Antitumor Activity of Quinazolinone Alkaloids Inspired by Marine Natural Products

 , ,
, ,  , ,
, ,  , ,
, ,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
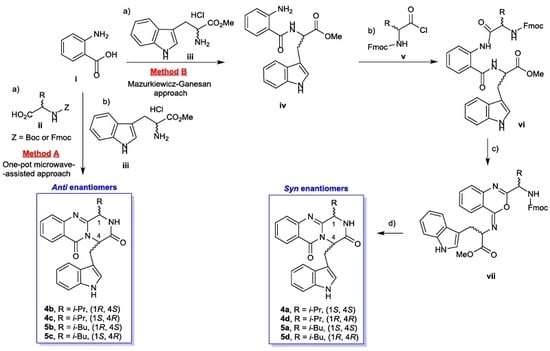
2.1. Synthesis of Pyrazinoquinazoline Alkaloids
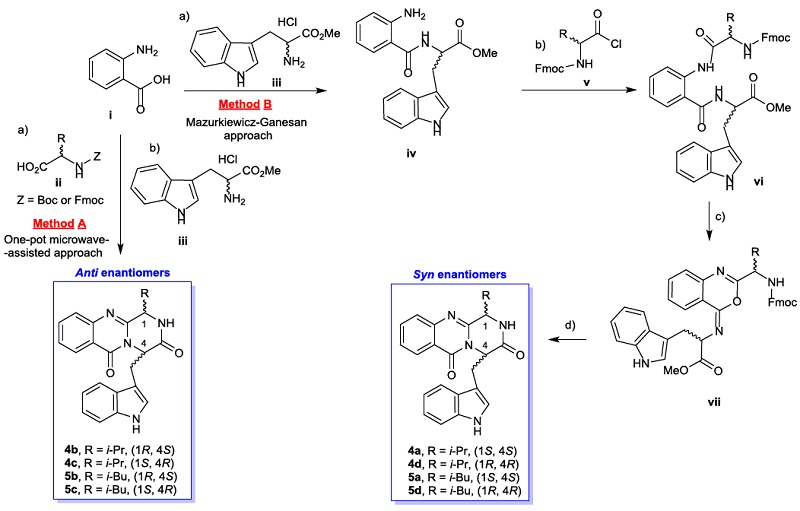
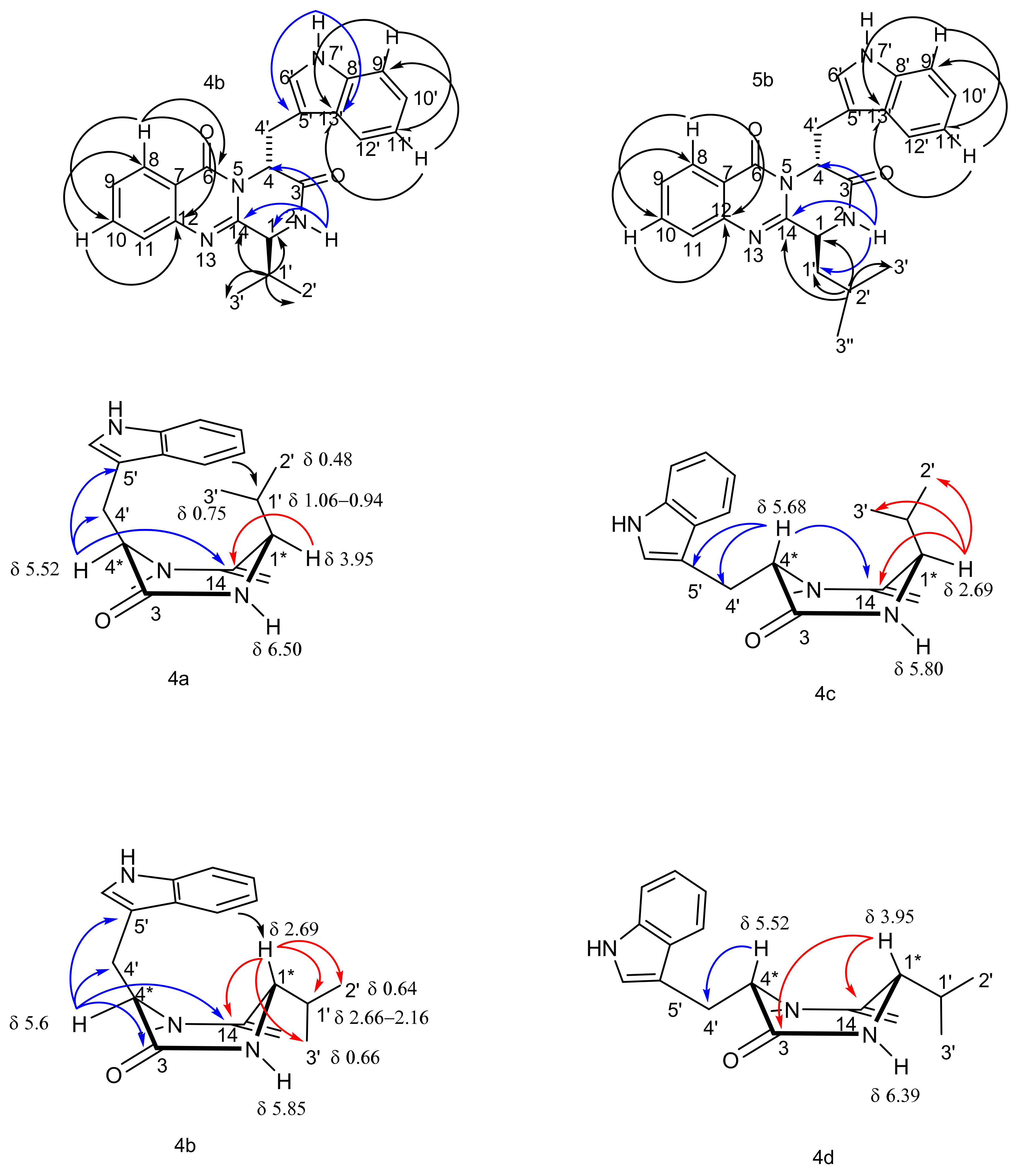
2.2. Structure Elucidation
2.3. Tumor Cell Growth Inhibitory Activity
3. Materials and Methods
3.1. General Procedure
3.2. General Conditions for the Synthesis of 4-(1H-Indol-3-ylmethyl)-1-isopropyl-2H-pyrazino[2,1-b]quinazoline-3,6-(1H,4H)-diones (4b/4c)
3.2.1. (1R,4S)-4-(1H-Indol-3-ylmethyl)-1-isopropyl-2H-pyrazino[2,1-b]quinazoline-3,6-(1H,4H)-dione (4b)
3.2.2. (1S,4R)-4-(1H-Indol-3-ylmethyl)-1-isopropyl-2H-pyrazino[2,1-b]quinazoline-3,6-(1H,4H)-dione (4c)
3.3. General Condition for the Synthesis of 4-(1H-Indol-3-ylmethyl)-1-isobutyl-2H-pyrazino[2,1-b]quinazoline-3,6-(1H,4H)-diones (5b/5c)
3.3.1. (1R,4S)-4-(1H-Indol-3-ylmethyl)-1-isobutyl-2H-pyrazino[2,1-b]quinazoline-3,6-(1H,4H)-dione (5b)
3.3.2. (1S,4R)-4-(1H-Indol-3-ylmethyl)-1-isobutyl-2H-pyrazino[2,1-b]quinazoline-3,6-(1H,4H)-dione (5c)
3.4. General Condition for the Synthesis of Compounds 4a and 5a
3.4.1. Synthesis of N-(2-Aminobenzoyl)-l-tryptophan methyl ester (iv-a)
3.4.2. Synthesis of N-[9H-Fluoren-9-ylmethoxy)carbonyl]-l-valinyl-2-aminobenzoyl-l-tryptophan methyl ester (vi-a)
3.4.3. Synthesis of (1S,4S)-4-(1H-Indol-3-ylmethyl)-1-isopropyl-2H-pyrazino[2,1-b]quinazolin-3,6-(1H, 4H)-dione (4a)
3.4.4. Synthesis of N-[9H-Fluoren-9-ylmethoxy)carbonyl]-l-methylpentanyl-2H-aminobenzoyl-l-tryptophan methyl ester (vi-b)
3.4.5. Synthesis of (1S,4S)-4-(1H-Indol-3-ylmethyl)-1-isobutyl-2H-pyrazino[2,1-b]quinazolin-3,6-(1H,4H)-dione (5a)
3.5. General Condition for the Synthesis Compound 4d and 5d
3.5.1. Synthesis of N-(2-Aminobenzoyl)-d-tryptophan methyl ester (iv-b)
3.5.2. Synthesis of N-[9H-Fluoren-9-ylmethoxy)carbonyl]-d-valinyl-2-aminobenzoyl-d-tryptophan methyl ester (vi-c)
3.5.3. Synthesis of (1R,4R)-4-(1H-Indol-3-ylmethyl)-1-isopropyl-2H-pyrazino[2,1-b]quinazolin-3,6-(1H,4H)-dione (4d)
3.5.4. Synthesis of N-[9H-Fluoren-9-ylmethoxy)carbonyl]-d-methylpentyl-2-aminobenzoyl-d-tryptophan methyl ester (vi-d)
