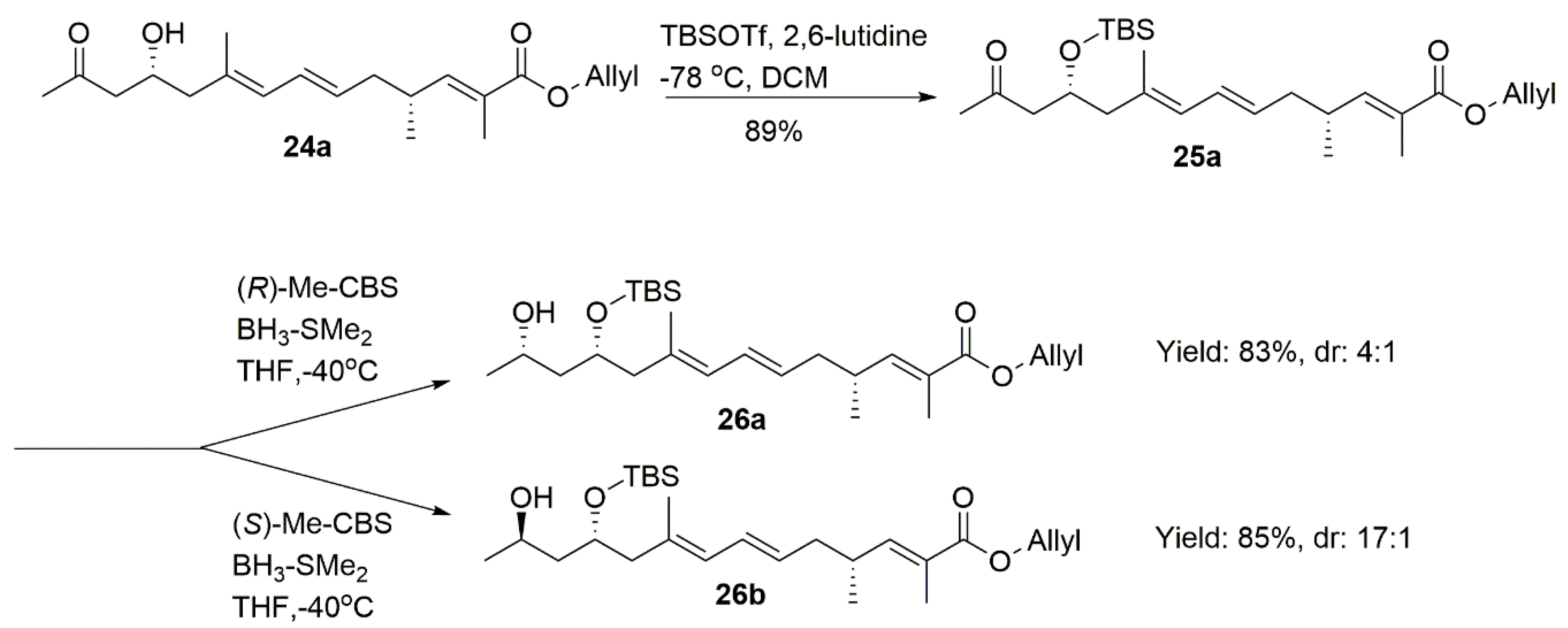
4.18. Allyl (2E,4R,6E,8E,11R)-11-((Tert-Butyldimethylsilyl)oxy)-2,4,9-Trimethyl-13-Oxotetradeca-2,6,8-Trienoate (25a)
To a stirred solution of compound 24a (132 mg, 0.4 mmol) in dry DCM (3 mL) at −78 °C under argon, 2,6-lutidine (0.23 mL, 2 mmol) and TBSOTf (0.35 mL, 1.5 mmol) were added sequentially. After 1 h, the reaction mixture was quenched with aqueous NaHCO3 (5 mL) and warmed up to room temperature before being extracted with DCM (2 × 10 mL). The combined organic extracts were washed with 1 M HCl and brine, dried over Na2SO4, and concentrated in vacuum. Purification by column chromatography (PE/EA = 40:1) created 25a (160 mg, 89%) as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 6.60 (dd, J = 10.0, 1.3 Hz, 1H), 6.19 (dd, J = 15.0, 10.8 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.75 (d, J = 10.8 Hz, 1H), 5.48 (dt, J = 14.8, 7.3 Hz, 1H), 5.33 (dd, J = 17.2, 1.5 Hz, 1H), 5.23 (dd, J = 10.4, 1.2 Hz, 1H), 4.64 (d, J = 5.6 Hz, 2H), 4.38–4.20 (m, 1H), 2.67–2.41 (m, 3H), 2.29–2.20 (m, 1H), 2.19–2.00 (m, 6H), 1.84 (d, J = 1.1 Hz, 3H), 1.73 (s, 3H), 1.01 (d, J = 6.7 Hz, 3H), 0.85 (s, 9H), 0.03 (d, J = 8.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 208.04, 168.10, 147.75, 133.24, 132.74, 130.28, 128.31, 128.07, 126.53, 117.86, 68.06, 65.29, 50.86, 48.64, 40.00, 33.93, 31.85, 25.96, 19.67, 18.12, 17.30, 12.74, −4.41, −4.71. = −40.90, (c 0.26, CHCl3). HRMS (ESI+): calcd. for C26H44O4Si [M + H]+, 449.3082, found 449.3078.
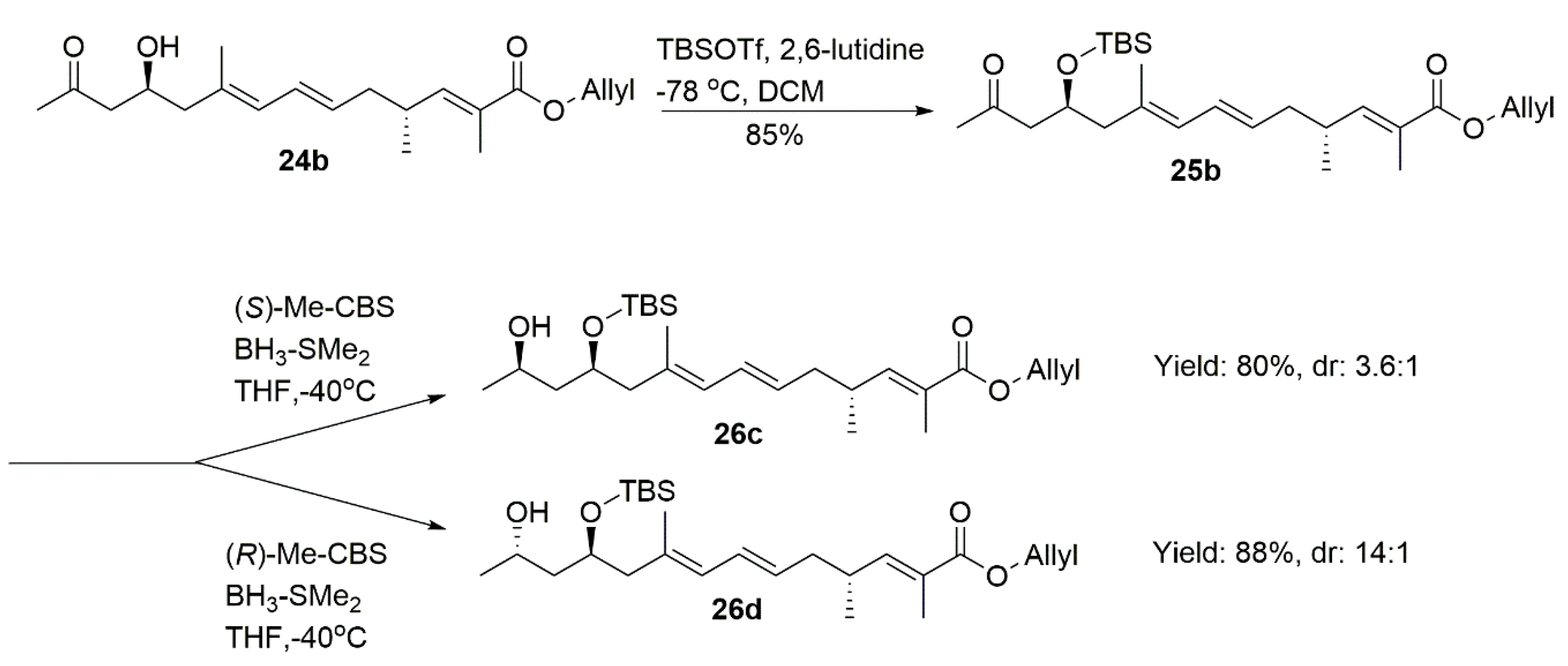
4.19. Allyl (2E,4R,6E,8E,11S)-11-((Tert-Butyldimethylsilyl)oxy)-2,4,9-Trimethyl-13-Oxotetradeca-2,6,8-Trienoate (25b)
The procedure was identical to 25a. Compound 25b (160 mg, 85%) was obtained as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 6.60 (dd, J = 10.0, 1.3 Hz, 1H), 6.19 (dd, J = 15.0, 10.8 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.74 (d, J = 10.8 Hz, 1H), 5.48 (dt, J = 14.8, 7.3 Hz, 1H), 5.32 (dd, J = 17.2, 1.5 Hz, 1H), 5.23 (dd, J = 10.4, 1.3 Hz, 1H), 4.64 (d, J = 5.5 Hz, 2H), 4.35–4.23 (m, 1H), 2.66–2.42 (m, 3H), 2.23 (dd, J = 13.2, 5.7 Hz, 1H), 2.19–2.05 (m, 6H), 1.84 (d, J = 1.2 Hz, 3H), 1.73 (s, 3H), 1.01 (d, J = 6.7 Hz, 3H), 0.84 (s, 9H), 0.02 (d, J = 7.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 208.07, 168.09, 147.76, 133.25, 132.74, 130.27, 128.31, 128.04, 126.52, 117.85, 68.09, 65.28, 50.84, 48.60, 40.04, 33.92, 31.84, 25.95, 19.71, 18.11, 17.33, 12.73, −4.42, −4.74. = −17.13, (c 0.51, CHCl3). HRMS (ESI+): calcd. for C26H44O4Si [M + H]+, 449.3082, found 449.3079.
4.20. Allyl (2E,4R,6E,8E,11R,13S)-11-((Tert-Butyldimethylsilyl)Oxy)-13-Hydroxy-2,4,9-Trimethyltetradeca-2,6,8-Trienoate (26a)
