
Penicibrocazines A–E, Five New Sulfide Diketopiperazines from the Marine-Derived Endophytic Fungus Penicillium brocae


Abstract
:1. Introduction

2. Results and Discussion
2.1. Structure Elucidation of the New Compounds

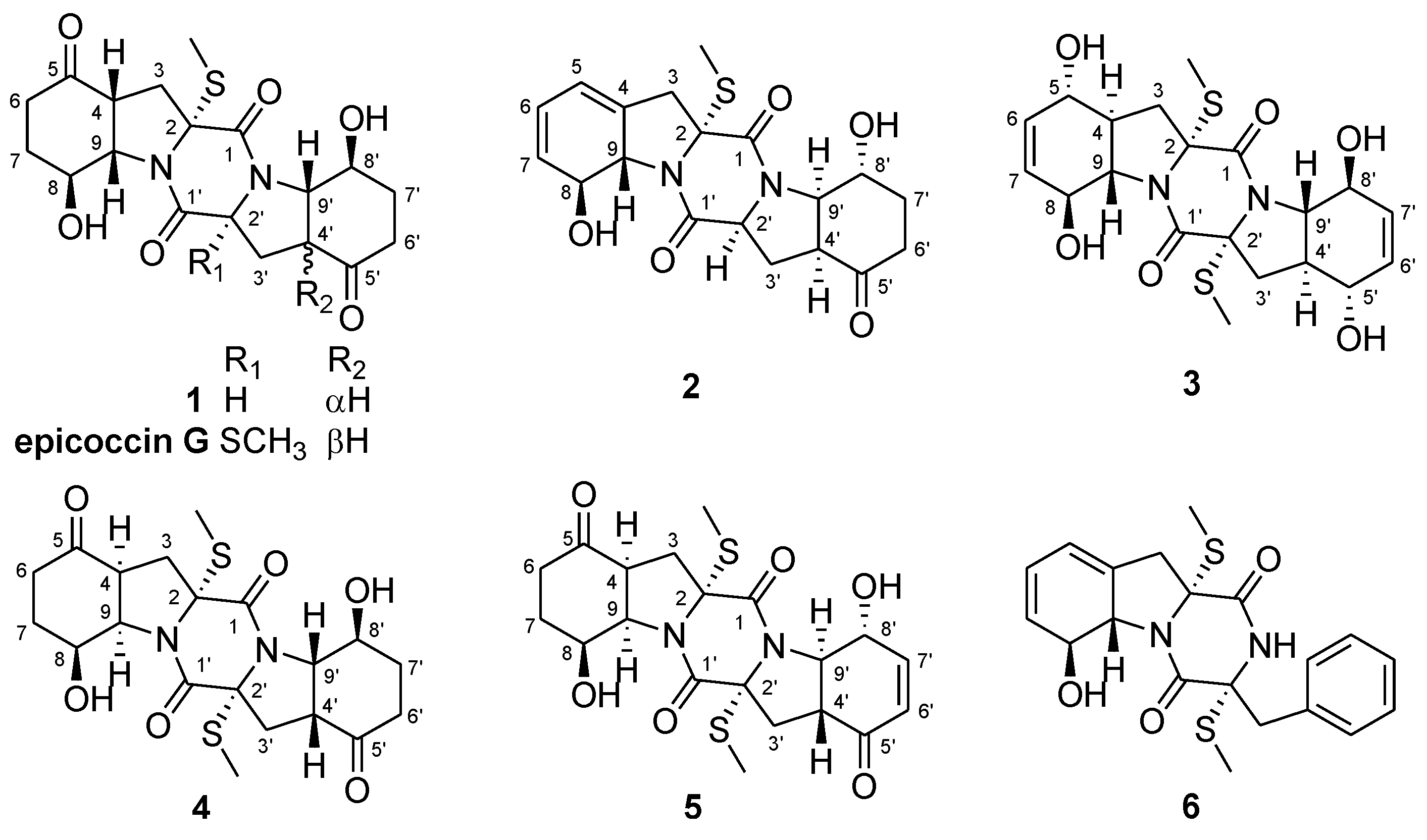{kind=link}
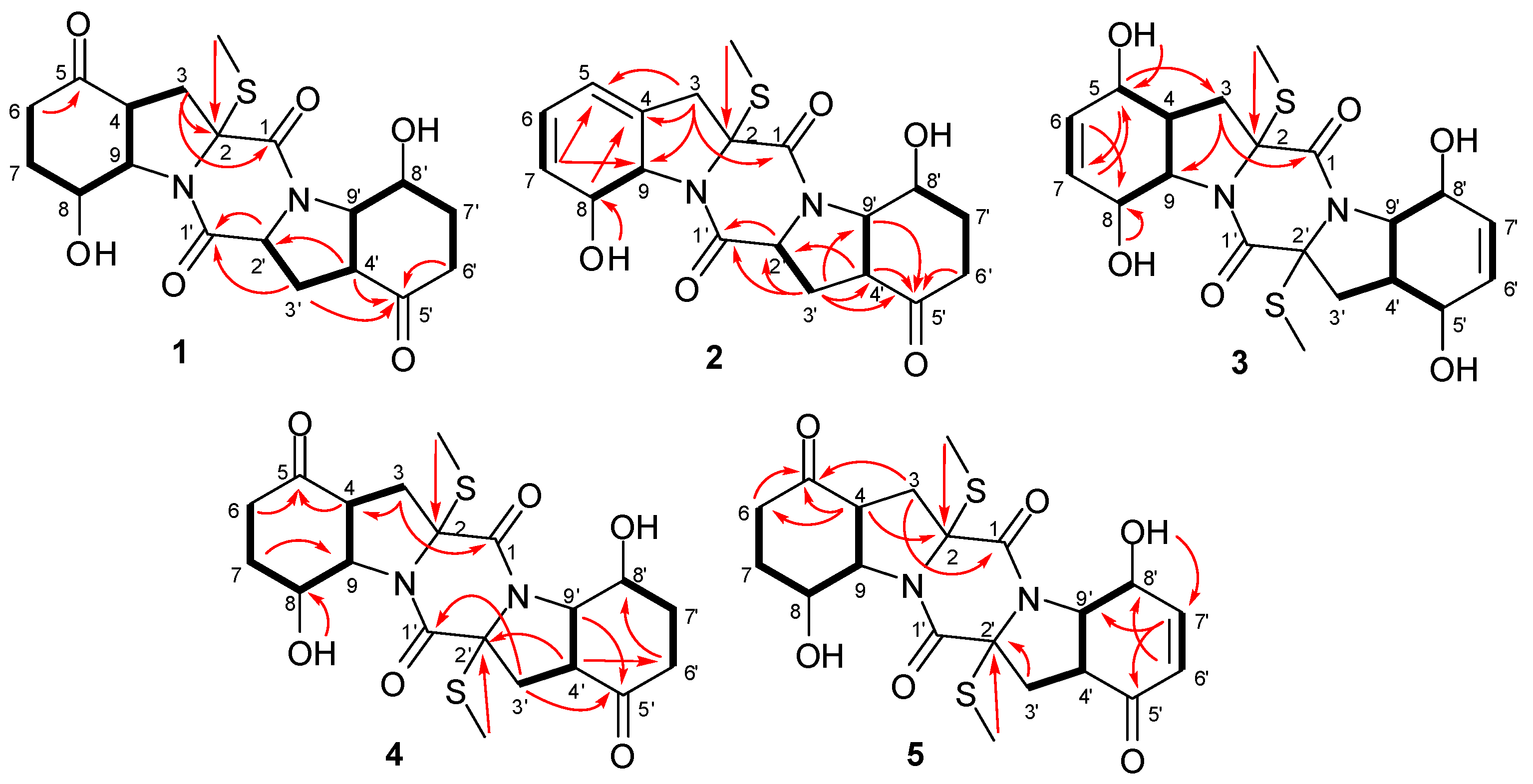{kind=link}
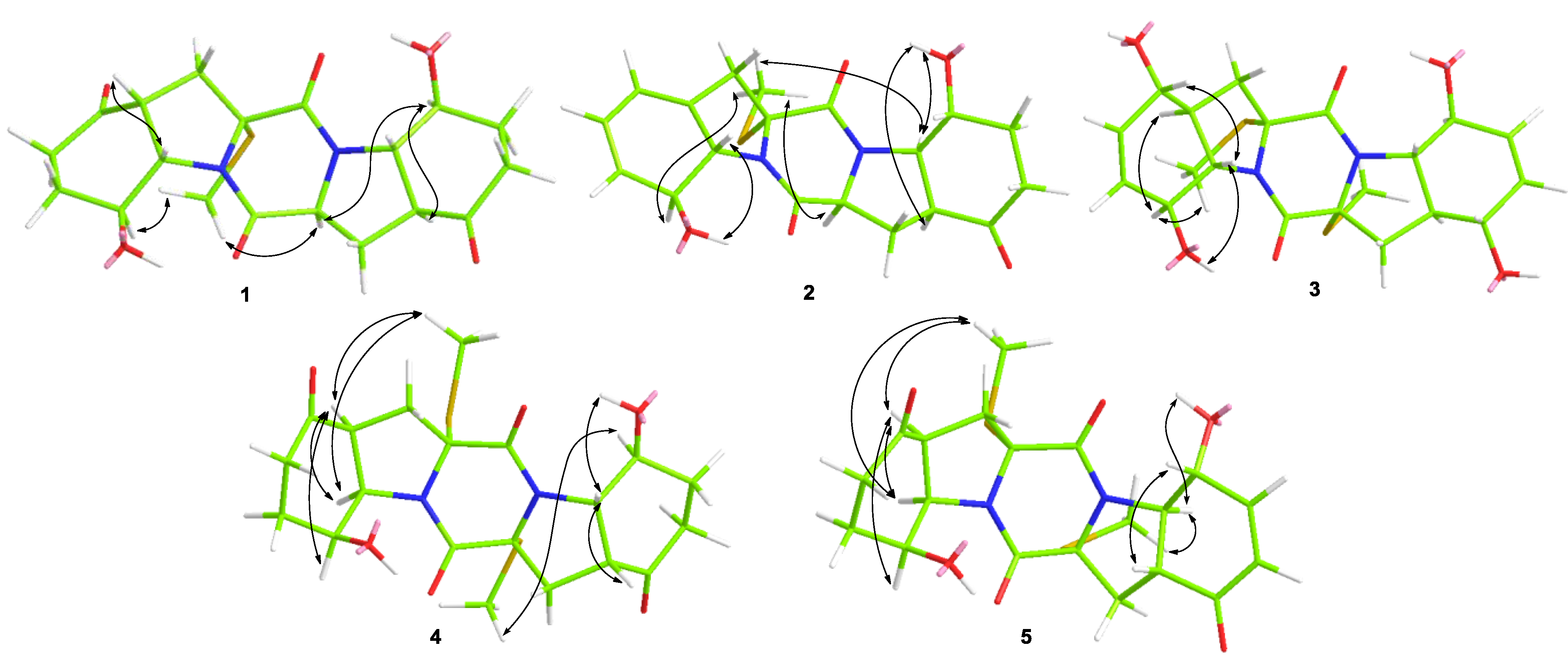{kind=link}
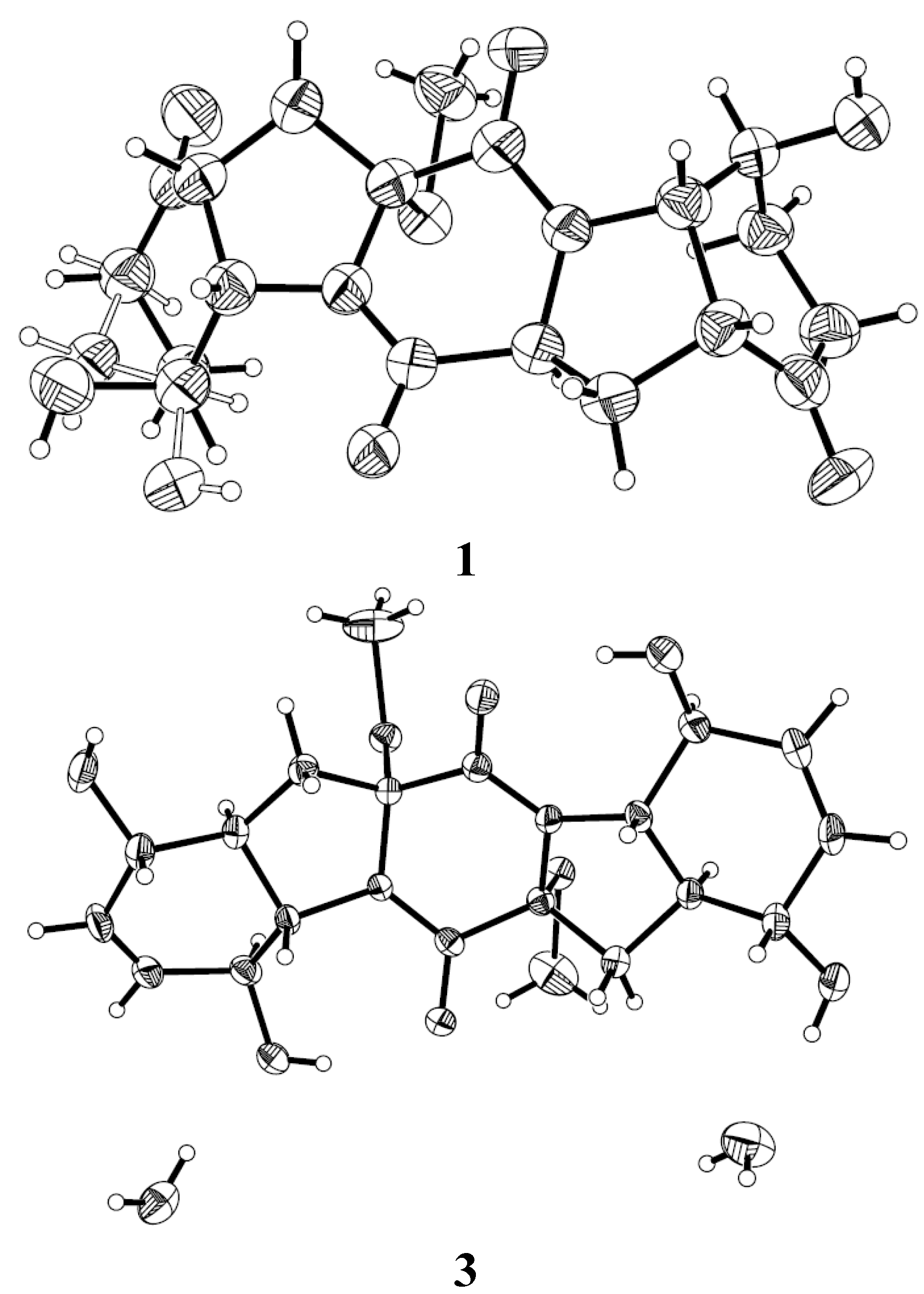{kind=link}
| Position | 1 a | 2 b | 3 b | 4 b | 5 c |
|---|---|---|---|---|---|
| 3α | 2.38, m (overlap) | 2.89, d (16.0) | 2.48, m (overlap) | 2.28, m (overlap) | 2.63, m (overlap) |
| 3β | 2.32, m (overlap) | 3.08, d (16.0) | 2.08, t (12.7) | 2.22, m (overlap) | 3.07, d (13.8) |
| 4 | 3.30, dd (8.7, 8.0) | 2.30, m | 2.96, t (8.0) | 3.14, t (8.6) | |
| 5 | 5.93, d (2.3) | 4.08, d (8.8) | |||
| 6 | 2.42, m (overlap) | 5.89, dd (9.6, 2.3) | 5.55,d (10.0) | α 2.77, dd (13.3, 7.3) β 2.27, m (overlap) | 2.33, m (overlap) |
| 7 | α 2.40, m (overlap) β 1.84, td (12.2, 6.2) | 5.59, d (9.6) | 5.69, d (10.0) | 1.90, m | 2.00, m |
| 8 | 4.15, dd (13.1, 6.2) | 4.53, d (13.4) | 4.16, d (8.2) | 4.33, brs | 4.27, brs |
| 9 | 3.57, dd (13.1, 8.0) | 4.69, d (13.4) | 3.43, dd (8.2, 11.9) | 4.29, d (8.0) | 4.51, br m |
| 2' | 4.57, dd (10.4, 7.1) | 4.45, dd (10.3, 7.0) | |||
| 3'α | 2.02, m (overlap) | 2.56, m (overlap) | 2.48, m (overlap) | 2.21, m (overlap) | 2.33, m (overlap) |