3.5.5. Synthesis of (1R,4R)-4-(1H-Indol-3-ylmethyl)-1-isobutyl-2H-pyrazino[2,1-b]quinazolin-3,6-(1H,4H)-dione (5d)
3.6. Screening Test for Antitumor and Anti-P-Glycoprotein Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Tripathy, U.P. Quinazolone: A molecule of significant pharmacological and biological activity. Res. J. Pharm. Technol. 2013, 6, 849–855. [Google Scholar]
- Eguchi, S. Quinazoline alkaloids and related chemistry. In Bioactive Heterocycles i; Eguchi, S., Ed.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 113–156. [Google Scholar]
- Peng, J.; Lin, T.; Wang, W.; Xin, Z.; Zhu, T.; Gu, Q.; Li, D. Antiviral alkaloids produced by the mangrove-derived fungus cladosporium sp. Pjx-41. J. Nat. Prod. 2013, 76, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Debbab, A.; Mándi, A.; Wray, V.; Schulz, B.; Müller, W.E.G.; Kassack, M.; Lin, W.; Kurtán, T.; Proksch, P.; et al. Alkaloids from the sponge-associated fungus Aspergillus sp. Eur. J. Org. Chem. 2013, 2013, 894–906. [Google Scholar] [CrossRef]
- He, F.; Han, Z.; Peng, J.; Qian, P.Y.; Qi, S.H. Antifouling indole alkaloids from two marine derived fungi. Nat. Prod. Commun. 2013, 8, 329–332. [Google Scholar] [PubMed]
- Rodrigues, B.S.F.; Sahm, B.D.B.; Jimenez, P.C.; Pinto, F.C.L.; Mafezoli, J.; Mattos, M.C.; Rodrigues-Filho, E.; Pfenning, L.H.; Abreu, L.M.; Costa-Lotufo, L.V.; et al. Bioprospection of cytotoxic compounds in fungal strains recovered from sediments of the brazilian coast. Chem. Biodivers. 2015, 12, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Afiyatullov, S.S.; Zhuravleva, O.I.; Antonov, A.S.; Kalinovsky, A.I.; Pivkin, M.V.; Menchinskaya, E.S.; Aminin, D.L. New metabolites from the marine-derived fungus Aspergillus fumigatus. Nat. Prod. Commun. 2012, 7, 497–500. [Google Scholar] [PubMed]
- Shao, C.L.; Xu, R.F.; Wei, M.Y.; She, Z.G.; Wang, C.Y. Structure and absolute configuration of fumiquinazoline l, an alkaloid from a gorgonian-derived Scopulariopsis sp. Fungus. J. Nat. Prod. 2013, 76, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, X.M.; Meng, L.H.; Wang, B.G. N-formyllapatin A, a new N-formylspiroquinazoline derivative from the marine-derived fungus Penicillium adametzioides As-53. Phytochem. Lett. 2014, 10, 145–148. [Google Scholar] [CrossRef]