To a solution of (R)-Me-CBS (1 M in toluene, 0.05 mL, 0.05 mmol) in dry THF (5 mL) was slowly added BH3·DMS (12 uL, 0.13 mmol) at −40 °C. After being stirred for 30 min at the same temperature, a solution of 25a (57 mg, 0.13 mmol) in THF (2 mL) was slowly added. After being stirred for 2 h at −40 °C, the mixture was diluted with MeOH. The resulting mixture was concentrated and extracted with DCM (30 mL × 3). The combined organic layers were washed with brine, dried, filtrated, and concentrated. The residue was purified by column chromatography on silica gel (PE/EA = 15:1) to give a mixture of 26a and 26b (4:1, 48 mg, 83%) as a colorless oil (pure major isomer 26a can be obtained by repeating the purification on silica gel).
1H NMR (400 MHz, CDCl3) δ 6.60 (d, J = 9.9 Hz, 1H), 6.18 (dd, J = 14.7, 11.0 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.77 (d, J = 10.8 Hz, 1H), 5.49 (dt, J = 14.8, 7.3 Hz, 1H), 5.32 (dd, J = 17.2, 1.4 Hz, 1H), 5.23 (dd, J = 10.5, 1.0 Hz, 1H), 4.63 (d, J = 5.5 Hz, 2H), 4.27–4.06 (m, 2H), 2.66–2.47 (m, 1H), 2.35 (dd, J = 13.4, 6.3 Hz, 1H), 2.23 (dd, J = 13.3, 7.6 Hz, 1H), 2.16 (dd, J = 20.8, 13.7 Hz, 2H), 1.84 (s, 3H), 1.71 (s, 3H), 1.61 (ddd, J = 14.0, 10.0, 3.7 Hz, 1H), 1.48 (ddd, J = 14.5, 9.4, 3.7 Hz, 1H), 1.14 (d, J = 6.2 Hz, 3H), 0.99 (d, J = 11.9 Hz, 3H), 0.88 (s, 9H), 0.07 (d, J = 18.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 168.12, 147.74, 133.01, 132.72, 130.26, 128.29, 127.95, 126.52, 117.86, 70.36, 65.29, 64.58, 46.98, 43.26, 40.03, 33.91, 25.95, 23.98, 19.68, 18.06, 17.15, 12.76, −4.51, −4.70. = −17.67, (c 0.1, CHCl3). HRMS (ESI+): calcd. for C26H46O4Si [M + H]+, 451.3238, found 451.3241.
4.21. Allyl (2E,4R,6E,8E,11R,13R)-11-((Tert-Butyldimethylsilyl)Oxy)-13-Hydroxy-2,4,9-Trimethyltetradeca-2,6,8-Trienoate (26b)
To a solution of (S)-Me-CBS (1 M in toluene, 0.1 mL, 0.1 mmol) in dry THF (5 mL), BH3·DMS (24 uL, 0.25 mmol) was slowly added at −40 °C. After being stirred for 30 min at the same temperature, a solution of 25a (110 mg, 0.24 mmol) in THF (2 mL) was slowly added. After being stirred for 2 h at −40 °C, the mixture was diluted with MeOH. The resulting mixture was concentrated and extracted with DCM (30 mL × 3). The combined organic layers were washed with brine, dried, filtrated, and concentrated. The residue was purified by column chromatography on silica gel (PE/EA = 15:1) to create 26b (92 mg, 85%) as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 6.60 (dd, J = 10.0, 1.3 Hz, 1H), 6.19 (dd, J = 15.0, 10.8 Hz, 1H), 6.04–5.92 (m, 1H), 5.77 (d, J = 10.8 Hz, 1H), 5.51 (dd, J = 14.9, 7.4 Hz, 1H), 5.33 (dd, J = 17.2, 1.5 Hz, 1H), 5.23 (dd, J = 10.4, 1.3 Hz, 1H), 4.64 (d, J = 5.5 Hz, 2H), 4.01 (dq, J = 13.0, 4.3 Hz, 1H), 3.96–3.83 (m, 1H), 3.18 (s, 1H), 2.66–2.50 (m, 1H), 2.32 (dd, J = 13.1, 4.6 Hz, 1H), 2.12 (dt, J = 13.2, 7.9 Hz, 3H), 1.84 (d, J = 1.1 Hz, 3H), 1.72 (s, 3H), 1.59–1.52 (m, 1H), 1.47–1.37 (m, 1H), 1.14 (d, J = 6.2 Hz, 3H), 1.02 (d, J = 6.7 Hz, 3H), 0.89 (s, 9H), 0.12 (d, J = 8.4 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 168.12, 147.73, 132.79, 132.72, 130.34, 128.23, 127.93, 126.54, 117.86, 72.22, 67.43, 65.29, 49.26, 44.82, 40.01, 33.91, 25.95, 23.70, 19.70, 18.03, 17.28, 12.75, −3.77, −4.55. = −40.33, (c 0.35, CHCl3). HRMS (ESI+): calcd. for C26H46O4Si [M + H]+, 451.3238, found 451.3241.