| 3'β | 2.92, dd (13.1, 7.1) | 2.04, ddd (12.4, 10.3, 8.3) | 2.08, t (12.7) | 2.28, m (overlap) | 2.53, dd (12.9, 4.5) |
| 4' | 3.24, dd (12.8, 6.7) | 3.12, m (overlap) | 2.30, m | 3.40, m | 3.47, td (13.0, 4.5) |
| 5' | 4.08, d (8.8) | ||||
| 6' | α 2.36, m (overlap) β 2.56, ddd (17.5, 6.3, 1.8) | α 2.60, m (overlap) β 2.28, dt (16.4, 5.6) | 5.55, d (10.0) | α 2.59, ddd (18.2, 12.4, 5.2) β 2.27, m (overlap) | 6.01, d (10.0) |
| 7' | α 2.12, m β 2.04, m (overlap) | α 1.74, m β 1.95, dt (12.5, 5.6) | 5.69, d (10.0) | α 1.58, m β 2.27, m (overlap) | 6.86, d (10.0) |
| 8' | 3.66, m | 4.36, m (overlap) | 4.16, d (8.2) | 4.00, dd (13.2, 7.4) | 4.60, d (8.2) |
| 9' | 4.45, dd (12.8, 8.2) | 4.36, m (overlap) | 3.43, dd (8.2, 11.9) | 3.65,dd (13.2, 8.8) | 3.86, dd (13.0, 8.2) |
| 2-SMe | 2.09, s | 2.10, s | 2.15, s | 1.93, s | 2.09, s |
| 2'-SMe | 2.15, s | 2.07, s | 2.22, s | ||
| 5-OH | 5.31, brs | ||||
| 5'-OH | 5.31, brs | ||||
| 8-OH | 5.68, s | 5.87, s | 5.36, brs | 4.83, brs | |
| 8'-OH | 5.41, brs | 5.87, s | 5.89, s | 6.21, s |

| Position | 1 a | 2 b | 3 b | 4 b | 5 c |
|---|---|---|---|---|---|
| 1 | 165.9, C | 163.7, C | 168.5, C | 169.0, C | 166.9, C |
| 2 | 74.0, C | 74.7, C | 72.4, C | 71.1, C | 72.4, C |
| 3 | 31.6, CH2 | 38.4, CH2 | 35.0, CH2 | 31.2, CH2 | 35.5, CH2 |
| 4 | 48.1, CH | 133.8, CH | 43.3, CH | 43.8, CH | 45.5, CH |