- Zin, W.W.M.; Prompanya, C.; Buttachon, S.; Kijjoa, A. Bioactive secondary metabolites from a Thai collection of soil and marine-derived fungi of the genera Neosartorya and Aspergillus. Curr. Drug Deliv. 2016, 13, 378–388. [Google Scholar] [PubMed]
- Prata-Sena, M.; Ramos, A.A.; Buttachon, S.; Castro-Carvalho, B.; Marques, P.; Dethoup, T.; Kijjoa, A.; Rocha, E. Cytotoxic activity of secondary metabolites from marine-derived fungus Neosartorya siamensis in human cancer cells. Phytother. Res. 2016, 30, 1862–1871. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; You, M.; Chung, B.K.; Oh, D.-C.; Oh, K.-B.; Shin, J. Alkaloidal metabolites from a marine-derived Aspergillus sp. Fungus. J. Nat. Prod. 2015, 78, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wei, X.; Kim, E.L.; Lin, X.; Yang, X.W.; Zhou, X.; Yang, B.; Jung, J.H.; Liu, Y. Erratum: Fumigatosides A–D, four new glucosidated pyrazinoquinazoline indole alkaloids from a jellyfish-derived fungus Aspergillus fumigatus (org. Lett. (2014)). Org. Lett. 2014, 16, 2574. [Google Scholar] [CrossRef] [PubMed]
- Martine, D.; Isabelle, B. Survey of recent literature related to the biologically active 4(3H)-quinazolinones containing fused heterocycles. Curr. Med. Chem. 2013, 20, 794–814. [Google Scholar] [CrossRef]
- Penn, J.; Purcell, M.; Mantle, P.G. Biosynthesis of glyantrypine by aspergillus clavatus. FEMS Microbiol. Lett. 1992, 92, 229–233. [Google Scholar] [CrossRef]
- Takahashi, C.; Matsushita, T.; Doi, M.; Minoura, K.; Shingu, T.; Kumeda, Y.; Numata, A. Fumiquinazolines A–G, novel metabolites of a fungus separated from a pseudolabrus marine fish. J. Chem. Soc. Perkin Trans. 1 1995, 2345–2353, 2345–2353. [Google Scholar] [CrossRef]
- Wong, S.M.; Musza, L.L.; Kydd, H.G.C.; Kullnig, R.; Gillum, A.M.; Cooper, R. Fiscalins: New substance P inhibitors produced by the fungus neosartorya fischeri taxonomy, fermentation, structures, and biological properties. J. Antibiot. 1993, 46, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Avendaño, C.; Caballero, E.; Méndez-Vidal, C.; De Quesada, A.R.; Menéndez, J.C. MDR reversal by deprenylated tetracyclic and hexacyclic analogues of N-acetylardeemin: Confirmation of the ardeemin pharmacophore. Lett. Drug Des. Discov. 2006, 3, 369–377. [Google Scholar] [CrossRef]