4.22. Allyl (2E,4R,6E,8E,11S,13R)-11-((Tert-Butyldimethylsilyl)Oxy)-13-Hydroxy-2,4,9-Trimethyltetradeca-2,6,8-Trienoate (26c)
To a solution of (S)-Me-CBS (1 M in toluene, 0.05 mL, 0.05 mmol) in dry THF (5 mL), BH3·DMS (12 uL, 0.13 mmol) was slowly added at −40 °C. After being stirred for 30 min at the same temperature, a solution of 25b (55 mg, 0.12 mmol) in THF (2 mL) was slowly added. After being stirred for 2 h at −40 °C, the mixture was diluted with MeOH. The resulting mixture was concentrated and extracted with DCM (30 mL × 3). The combined organic layers were washed with brine, dried, filtrated, and concentrated. The residue was purified by column chromatography on silica gel (PE/EA = 15:1) to give a mixture of 26c and 26d (3.6:1, 44 mg, 80%) as a colorless oil (pure major isomer 26c can be obtained by repeating the purification on silica gel).
1H NMR (400 MHz, CDCl3) δ 6.60 (dd, J = 10.0, 1.3 Hz, 1H), 6.19 (dd, J = 15.0, 10.9 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.77 (d, J = 10.8 Hz, 1H), 5.49 (dt, J = 14.8, 7.3 Hz, 1H), 5.32 (dd, J = 17.2, 1.5 Hz, 1H), 5.23 (dd, J = 10.4, 1.2 Hz, 1H), 4.64 (d, J = 5.5 Hz, 2H), 4.23–4.07 (m, 2H), 3.36 (s, 1H), 2.67–2.49 (m, 1H), 2.35 (dd, J = 13.3, 6.2 Hz, 1H), 2.29–2.21 (m, 1H), 2.14 (t, J = 7.0 Hz, 2H), 1.84 (d, J = 1.1 Hz, 3H), 1.71 (s, 3H), 1.64–1.56 (m, 1H), 1.49 (ddd, J = 14.5, 9.5, 3.7 Hz, 1H), 1.14 (d, J = 6.2 Hz, 3H), 1.01 (d, J = 6.6 Hz, 3H), 0.88 (s, 9H), 0.06 (d, J = 20.4 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 168.09, 147.74, 133.01, 132.71, 130.27, 128.27, 127.94, 126.52, 117.86, 70.43, 65.29, 64.58, 46.92, 43.25, 40.04, 33.93, 25.95, 23.98, 19.73, 18.06, 17.21, 12.73, −4.52, −4.71. = −14.83, (c 0.1, CHCl3). HRMS (ESI+): calcd. for C26H46O4Si [M + H]+, 451.3238, found 451.3240.
4.23. Allyl (2E,4R,6E,8E,11S,13S)-11-((Tert-Butyldimethylsilyl)Oxy)-13-Hydroxy-2,4,9-Trimethyltetradeca-2,6,8-Trienoate (26d)
To a solution of (R)-Me-CBS (1 M in toluene, 0.1 mL, 0.1 mmol) in dry THF (5 mL), BH3·DMS (24 uL, 0.25 mmol) was slowly added at −40 °C. After being stirred for 30 min at the same temperature, a solution of 25b (113 mg, 0.25 mmol) in THF (2 mL) was slowly added. After being stirred for 2 h at −40 °C, the mixture was diluted with MeOH. The resulting mixture was concentrated and extracted with DCM (30 mL × 3). The combined organic layers were washed with brine, dried, filtrated, and concentrated. The residue was purified by column chromatography on silica gel (PE/EA = 15:1) to provide 26d (100 mg, 88%) as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 6.60 (dd, J = 10.0, 1.3 Hz, 1H), 6.19 (dd, J = 15.0, 10.8 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.76 (d, J = 10.8 Hz, 1H), 5.50 (dt, J = 14.8, 7.3 Hz, 1H), 5.32 (dd, J = 17.2, 1.5 Hz, 1H), 5.23 (dd, J = 10.4, 1.3 Hz, 1H), 4.64 (d, J = 5.5 Hz, 2H), 4.11–3.98 (m, 1H), 3.95–3.82 (m, 1H), 3.12 (s, 1H), 2.67–2.51 (m, 1H), 2.32 (dd, J = 13.2, 4.7 Hz, 1H), 2.19–2.06 (m, 3H), 1.84 (d, J = 1.2 Hz, 3H), 1.71 (s, 3H), 1.56 (dt, J = 14.4, 3.2 Hz, 1H), 1.46–1.38 (m, 1H), 1.14 (d, J = 6.2 Hz, 3H), 1.02 (d, J = 6.7 Hz, 3H), 0.89 (s, 9H), 0.11 (d, J = 8.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 168.10, 147.74, 132.78, 132.71, 130.32, 128.22, 127.91, 126.53, 117.84, 72.26, 67.41, 65.28, 49.21, 44.81, 40.03, 33.90, 25.95, 23.70, 19.72, 18.02, 17.33, 12.74, −3.78, −4.56. = −15.29, (c 0.63, CHCl3). HRMS (ESI+): calcd. for C26H46O4Si [M + H]+, 451.3238, found 451.3228.
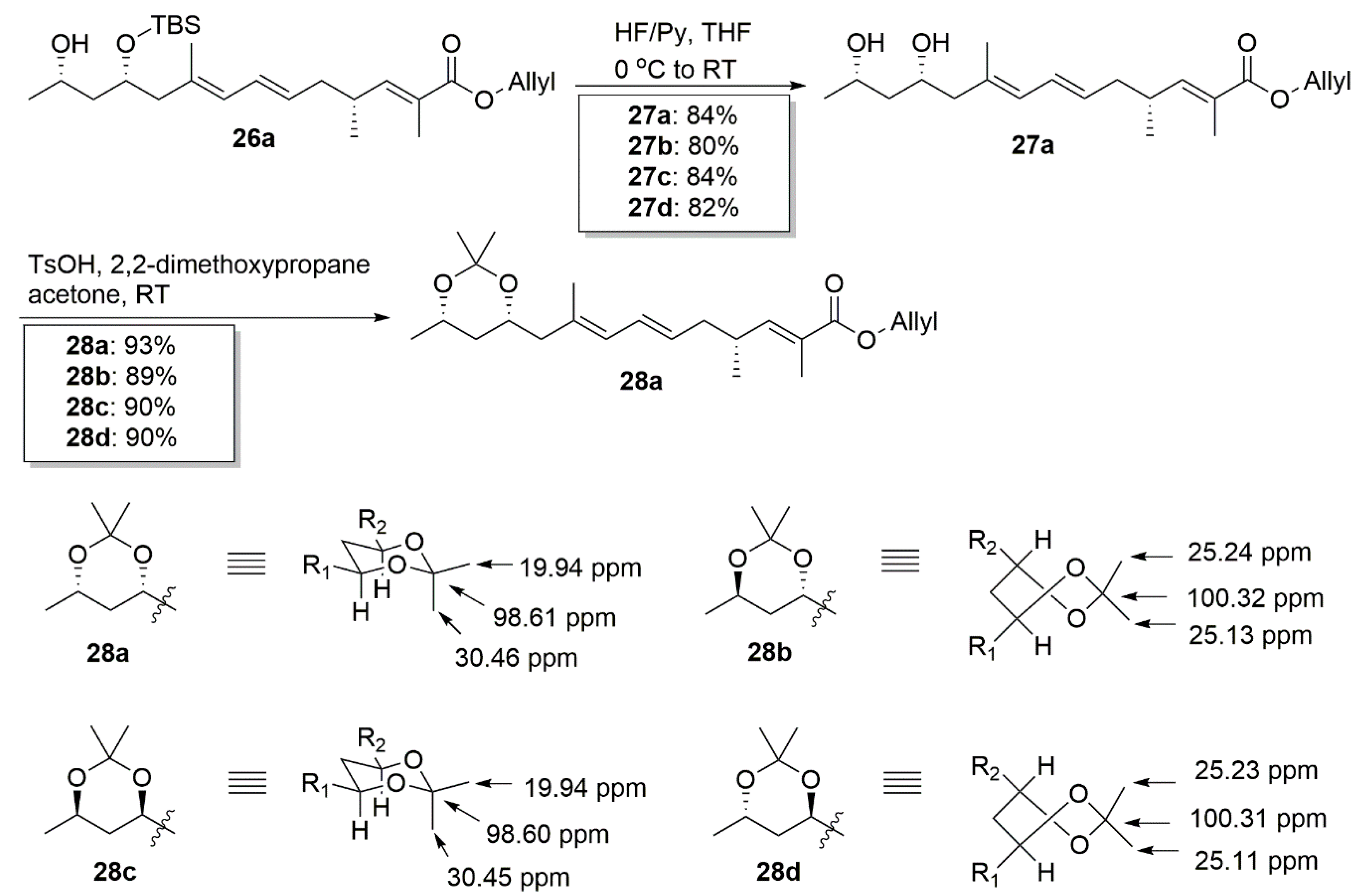
4.24. Synthetic Procedure of 27a–27d