| 5 | 206.5, C | 118.7, CH | 68.8, CH | 207.4, C | 207.4, C |
| 6 | 37.1, CH2 | 123.3, CH | 133.3, CH | 34.2, CH2 | 34.8, CH2 |
| 7 | 30.8, CH2 | 130.0, CH | 129.9, CH | 25.8, CH2 | 27.4, CH2 |
| 8 | 71.0, CH | 73.8, CH | 71.3, CH | 64.7, CH | 70.2, CH |
| 9 | 69.9, CH | 68.0, CH | 67.8, CH | 63.3, CH | 67.2, CH |
| 1' | 169.6, C | 169.0, C | 168.5, C | 165.2, C | 170.4, C |
| 2' | 59.6, CH | 58.8, CH | 72.4, C | 72.7, C | 73.9, C |
| 3' | 27.1, CH2 | 29.2, CH2 | 35.0, CH2 | 31.0, CH2 | 32.5, CH2 |
| 4' | 46.9, CH | 46.0, CH | 43.3, CH | 47.0, CH | 47.4, CH |
| 5' | 207.9, C | 209.6, C | 68.8, CH | 206.8, C | 196.4, C |
| 6' | 36.2, CH2 | 34.1, CH2 | 133.3, CH | 36.7, CH2 | 129.1, CH |
| 7' | 26.7, CH2 | 26.6, CH2 | 129.9, CH | 33.7, CH2 | 152.6, CH |
| 8' | 72.2, CH | 64.0, CH | 71.3, CH | 71.1, CH | 73.8, CH |
| 9' | 68.1, CH | 65.0, CH | 67.8, CH | 69.0, CH | 69.8, CH |
| 2-SMe | 14.5, CH3 | 13.2, CH3 | 14.3, CH3 | 14.1, CH3 | 14.9, CH3 |
| 2'-SMe | 14.3, CH3 | 14.3, CH3 | 14.9, CH3 |


2.2. Biological Activities of the Isolated Compounds
3. Experimental Section
3.1. General
3.2. Fungal Material
3.3. Fermentation
3.4. Extraction and Isolation
3.5. X-Ray Crystallographic Analysis of Compounds 1 and 3 [15]
3.6. Cytotoxicity Assay
3.7. Antimicrobial Assay