- Hayashi, D.; Tsukioka, N.; Inoue, Y.; Matsubayashi, Y.; Iizuka, T.; Higuchi, K.; Ikegami, Y.; Kawasaki, T. Synthesis and ABCG2 inhibitory evaluation of 5-N-acetylardeemin derivatives. Bioorg. Med. Chem. 2015, 23, 2010–2023. [Google Scholar] [CrossRef] [PubMed]
- Buttachon, S.; Chandrapatya, A.; Manoch, L.; Silva, A.; Gales, L.; Bruyère, C.; Kiss, R.; Kijjoa, A. Sartorymensin, a new indole alkaloid, and new analogues of tryptoquivaline and fiscalins produced by Neosartorya siamensis (KUFC 6349). Tetrahedron 2012, 68, 3253–3262. [Google Scholar] [CrossRef]
- Bessa, L.J.; Buttachon, S.; Dethoup, T.; Martins, R.; Vasconcelos, V.; Kijjoa, A.; da Costa, P.M. Neofiscalin A and fiscalin C are potential novel indole alkaloid alternatives for the treatment of multidrugresistant gram-positive bacterial infections. FEMS Microbiol. Lett. 2016, 363. [Google Scholar] [CrossRef] [PubMed]
- Cledera, P.; Avendaño, C.; Menéndez, J.C. A new route toward 4-substituted pyrazino[2,1-b]quinazoline-3,6-dione systems. Total synthesis of glyantrypine. J. Org. Chem. 2000, 65, 1743–1749. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ganesan, A. Total Synthesis of the quinazoline alkaloids (−)-Fumiquinazoline G and (−)-Fiscalin B. J. Org. Chem. 1998, 63, 2432–2433. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ganesan, A. Total synthesis of the fumiquinazoline alkaloids: Solution-phase studies. J. Org. Chem. 2000, 65, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Ye, P.; Zhang, B.; Bi, G.; Sargent, K.; Yu, L.; Yohannes, D.; Baldino, C.M. Three-component one-pot total syntheses of glyantrypine, Fumiquinazoline F, and Fiscalin B promoted by microwave irradiation. J. Org. Chem. 2005, 70, 6339–6345. [Google Scholar] [CrossRef] [PubMed]
- Kantharaju; Patil, B.S.; Suresh Babu, V.V. Synthesis of fmoc-amino acid chlorides assisted by ultrasonication, a rapid approach. Int. J. Pept. Res. Ther. 2002, 9, 227–229. [Google Scholar]