To a stirred solution of 26a (35 mg) in dry THF (2 mL), the HF/Py complex (0.7 mL) was added at 0 °C. After being stirred for 1 h, the mixture was quenched with a saturated aqueous solution of NaHCO3 and extracted with DCM (10 mL × 3). The combined organic layers were washed with 1 M HCl, brine, dried over Na2SO4, filtrated, and concentrated. The residue was purified by a column chromatography on silica gel (PE/EA = 2:1) to create 27a (22 mg, 84%) as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 6.64–6.56 (m, 1H), 6.23 (dd, J = 15.0, 10.8 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.84 (d, J = 10.8 Hz, 1H), 5.54 (dt, J = 14.8, 7.3 Hz, 1H), 5.32 (dd, J = 17.2, 1.4 Hz, 1H), 5.23 (dd, J = 10.5, 0.9 Hz, 1H), 4.64 (d, J = 5.5 Hz, 2H), 4.13–4.02 (m, 1H), 4.02–3.92 (m, 1H), 2.65–2.51 (m, 1H), 2.24–2.10 (m, 4H), 1.84 (d, J = 1.3 Hz, 3H), 1.75 (s, 3H), 1.61–1.55 (m, 1H), 1.55–1.47 (m, 1H), 1.20 (d, J = 6.2 Hz, 3H), 1.02 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.11, 147.65, 132.71, 132.58, 130.99, 128.31, 128.00, 126.60, 117.87, 70.45, 68.91, 65.30, 48.84, 44.80, 40.00, 33.83, 24.07, 19.72, 16.88, 12.75. = −29.76, (c 0.34, CHCl3). HRMS (ESI+): calcd. for C20H32O4 [M + Na]+,337.2373, found 337.2375.
Compound 27b (20.9 mg, 80%) was obtained as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.60 (dd, J = 10.0, 1.3 Hz, 1H), 6.23 (dd, J = 15.0, 10.8 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.85 (d, J = 10.7 Hz, 1H), 5.54 (dt, J = 14.8, 7.3 Hz, 1H), 5.32 (dd, J = 17.2, 1.5 Hz, 1H), 5.23 (dd, J = 10.4, 1.2 Hz, 1H), 4.64 (d, J = 5.5 Hz, 2H), 4.21–4.12 (m, 1H), 4.08 (ddd, J = 12.4, 7.8, 5.3 Hz, 1H), 2.65–2.51 (m, 1H), 2.22–2.18 (m, 2H), 2.16 (d, J = 7.2 Hz, 2H), 1.84 (d, J = 1.3 Hz, 3H), 1.76 (s, 3H), 1.67–1.54 (m, 2H), 1.23 (d, J = 6.3 Hz, 3H), 1.02 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.11, 147.65, 133.00, 132.71, 130.92, 128.15, 128.03, 126.59, 117.86, 66.73, 65.47, 65.30, 48.14, 44.09, 40.01, 33.84, 23.69, 19.72, 16.81, 12.75. = −14.00, (c 0.05, CHCl3). HRMS (ESI+): calcd. for C20H32O4 [M + Na]+,337.2373, found 337.2377.
Compound 27c (22 mg, 84%) was obtained as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.60 (dd, J = 10.0, 1.3 Hz, 1H), 6.23 (dd, J = 14.9, 10.8 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.84 (d, J = 10.8 Hz, 1H), 5.54 (dt, J = 14.9, 7.3 Hz, 1H), 5.33 (dd, J = 17.2, 1.5 Hz, 1H), 5.23 (dd, J = 10.4, 1.2 Hz, 1H), 4.64 (d, J = 5.5 Hz, 2H), 4.10–4.02 (m, 1H), 4.01–3.94 (m, 1H), 2.62–2.52 (m, 1H), 2.20–2.11 (m, 4H), 1.84 (d, J = 1.3 Hz, 3H), 1.75 (s, 3H), 1.61–1.55 (m, 1H), 1.54–1.46 (m, 1H), 1.20 (d, J = 6.2 Hz, 3H), 1.02 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.12, 147.64, 132.71, 132.57, 130.99, 128.32, 128.01, 126.60, 117.87, 70.45, 68.91, 65.31, 48.84, 44.81, 39.99, 33.85, 24.07, 19.70, 16.88, 12.75. = −29.37, (c 0.32, CHCl3). HRMS (ESI+): calcd. for C20H32O4 [M + Na]+,337.2373, found 337.2374.
Compound 27d (21.4 mg, 82%) was obtained as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.23 (dd, J = 15.0, 10.8 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.86 (d, J = 10.8 Hz, 1H), 5.54 (dt, J = 14.8, 7.3 Hz, 1H), 5.33 (dd, J = 17.2, 1.5 Hz, 1H), 5.23 (dd, J = 10.4, 1.3 Hz, 1H), 4.64 (d, J = 5.5 Hz, 2H), 4.21–4.11 (m, 1H), 4.12–4.02 (m, 1H), 2.65–2.51 (m, 1H), 2.23–2.18 (m, 2H), 2.15 (t, J = 7.2 Hz, 2H), 1.84 (d, J = 1.3 Hz, 3H), 1.76 (s, 3H), 1.65–1.56 (m, 2H), 1.23 (d, J = 6.3 Hz, 3H), 1.02 (d, J = 6.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.12, 147.64, 132.99, 132.72, 130.95, 128.18, 128.04, 126.60, 117.87, 66.72, 65.48, 65.31, 48.14, 44.10, 39.99, 33.86, 23.69, 19.70, 16.82, 12.76. = −17.62, (c 0.32, CHCl3). HRMS (ESI+): calcd. for C20H32O4 [M + Na]+, 337.2373, found 337.2374.