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Deffieux, G.; Baute, M.A.; Baute, R.; Filleau, M.J. New antibiotics from the fungus Epicoccum nigrum II. Epicorazine A: Structure elucidation and absolute configuration. J. Antibiot. 1978, 31, 1102–1105. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.J.; Sun, B.D.; Gao, H.; Chen, X.L.; Liu, S.C.; Yao, X.S.; Liu, X.Z.; Che, Y.S. Diketopiperazines from the cordyceps-colonizing fungus Epicoccum nigrum. J. Nat. Prod. 2009, 72, 2115–2119. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.X.; Jensen, P.R.; Williams, P.G.; Fenical, W. Isolation and structure assignments of rostratins A-D, cytotoxic disulfides produced by the marine-derived fungus Exserohilum rostratum. J. Nat. Prod. 2004, 67, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.H.; Li, X.M.; Lv, C.T.; Li, C.S.; Xu, G.M.; Huang, C.G.; Wang, B.G. Sulfur-containing cytotoxic curvularin macrolides from Penicillium sumatrense MA-92, a fungus obtained from the rhizosphere of the mangrove Lumnitzera racemosa. J. Nat. Prod. 201 2013, 76, 2145–2149. [Google Scholar] [CrossRef]
- An, C.Y.; Li, X.M.; Luo, H.; Li, C.S.; Wang, M.H.; Xu, G.M.; Wang, B.G. 4-Phenyl-3,4-dihydroquinolone derivatives from Aspergillus nidulans MA-143, an endophytic fungus isolated from the mangrove plant Rhizophora stylosa. J. Nat. Prod. 201 2013, 76, 1896–1901. [Google Scholar] [CrossRef]
- Wang, M.H.; Li, X.M.; Li, C.S.; Ji, N.Y.; Wang, B.G. Secondary metabolites from Penicillium pinophilum SD-272, a marine sediment-derived fungus. Mar. Drugs 2013, 11, 2230–2238. [Google Scholar] [CrossRef] [PubMed]