- Hernández, F.; Buenadicha, F.L.; Avendao, C.; Söllhuber, M. 1-alkyl-2,4-dihydro-1h-pyrazino[2,1-b]quinazoline-3,6-diones as glycine templates. Synthesis of Fiscalin B. Tetrahedron Asymmetry 2002, 12, 3387–3398. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Rodrigues, V.; Oliveira, A.; Correia-da-Silva, M.; Pinto, M.; Lima, R.T.; Sousa, E.; Vasconcelos, M.H. A novel curcumin derivative which inhibits P-glycoprotein, arrests cell cycle and induces apoptosis in multidrug resistance cells. Bioorg. Med. Chem. 2017, 25, 581–596. [Google Scholar] [CrossRef] [PubMed]
- Han, X.X.; Xu, X.Y.; Cui, C.B.; Gu, Q.Q. Alkaloidal compounds produced by a marine-derived fungus, aspergillus fumigatus H1-04, and their antitumor activities. J. Chin. Med. Chem. 2007, 4, 232. [Google Scholar]
- Yu, G.; Zhou, G.; Zhu, M.; Wang, W.; Zhu, T.; Gu, Q.; Li, D. Neosartoryadins A and B, fumiquinazoline alkaloids from a mangrove-derived fungus neosartorya udagawae HDN13-313. Org. Lett. 2016, 18, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Kijjoa, A.; Santos, S.; Dethoup, T.; Manoch, L.; Almeida, A.P.; Vasconcelos, M.H.; Silva, A.; Gales, L.; Herz, W. Sartoryglabrins, analogs of ardeemins, from neosartorya glabra. Nat. Prod. Commun. 2011, 6, 807–812. [Google Scholar] [PubMed]
- Palmeira, A.; Vasconcelos, M.H.; Paiva, A.; Fernandes, M.X.; Pinto, M.; Sousa, E. Dual inhibitors of p-glycoprotein and tumor cell growth: (Re)discovering thioxanthones. Biochem. Pharmacol. 2012, 83, 57–68. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
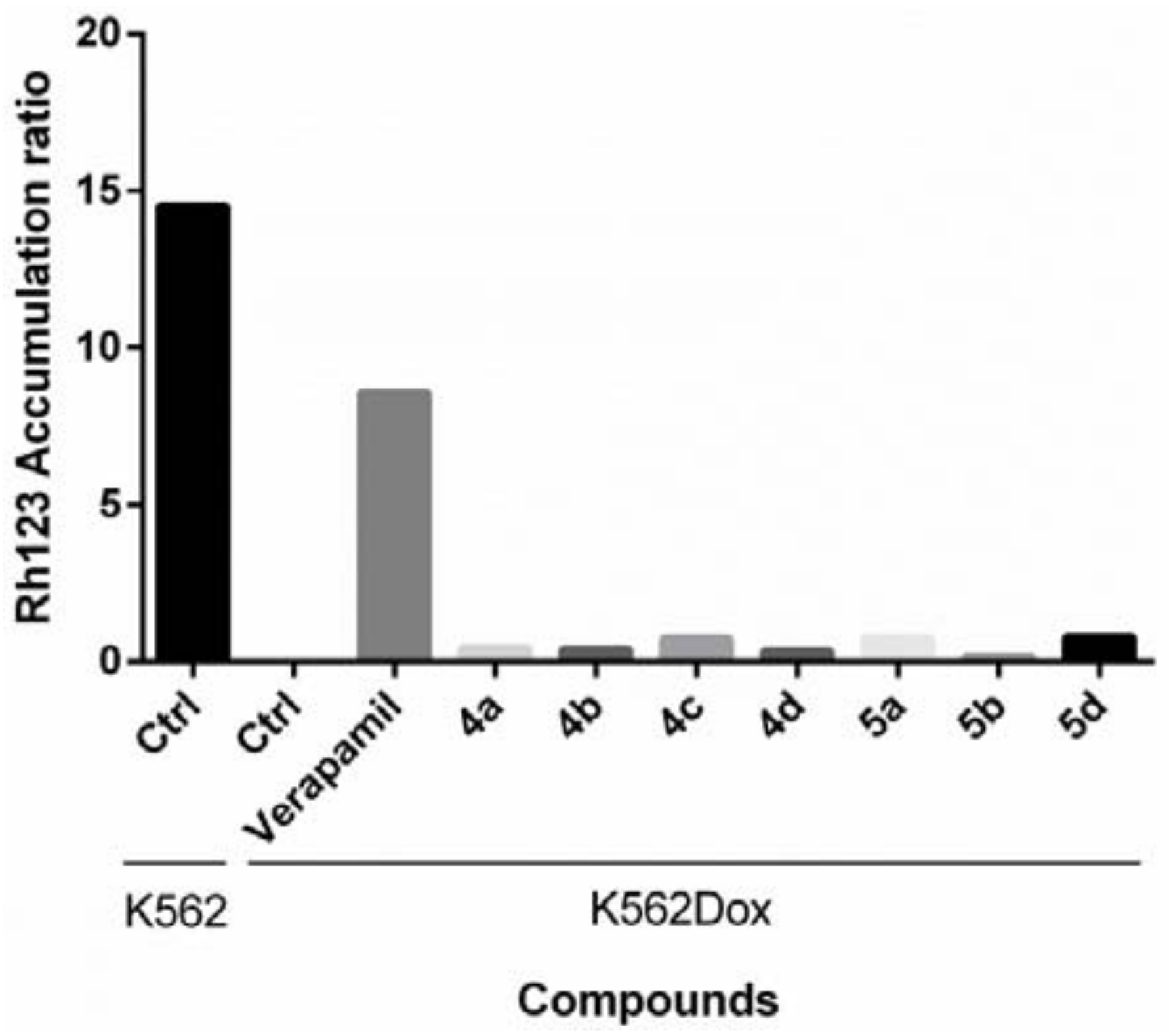{kind=link}
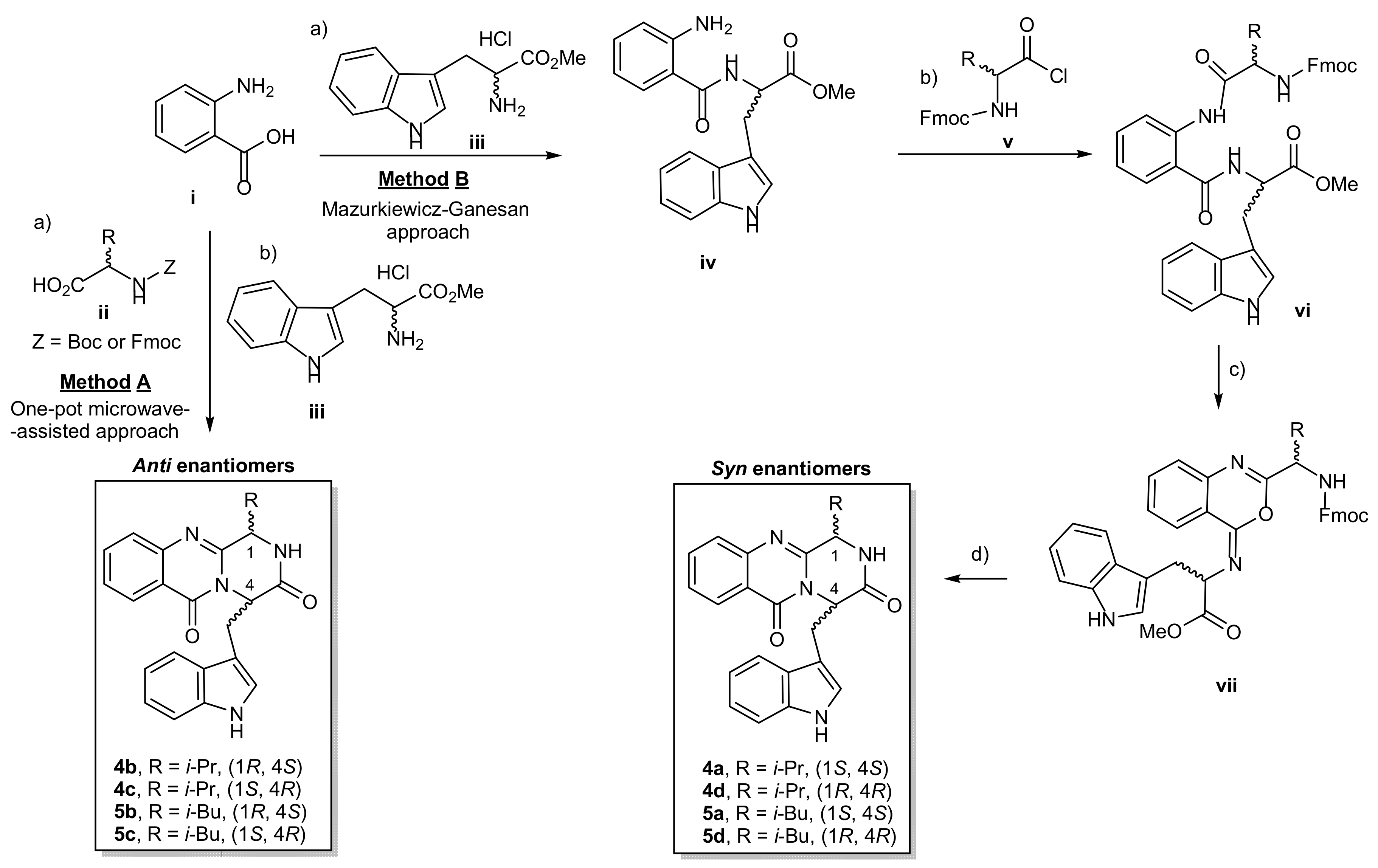{kind=link}

| Entry | R a | Tryptophan | % Anti Compound | % Syn Compound | [α]D b | Enantiomeric Ratio c | Purity (%) d |
|---|---|---|---|---|---|---|---|
| 1 | l-valine | l-tryptophan | 10 (4b) | - | +63.8 | 40:60 | 87 |
| 2 | d-valine | l-tryptophan | 14 (4b) | - | +65.2 | 39:61 | 89 |
| 3 | l-valine | d-tryptophan | 8 (4c) | - | −248.1 | 62:37 | 90 |
| 4 | d-valine | d-tryptophan | 10 (4c) | - | −100.0 | 63:37 | 91 |
| 5 | l-leucine | l-tryptophan | 7 (5b) | - | +44.9 | 67:33 | 94 |
| 6 | d-leucine | l-tryptophan | 10 (5b) | - | +89.7 | 50:50 | 94 |
| 7 | l-leucine | d-tryptophan | 8 (5c) | - | −61.7 | 31:69 | 90 |
| 8 | d-leucine | d-tryptophan | 10 (5c) | - | −142.9 | 46:54 | 86 |
| 9 * | l-valine | l-tryptophan | - | 28 (4a) | +300.5 | 100:0 | 94 |