4.25. Synthetic Procedure of 28a–28d
To a stirred solution of compound 27a (22 mg, 0.065 mmol) in dry acetone (2 mL) under argon, PTSA· H2O (1.14 mg, 0.006 mmol) and 2,2-dimethylpropane (0.08 mL, 0.65 mmol) were added sequentially. After 1 h, the reaction mixture was quenched with a saturated aqueous solution of NaHCO3 and extracted with DCM (2 × 10 mL). The combined organic extracts were washed with water and brine, dried over Na2SO4, and concentrated in vacuum. Purification by column chromatography (PE/EA = 20:1) provided 28a (23 mg, 93%) as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 6.61 (dd, J = 9.9, 1.3 Hz, 1H), 6.23 (dd, J = 15.0, 10.8 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.80 (d, J = 10.8 Hz, 1H), 5.50 (dt, J = 14.8, 7.3 Hz, 1H), 5.32 (dd, J = 17.2, 1.5 Hz, 1H), 5.23 (dd, J = 10.4, 1.3 Hz, 1H), 4.64 (d, J = 5.5 Hz, 2H), 4.04–3.88 (m, 2H), 2.65–2.50 (m, 1H), 2.30 (dd, J = 13.6, 5.9 Hz, 1H), 2.15 (t, J = 7.1 Hz, 2H), 2.06 (dd, J = 13.6, 7.0 Hz, 1H), 1.84 (d, J = 1.1 Hz, 3H), 1.74 (s, 3H), 1.49–1.42 (m, 4H), 1.41–1.34 (m, 4H), 1.15 (d, J = 6.1 Hz, 3H), 1.02 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.11, 147.77, 133.12, 132.72, 129.98, 128.39, 127.15, 126.52, 117.83, 98.61, 67.97, 65.34, 65.27, 46.85, 40.00, 38.60, 33.90, 30.46, 22.38, 19.94, 19.69, 17.34, 12.74. = −30.22, (c 0.26, CHCl3). HRMS (ESI+): calcd. for C23H36O4 [M + Na]+,377.2686, found 377.2689.
Compound 28b (20.8 mg, 89%) was obtained as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.61 (dd, J = 10.0, 1.3 Hz, 1H), 6.23 (dd, J = 15.0, 10.8 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.81 (d, J = 10.7 Hz, 1H), 5.49 (dt, J = 14.8, 7.3 Hz, 1H), 5.33 (dd, J = 17.2, 1.5 Hz, 1H), 5.23 (dd, J = 10.4, 1.3 Hz, 1H), 4.64 (d, J = 5.5 Hz, 2H), 4.04–3.88 (m, 2H), 2.64–2.51 (m, 1H), 2.31 (dd, J = 14.1, 6.9 Hz, 1H), 2.22–2.07 (m, 3H), 1.84 (d, J = 1.3 Hz, 3H), 1.74 (s, 3H), 1.63–1.57 (m, 2H), 1.35 (s, 6H), 1.18 (d, J = 6.3 Hz, 3H), 1.02 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.13, 147.79, 133.39, 132.74, 129.93, 128.43, 126.83, 126.52, 117.84, 100.32, 65.40, 65.28, 62.94, 46.16, 40.00, 39.83, 33.93, 25.24, 25.13, 21.86, 19.70, 17.07, 12.75. = −47.32, (c 0.11, CHCl3). HRMS (ESI+): calcd. for C23H36O4 [M + Na]+, 377.2686, found 377.2684.
Compound 28c (22 mg, 90%) was obtained as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.61 (dd, J = 9.9, 1.3 Hz, 1H), 6.23 (dd, J = 15.0, 10.8 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.80 (d, J = 10.8 Hz, 1H), 5.50 (dt, J = 14.8, 7.3 Hz, 1H), 5.32 (dd, J = 17.2, 1.5 Hz, 1H), 5.23 (dd, J = 10.4, 1.2 Hz, 1H), 4.64 (d, J = 5.5 Hz, 2H), 4.02–3.88 (m, 2H), 2.66–2.50 (m, 1H), 2.30 (dd, J = 13.6, 5.9 Hz, 1H), 2.15 (t, J = 7.1 Hz, 2H), 2.06 (dd, J = 13.7, 7.0 Hz, 1H), 1.84 (d, J = 1.2 Hz, 3H), 1.74 (s, 3H), 1.49–1.42 (m, 4H), 1.41–1.34 (m, 4H), 1.15 (d, J = 6.1 Hz, 3H), 1.02 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.10, 147.76, 133.12, 132.72, 129.96, 128.39, 127.11, 126.51, 117.83, 98.60, 67.97, 65.34, 65.27, 46.81, 40.00, 38.60, 33.89, 30.45, 22.38, 19.94, 19.70, 17.35, 12.73. = −25.76, (c 0.69, CHCl3). HRMS (ESI+): calcd. for C23H36O4 [M + Na]+, 377.2686, found 377.2685.
Compound 28d (21.5 mg, 90%) was obtained as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.61 (dd, J = 10.0, 1.3 Hz, 1H), 6.23 (dd, J = 15.0, 10.8 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.49 (dt, J = 14.8, 7.3 Hz, 1H), 5.32 (dd, J = 17.2, 1.5 Hz, 1H), 5.23 (dd, J = 10.4, 1.3 Hz, 1H), 4.64 (d, J = 5.5 Hz, 2H), 4.04–3.86 (m, 2H), 2.65–2.48 (m, 1H), 2.31 (dd, J = 14.1, 7.0 Hz, 1H), 2.20–2.06 (m, 3H), 1.84 (d, J = 1.3 Hz, 3H), 1.74 (s, 3H), 1.68–1.57 (m, 2H), 1.35 (s, 6H), 1.18 (d, J = 6.3 Hz, 3H), 1.02 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.11, 147.78, 133.38, 132.73, 129.90, 128.42, 126.79, 126.51, 117.83, 100.31, 65.38, 65.27, 62.93, 46.13, 40.00, 39.84, 33.92, 25.23, 25.11, 21.85, 19.70, 17.07, 12.74. = −17.71, (c 0.23, CHCl3). HRMS (ESI+): calcd. for C23H36O4 [M + Na]+, 377.2686, found 377.2692.