- Li, C.S.; Li, X.M.; Gao, S.S.; Lu, Y.H.; Wang, B.G. Cytotoxic anthranilic acid derivatives from deep sea sediment-derived fungus Penicillium paneum SD-44. Mar. Drugs 2013, 11, 3068–3076. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Li, X.M.; Li, C.S.; Wang, B.G. Nigerasterols A and B, antiproliferative sterols from the mangrove-derived endophytic fungus Aspergillus niger MA-132. Helv. Chim. Acta 2013, 96, 1055–1061. [Google Scholar] [CrossRef]
- Du, F.Y.; Li, X.M.; Zhang, P.; Li, C.S.; Wang, B.G. Cyclodepsipeptides and O-containing heterocyclic metabolites from Beauveria felina EN-135, a marine-derived entomopathogenic fungus. Mar. Drugs 2014, 1, 2816–2826. [Google Scholar] [CrossRef]
- Meng, L.H.; Li, X.M.; Lv, C.T.; Huang, C.G.; Wang, B.G. Brocazines A–F, cytotoxic bisthiodiketopiperazine derivatives from Penicillium brocae MA-231, an endophytic fungus derived from the marine mangrove plant Avicennia marina. J. Nat. Prod. 201 2014, 77, 1921–1927. [Google Scholar] [CrossRef]
- Wang, J.M.; Ding, G.Z.; Fang, L.; Dai, J.G.; Yu, S.S.; Wang, Y.H.; Chen, X.G.; Ma, S.G.; Qu, J.; Xu, S.; et al. Thiodiketopiperazines produced by the endophytic fungus Epicoccum nigrum. J. Nat. Prod. 2010, 73, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Jiang, N.; Ma, J.; Yu, S.S.; Tan, R.X.; Dai, J.G.; Si, Y.K.; Ding, G.Z.; Ma, S.G.; Qu, J.; et