| 10 * | d-valine | d-tryptophan | - | 21(4d) | −210.5 | 7:93 | 89 |
| 11 * | l-leucine | l-tryptophan | - | 28 (5a) | +81.8 | 90:10 | 89 |
| 12 * | d-leucine | d-tryptophan | - | 40 (5d) | −186.0 | 17:83 | 85 |
| Position | δH (J in Hz) | |||
|---|---|---|---|---|
| 4a | 4b | 4c | 4d | |
| H-1 | 3.95, dd (8.4,3.5) | 2.69, d (2.4) | 2.69, d (2.4) | 3.95, dd (8.5, 3.6) |
| H-2 | 6.50, d (3.0) | 5.85, br | 5.80, br | 6.39, d (3.2) |
| H-4 | 5.52, dd (6.3, 3.8) | 5.68, dd (5.0, 2.8) | 5.67, dd (5.0, 2.8) | 5.52, dd (6.3, 3.8) |
| H-8 | 8. 38, dd (8.0, 1.1) | 8. 37, dd (8.0, 1.1) | 8. 37, dd (8.0, 1.1) | 8. 38, dd (8.0, 1.2) |
| H-9 | 7.54, ddd (8.2, 7.2, 1.2) | 7.54, t (3.0) | 7.54, t (5.2) | 7.54, ddd (8.1, 7.3, 1.1) |
| H-10 | 7.79, ddd (8.7, 7.7, 1.9) | 7.77, ddd (7.9, 7.7, 1.9) | 7.77, ddd (8.9, 6.2, 1.9) | 7.79, ddd (8.5, 7.1, 1.2) |
| H-11 | 7.62, d (7.7) | 7.57, d (3.4) | 7.57, d (3.5) | 7.62, d (7.7) |
| H-1′ | 1.06–0.94, m | 2.66–2.61, m | 2.66–2.61, m | 1.06–0.94, m |
| H-2′ | 0.48, d (6.6) | 0.63, d (2.3) | 0.63, d (4.0) | 0.48, d (6.6) |
| H-3′ | 0.75, d (6.8) | 0.66, d (3.7) | 0.65, d (4.3) | 0.75, d (6.8) |
| H-4′a | 3.72, dd (14.9, 3.7) | 3.69, dd (15.0, 5.3) | 3.69, dd (17.8, 4.0) | 3.72, dd (14.8, 3.7) |
| H-4′b | 3.80, dd (14.9, 6.3) | 3.77, dd (15.0, 2.9) | 3.77, dd (15.0, 2.8) | 3.80, dd (14.8, 6.4) |
| H-6′ | 6.89, d (2.4) | 6.60, d (2.3) | 6.60, d (2.4) | 6.90, d (2.3) |
| H-7′ | 8.06, br | 8.03, br | 8.03, br | 8.03, br |
| H-9′ | 7.28, d (8.4) | 7.28, d (8.2) | 7.28, d (8.2) | 7.28, d (7.8) |
| H-10′ | 7.10, ddd (8.6, 7.6, 1.0) | 7.13, t (7.1) | 7.13, t (7.1) | 7.10, ddd (8.5, 7.5 0.9) |
| H-11′ | 6.93, ddd (8.0, 7.1, 1.0) | 6.93, t (7.5) | 6.93, t (7.5) | 6.94, ddd (8.2, 7.2, 1.0) |
| H-12′ | 7.49, d (7.9) | 7.44, d (8.0) | 7.44, d (8.1) | 7.49, d (7.9) |
| 5a | 5b | 5c | 5d | |
| H-1 | 4.32, dt (10.7, 3.2) | 2.73, dd (9.7, 3.4) | 2.72, d (9.7) | 4.31, dt (11.0, 3.2) |
| H-2 | 6.45, d (2.5) | 5.75, br | 5.75, br | 6.21, d (2.5) |
| H-4 | 5.54, dd (5.2, 3.3) | 5.68, dd (5.2, 3.0) | 5.68, dd (5.0, 2.9) | 5.55, dd (5.2, 3.3) |
| H-8 | 8. 39, dd (8.0, 1.2) | 8. 37, d (8.0) | 8. 37, d (8.0) | 8. 39, dd (8.0, 1.1) |
| H-9 | 7.41, d (8.0) | 7.55, d (8.1) | 7.55, d (8.0) | 7.42, d (8.0) |