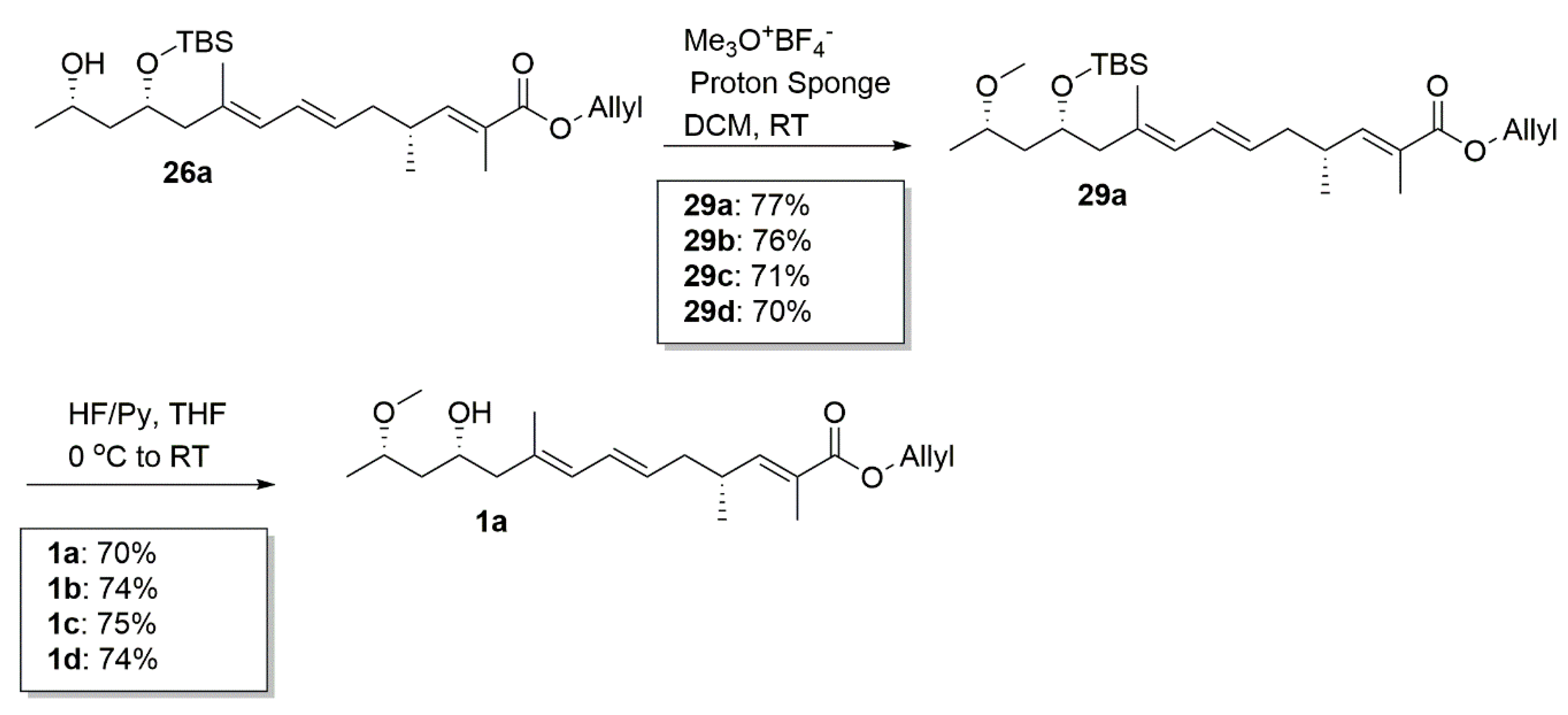
4.26. Synthetic Procedure of 29a–29d
To a stirred solution of compound 26a (48 mg, 0.11 mmol) in dry DCM (3 mL) and 4A molecular sieve under argon, the Proton Sponge (107 mg, 0.5 mmol) and trimethyloxonium tetrafluoroborate (60 mg, 0.4 mmol) were added sequentially. After 1 h, the reaction mixture was quenched with 1 M HCl (10 mL) and extracted with DCM (2 × 10 mL). The combined organic extracts were washed with water and brine, dried over Na2SO4, and concentrated in vacuum. Purification by column chromatography (PE/EA = 40:1) created 29a (38 mg, 77%) as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 6.60 (d, J = 9.9 Hz, 1H), 6.20 (dd, J = 15.0, 10.8 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.75 (d, J = 10.8 Hz, 1H), 5.47 (dt, J = 14.8, 7.3 Hz, 1H), 5.28 (ddd, J = 13.8, 11.6, 1.3 Hz, 2H), 4.64 (d, J = 5.6 Hz, 2H), 4.00 (tdd, J = 8.7, 5.4, 3.2 Hz, 1H), 3.54–3.37 (m, 1H), 3.28 (d, J = 2.2 Hz, 3H), 2.71–2.44 (m, 1H), 2.36–2.21 (m, 1H), 2.21–2.01 (m, 3H), 1.84 (d, J = 1.2 Hz, 3H), 1.72 (s, 3H), 1.62–1.54 (m, 1H), 1.37–1.28 (m, 1H), 1.11 (d, J = 6.1 Hz, 3H), 1.01 (d, J = 6.6 Hz, 3H), 0.88 (s, 9H), 0.05 (d, J = 6.1 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 168.12, 147.85, 133.80, 132.75, 129.65, 128.52, 127.56, 126.47, 117.85, 73.13, 67.86, 65.27, 55.62, 49.18, 44.92, 39.99, 33.96, 26.08, 19.64, 19.38, 18.20, 17.36, 12.72, −3.96, −4.53. = −38.67, (c 0.05, CHCl3). HRMS (ESI+): calcd. for C27H48O4Si [M + H]+, 465.3395, found 465.3388.
Compound 29b (72 mg, 76%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 6.61 (dd, J = 9.9, 1.1 Hz, 1H), 6.20 (dd, J = 14.9, 10.9 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.77 (d, J = 10.8 Hz, 1H), 5.46 (dt, J = 14.8, 7.3 Hz, 1H), 5.33 (dd, J = 17.2, 1.5 Hz, 1H), 5.23 (dd, J = 10.4, 1.2 Hz, 1H), 4.64 (dd, J = 5.5, 1.1 Hz, 2H), 3.93–3.83 (m, 1H), 3.41 (dd, J = 12.5, 6.3 Hz, 1H), 3.29 (s, 3H), 2.64–2.47 (m, 1H), 2.28–2.05 (m, 4H), 1.84 (d, J = 1.3 Hz, 3H), 1.72 (s, 4H), 1.48–1.39 (m, 1H), 1.11 (d, J = 6.1 Hz, 3H), 1.01 (d, J = 6.7 Hz, 3H), 0.86 (s, 9H), 0.02 (d, J = 9.8 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 168.14, 147.87, 133.66, 132.73, 129.73, 128.47, 127.73, 126.45, 117.85, 74.24, 68.48, 65.28, 55.97, 48.38, 44.21, 40.01, 33.97, 26.01, 19.64, 19.43, 18.16, 17.33, 12.74, −4.15, −4.47. = −34.83, (c 0.38, CHCl3). HRMS (ESI+): calcd. for C27H48O4Si [M + H]+, 465.3395, found 465.3391.
Compound 29c (32 mg, 71%) was obtained as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.59 (d, J = 9.7 Hz, 1H), 6.18 (dd, J = 15.0, 10.9 Hz, 1H), 5.94 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.73 (d, J = 10.7 Hz, 1H), 5.45 (dt, J = 14.7, 7.2 Hz, 1H), 5.31 (dd, J = 17.2, 1.1 Hz, 1H), 5.21 (d, J = 10.5 Hz, 1H), 4.62 (d, J = 5.5 Hz, 2H), 3.98 (dt, J = 16.1, 6.0 Hz, 1H), 3.51–3.35 (m, 1H), 3.26 (s, 3H), 2.62–2.44 (m, 1H), 2.27 (ddd, J = 19.7, 13.8, 9.4 Hz, 1H), 2.18–2.01 (m, 3H), 1.82 (s, 3H), 1.70 (s, 3H), 1.60–1.52 (m, 1H), 1.37–1.25 (m, 1H), 1.09 (d, J = 6.1 Hz, 3H), 0.99 (d, J = 6.6 Hz, 3H), 0.86 (s, 9H), 0.03 (d, J = 6.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 168.11, 147.86, 133.79, 132.74, 129.66, 128.51, 127.55, 126.46, 117.84, 73.13, 67.86, 65.27, 55.63, 49.18, 44.91, 40.05, 33.97, 26.08, 19.71, 19.38, 18.20, 17.39, 12.72, −3.96, −4.54. = −26.17, (c 0.1, CHCl3). HRMS (ESI+): calcd. for C27H48O4Si [M + H]+, 465.3395, found 465.3389.
Compound 29d (72 mg, 70%) was obtained as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.61 (dd, J = 10.0, 1.2 Hz, 1H), 6.20 (dd, J = 15.0, 10.9 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.77 (d, J = 10.8 Hz, 1H), 5.46 (dt, J = 14.7, 7.2 Hz, 1H), 5.33 (dd, J = 17.2, 1.5 Hz, 1H), 5.23 (dd, J = 10.4, 1.2 Hz, 1H), 4.64 (d, J = 5.5 Hz, 2H), 3.94–3.79 (m, 1H), 3.51–3.36 (m, 1H), 3.29 (s, 3H), 2.70–2.50 (m, 1H), 2.25–2.07 (m, 4H), 1.85 (d, J = 1.3 Hz, 3H), 1.79–1.67 (m, 4H), 1.45 (m, 1H), 1.12 (d, J = 6.1 Hz, 3H), 1.01 (d, J = 6.6 Hz, 3H), 0.86 (s, 9H), 0.01 (d, J = 9.0 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 168.12, 147.87, 133.67, 132.75, 129.73, 128.47, 127.72, 126.47, 117.84, 74.25, 68.54, 65.28, 55.97, 48.39, 44.20, 40.07, 33.96, 26.02, 19.70, 19.44, 18.17, 17.37, 12.74, −4.15, −4.47. = −16.46, (c 0.41, CHCl3). HRMS (ESI+): calcd. for C27H48O4Si [M + H]+, 465.3395, found 465.3389.