al. Study on absolute configurations of α/α' chiral carbons of thiodiketopiperazines by experimental and calculated circular dichroism spectra. Tetrahedron 2013, 69, 1195–1201. [Google Scholar] [CrossRef]
- Kong, F.D.; Wang, Y.; Liu, P.P.; Dong, T.H.; Zhu, W.M. Thiodiketopiperazines from the marine-derived fungus Phoma sp. OUCMDZ-1847. J. Nat. Prod. 2014, 77, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, X.M.; Teuscher, F.; Li, D.L.; Diesel, A.; Ebel, R.; Proksch, P.; Wang, B.G. Chaetopyranin, a benzaldehyde derivative, and other related metabolites from Chaetomium globosum, an endophytic fungus derived from the marine red alga Polysiphonia urceolata. J. Nat. Prod. 2006, 69, 1622–1625. [Google Scholar] [CrossRef] [PubMed]
- Crystallographic Data of Compounds 1 and 3 have been Deposited in the Cambridge Crystallographic Data Centre as CCDC 1031878 for Compound 1 and CCDC 1031879 For Compound 3. Available online: http://www.ccdc.cam.ac.uk/data_request/cif (accessed on 31 October 2014).
- Sheldrick, G.M. SADABS, Software for Empirical Absorption Correction; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXTL, Structure Determination Software Programs; Bruker Analytical X-ray System Inc.: Madison, WI, USA, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXL-97 and SHELXS-97, Program for