| H-10 | 7.78, ddd (8.5, 7.2, 1.5) | 7.78, ddd (8.5, 7.1, 1.5) | 7.78, ddd (8.5, 7.1, 1.5) | 7.79, ddd (8.5, 7.1, 1.6) |
| H-11 | 7.57, d (7.8) | 7.60, d (7.8) | 7.60, d (7.9) | 7.59, d (7.8) |
| H-1′ | 2.84–2.44, m | 1.43–1.33, m | 1.44–1.36, m | 2.89–2.56, m |
| H-2′ | 2.08–2.00, m | 2.01, ddd (12.4, 12.5, 4.2) | 2.01, dd (13.0, 9.4) | 2.07–2.00, m |
| H-3′ | 0.75, d (6.0) | 0.77, d (6.4) | 0.77, d (6.3) | 0.74, d (6.0) |
| H-3″ | 0.49, d (6.1) | 0.28, d (6.5) | 0.28, d (6.4) | 0.49, d (6.0) |
| H-4′a | 3.75, dd (15.0, 3.3) | 3.65, dd (15.0, 5.3) | 3.65, dd (14.9, 5.2) | 3.75, dd (15.1, 3.4) |
| H-4′b | 3.83, dd (15.0, 5.3) | 3.77, dd (15.0, 2.6) | 3.78, dd (15.1, 2.8) | 3.84, dd (15.0, 5.3) |
| H-6′ | 6.68, d (2.3) | 6.64, d (2.3) | 6.64, d (2.0) | 6.68, d (2.4) |
| H-7′ | 8.08, br | 8.09, br | 8.07, br | 8.03, br |
| H-9′ | 7.28, d (8.1) | 7.29, d (8.2) | 7.29, d (8.2) | 7.29, d (8.3) |
| H-10′ | 7.12, ddd (8.5, 7.1, 1.1) | 7.13, t (7.6) | 7.13, t (8.0) | 7.12, ddd (8.1, 7.0, 1.1) |
| H-11′ | 6.97, ddd (8.5, 7.0, 1.1) | 6.98, t (7.5) | 6.98, t (7.5) | 6.93, ddd (8.0, 7.1, 1.0) |
| H-12′ | 7.53, ddd (8.1, 7.1, 1.0) | 7.50, d (8.0) | 7.50, d (8.0) | 7.53, ddd (8.2, 7.2, 1.1) |
| Position | δC, Type | |||||||
|---|---|---|---|---|---|---|---|---|
| 4a | 4b | 4c | 4d | 5a | 5b | 5c | 5d | |
| C-1 | 61.9, CH | 58.1 | 58.1 | 61.8 | 54.1, CH | 50.8 | 50.8 | 54.1 |
| C-3 | 167.8, CO | 169.5 | 169.5 | 167.8 | 167.6, CO | 169.4 | 169.4 | 167.5 |
| C-4 | 57.5, CH | 56.8 | 57.3 | 57.5 | 56.7, CH | 57.3 | 57.3 | 56.7 |
| C-6 | 161.4, CO | 160.9 | 160.9 | 161.0 | 161.0, CO | 160.8 | 160.8 | 161.1 |
| C-7 | 120.2, C | 120.2 | 120.2 | 120.2 | 120.4, C | 120.3 | 120.3 | 120.0 |
| C-8 | 126.8, CH | 126.9 | 126.9 | 126.7 | 126.9, CH | 126.9 | 126.9 | 126.8 |
| C-9 | 127.1, CH | 127.2 | 127.2 | 127.1 | 126.9, CH | 127.4 | 127.4 | 126.8 |
| C-10 | 134.7, CH | 134.7 | 134.7 | 134.7 | 134.8, CH | 134.6 | 134.6 | 134.7 |
| C-11 | 127.1, CH | 127.0 | 127.0 | 127.1 | 126.7, CH | 127.1 | 127.1 | 126.7 |
| C-12 | 146.8, C | 147.1 | 147.1 | 146.1 | 147.2, C | 147.0 | 147.0 | 147.2 |
| C-14 | 149.3, C | 150.3 | 150.3 | 149.8 | 151.3, C | 151.5 | 151.5 | 151.3 |
| C-1′ | 34.5, CH | 29.4 | 29.5 | 34.8 | 45.8, CH2 | 40.2 | 40.2 | 45.8 |