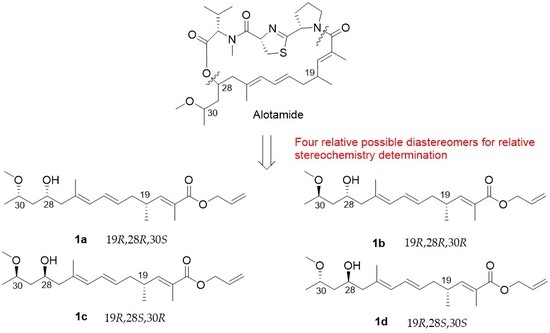
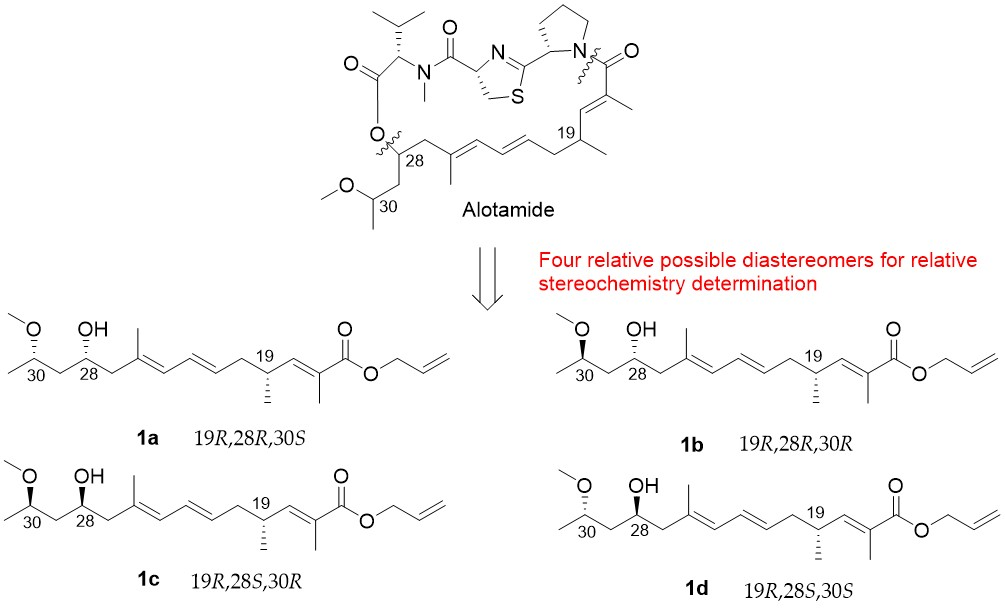
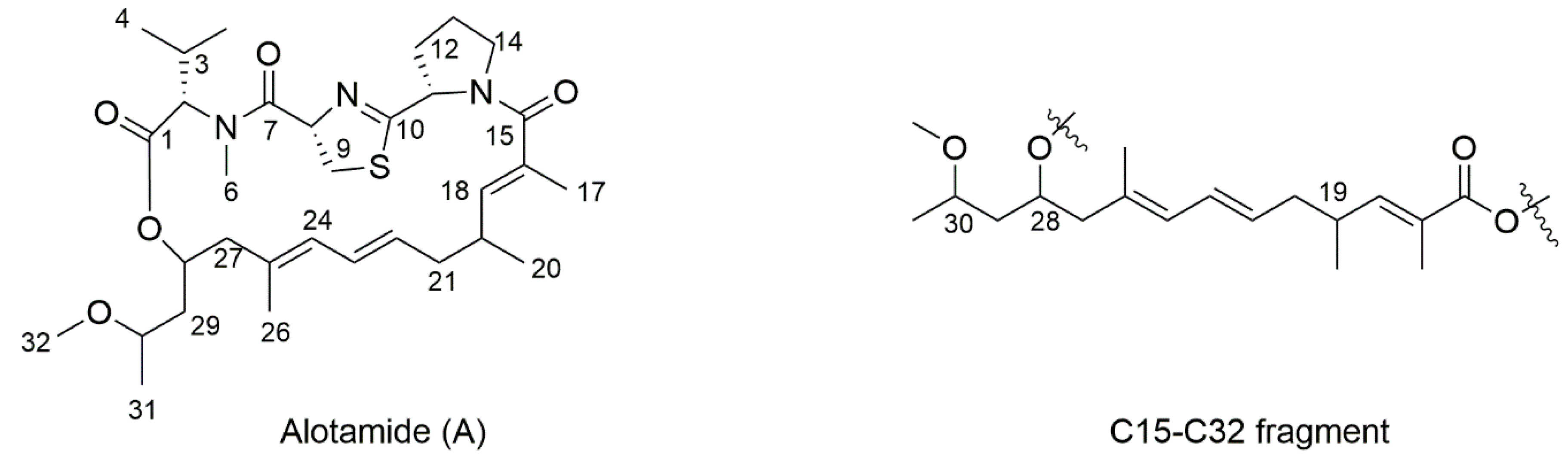
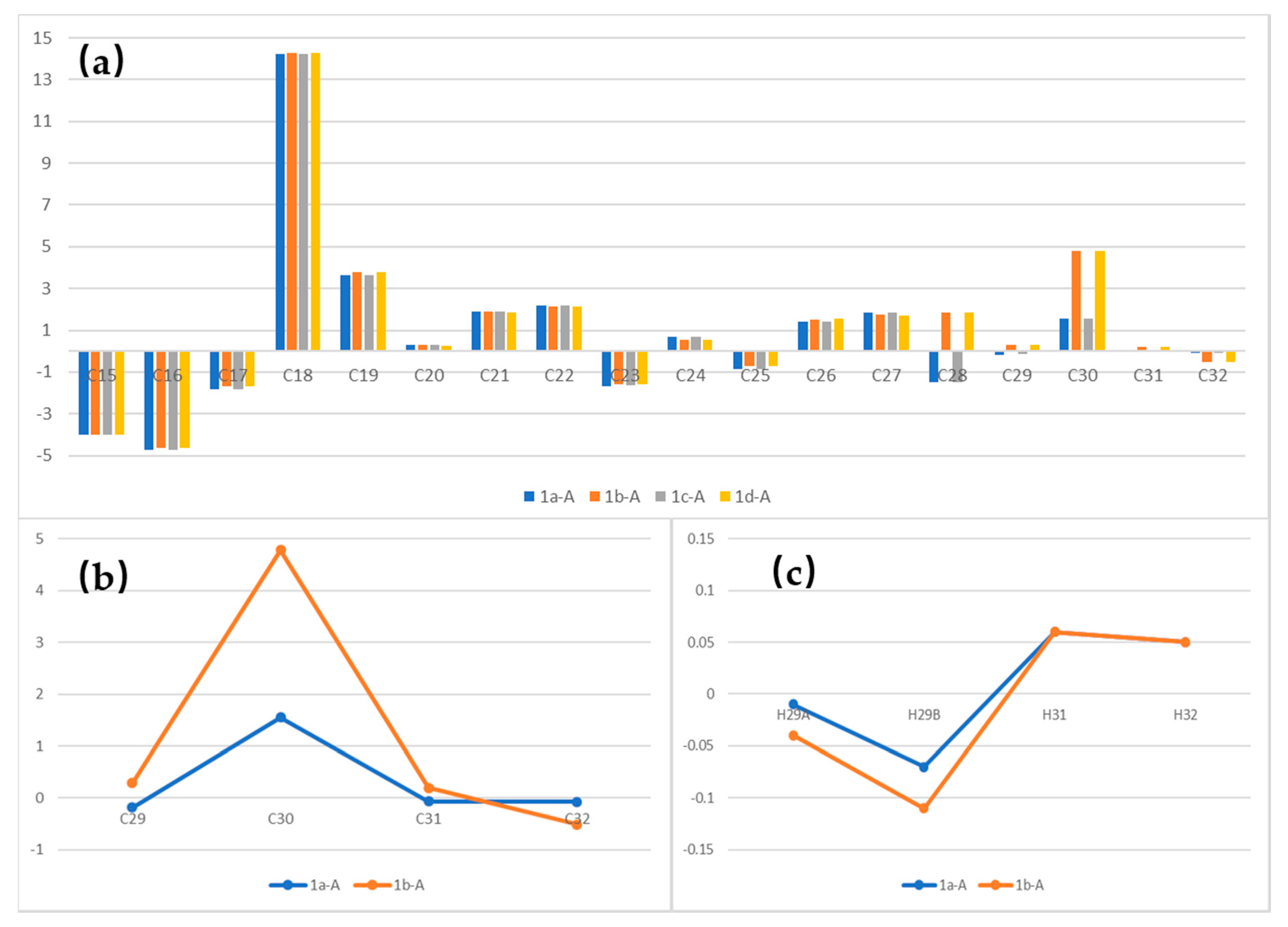
4.27. Synthetic Procedure of 1a–1d
To a stirred solution of 29a in dry THF (2 mL), the HF/Py complex (0.4 mL) was added at 0 °C. After being stirred for 1 h, the mixture was quenched with a saturated aqueous solution of NaHCO3 and extracted with DCM (10 mL × 3). The combined organic layers were washed with 1 M HCl, brine, dried over Na2SO4, filtrated, and concentrated. The residue was purified by column chromatography on silica gel (PE/EA = 4:1) to create 1a (20 mg, 70%) as a colorless oil.