X-ray Crystal Structure Solution and Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Bergeron, R.J.; Cavanaugh, P.F., Jr.; Kline, S.J.; Hughes, R.G., Jr.; Elliott, G.T.; Porter, C.W. Antineoplastic and antiherpetic activity of spermidine catecholamide iron chelators. Biochem. Biophys. Res. Commun. 1984, 121, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Al-Burtamani, S.K.S.; Fatope, M.O.; Marwah, R.G.; Onifade, A.K.; Al-Saidi, S.H. Chemical composition, antibacterial and antifungal activities of the essential oil of Haplophyllum tuberculatum from Oman. J. Ethnopharmacol. 2005, 96, 107–112. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, L.-H.; Zhang, P.; Li, X.-M.; Wang, B.-G. Penicibrocazines A–E, Five New Sulfide Diketopiperazines from the Marine-Derived Endophytic Fungus Penicillium brocae. Mar. Drugs 2015, 13, 276-287. https://doi.org/10.3390/md13010276
Meng L-H, Zhang P, Li X-M, Wang B-G. Penicibrocazines A–E, Five New Sulfide Diketopiperazines from the Marine-Derived Endophytic Fungus Penicillium brocae. Marine Drugs. 2015; 13(1):276-287. https://doi.org/10.3390/md13010276
Chicago/Turabian StyleMeng, Ling-Hong, Peng Zhang, Xiao-Ming Li, and Bin-Gui Wang. 2015. "Penicibrocazines A–E, Five New Sulfide Diketopiperazines from the Marine-Derived Endophytic Fungus Penicillium brocae" Marine Drugs 13, no. 1: 276-287. https://doi.org/10.3390/md13010276