| C-2′ | 18.0, CH3 | 14.8 | 14.8 | 18.1 | 23.9, CH | 24.1 | 24.1 | 23.9 |
| C-3″ | - | - | - | - | 20.4, CH3 | 19.7 | 19.7 | 20.4 |
| C-3′ | 19.6, CH3 | 18.8 | 18.8 | 19.8 | 22.9, CH3 | 23.3 | 23.3 | 22.9 |
| C-4′ | 27.4, CH2 | 27.4 | 27.4 | 27.4 | 26.4, CH2 | 27.1 | 27.1 | 26.4 |
| C-5′ | 110.2, C | 109.3 | 109.3 | 110.2 | 107.9, C | 109.7 | 109.7 | 107.9 |
| C-6′ | 123.5, CH | 123.6 | 123.6 | 123.6 | 123.6, CH | 123.6 | 123.6 | 123.6 |
| C-8′ | 136.0, C | 136.0 | 136.0 | 135.9 | 137.1, C | 136.1 | 136.1 | 137.1 |
| C-9′ | 110.9, CH | 111.1 | 111 | 111 | 110.2, CH | 111.1 | 111.1 | 110.2 |
| C-10′ | 122.3, CH | 122.6 | 122.6 | 122.4 | 122.2, CH | 122.8 | 122.8 | 122.2 |
| C-11′ | 119.9, CH | 120.0 | 120.0 | 119.9 | 119.9, CH | 120.2 | 120.2 | 119.9 |
| C-12′ | 118.7, CH | 118.7 | 118.7 | 118.7 | 119.0, CH | 118.8 | 118.8 | 119.1 |
| C-13′ | 127.8, C | 127.2 | 127.2 | 127.8 | 126.8, C | 127.1 | 127.1 | 126.8 |
| Compound | GI50 (µM) | |
|---|---|---|
| NCI-H460 | HCT-15 | |
| 4a | 81.33 ± 1.55 | 40.33 ± 3.12 |
| 4b | 70.20 ± 3.15 | 38.15 ± 0.29 |
| 4c | 57.62 ± 2.08 | 31.78 ± 1.21 |
| 4d | 60.10 ± 2.61 | 33.30 ± 1.37 |
| 5a | 32.52 ± 4.24 | 48.18 ± 2.51 |
| 5b | 41.52 ± 2.52 | 51.94 ± 4.26 |
| 5c | 31.19 ± 3.01 | 43.63 ± 0.25 |
| 5d | 36.47 ± 3.98 | 47.00 ± 1.47 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Long, S.; Resende, D.I.S.P.; Kijjoa, A.; Silva, A.M.S.; Pina, A.; Fernández-Marcelo, T.; Vasconcelos, M.H.; Sousa, E.; Pinto, M.M.M. Antitumor Activity of Quinazolinone Alkaloids Inspired by Marine Natural Products. Mar. Drugs 2018, 16, 261. https://doi.org/10.3390/md16080261
Long S, Resende DISP, Kijjoa A, Silva AMS, Pina A, Fernández-Marcelo T, Vasconcelos MH, Sousa E, Pinto MMM. Antitumor Activity of Quinazolinone Alkaloids Inspired by Marine Natural Products. Marine Drugs. 2018; 16(8):261. https://doi.org/10.3390/md16080261
Chicago/Turabian StyleLong, Solida, Diana I. S. P. Resende, Anake Kijjoa, Artur M. S. Silva, André Pina, Tamara Fernández-Marcelo, M. Helena Vasconcelos, Emília Sousa, and Madalena M. M. Pinto. 2018. "Antitumor Activity of Quinazolinone Alkaloids Inspired by Marine Natural Products" Marine Drugs 16, no. 8: 261. https://doi.org/10.3390/md16080261