1H NMR (400 MHz, CDCl3) δ 6.60 (dd, J = 9.9, 1.3 Hz, 1H), 6.23 (dd, J = 15.0, 10.8 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.83 (d, J = 10.8 Hz, 1H), 5.59–5.42 (m, 1H), 5.32 (dd, J = 17.2, 1.5 Hz, 1H), 5.22 (dd, J = 10.4, 1.2 Hz, 1H), 4.63 (d, J = 5.5 Hz, 2H), 4.10–3.98 (m, 1H), 3.69–3.61 (m, 1H), 3.34 (s, 3H), 2.57 (m, 1H), 2.17 (m, 4H), 1.84 (d, J = 1.3 Hz, 3H), 1.75 (s, 3H), 1.59–1.55 (m, 2H), 1.17 (d, J = 6.2 Hz, 3H), 1.01 (d, J = 6.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.11, 147.75, 133.68, 132.71, 130.36, 128.23, 127.58, 126.52, 117.85, 74.75, 66.38, 65.28, 56.34, 48.36, 42.82, 40.00, 33.89, 19.70, 18.94, 16.91, 12.73. = −25.83, (c 0.46, CHCl3). HRMS (ESI+): calcd. for C21H34O4 [M + Na]+, 373.2349, found 373.2346.
Compound 1b (40 mg, 74%) was obtained as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.60 (dd, J = 9.9, 1.1 Hz, 1H), 6.23 (dd, J = 15.0, 10.8 Hz, 1H), 5.95 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.82 (d, J = 10.8 Hz, 1H), 5.50 (dt, J = 14.8, 7.3 Hz, 1H), 5.32 (dd, J = 17.2, 1.5 Hz, 1H), 5.22 (dd, J = 10.4, 1.2 Hz, 1H), 4.63 (d, J = 5.5 Hz, 2H), 3.98–3.87 (m, 1H), 3.61–3.49 (m, 1H), 3.33 (s, 3H), 2.65–2.47 (m, 1H), 2.26–2.05 (m, 4H), 1.83 (s, 3H), 1.75 (s, 3H), 1.59–1.50 (m, 2H), 1.17 (d, J = 6.0 Hz, 3H), 1.01 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.10, 147.78, 133.68, 132.70, 130.13, 128.30, 127.42, 126.48, 117.82, 77.98, 69.74, 65.25, 55.89, 48.24, 43.29, 40.00, 33.88, 19.68, 19.20, 17.00, 12.71. = −29.17, (c 0.80, CHCl3). HRMS (ESI+): calcd. for C21H34O4 [M + Na]+, 373.2349, found 373.2348.
Compound 1c (18 mg, 75%) was obtained as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 6.60 (dd, J = 9.9, 1.3 Hz, 1H), 6.23 (dd, J = 15.0, 10.8 Hz, 1H), 5.96 (ddt, J = 17.1, 10.5, 5.6 Hz, 1H), 5.83 (d, J = 10.8 Hz, 1H), 5.59–5.42 (m, 1H), 5.32 (dd, J = 17.2, 1.5 Hz, 1H), 5.22 (dd, J = 10.4, 1.2 Hz, 1H), 4.63 (d, J = 5.5 Hz, 2H), 4.10–3.98 (m, 1H), 3.69–3.61 (m, 1H), 3.34 (s, 3H), 2.57 (m, 1H), 2.17 (m, 4H), 1.84 (d, J = 1.3 Hz, 3H), 1.75 (s, 3H), 1.59–1.55 (m, 2H), 1.17 (d, J = 6.2 Hz, 3H), 1.01 (d, J = 6.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.11, 147.74, 133.68, 132.71, 130.34, 128.24, 127.58, 126.52, 117.85, 74.74, 66.38, 65.28, 56.34, 48.36, 42.87, 39.98, 33.89, 19.68, 18.96, 16.91, 12.73. = −21.89, (c 0.52, CHCl3). HRMS (ESI+): calcd. for C21H34O4 [M + Na]+, 373.2349, found 373.2349.
Compound
1d (40 mg, 74%) was obtained as a colorless oil.
1H NMR (400 MHz, CDCl
3) δ 6.60 (dd,
J = 9.9, 1.1 Hz, 1H), 6.23 (dd,
J = 15.0, 10.8 Hz, 1H), 5.95 (ddt,
J = 17.1, 10.5, 5.6 Hz, 1H), 5.82 (d,
J = 10.8 Hz, 1H), 5.50 (dt,
J = 14.8, 7.3 Hz, 1H), 5.32 (dd,
J = 17.2, 1.5 Hz, 1H), 5.22 (dd,
J = 10.4, 1.2 Hz, 1H), 4.63 (d,
J = 5.5 Hz, 2H), 3.98–3.87 (m, 1H), 3.61–3.49 (m, 1H), 3.33 (s, 3H), 2.65–2.47 (m, 1H), 2.26–2.05 (m, 4H), 1.83 (s, 3H), 1.75 (s, 3H), 1.59–1.50 (m, 2H), 1.17 (d,
J = 6.0 Hz, 3H), 1.01 (d,
J = 6.7 Hz, 3H).
13C NMR (101 MHz, CDCl
3) δ 168.10, 147.78, 133.69, 132.70, 130.11, 128.31, 127.41, 126.48, 117.83, 77.96, 69.75, 65.26, 55.89, 48.22, 43.30, 39.96, 33.88, 19.66, 19.20, 17.04, 12.71.
= −38.68, (
c 0.76, CHCl
3). HRMS (ESI+): calcd. for C
21H
34O
4 [M + Na]
+, 373.2349, found 373.2350. Detailed NMR data tables of
1a–
1d are in the
Supplementary Material.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

