The Corrosion Behavior of Pure Iron under Solid Na2SO4 Deposit in Wet Oxygen Flow at 500 °C

Abstract
:
1. Introduction
2. Results
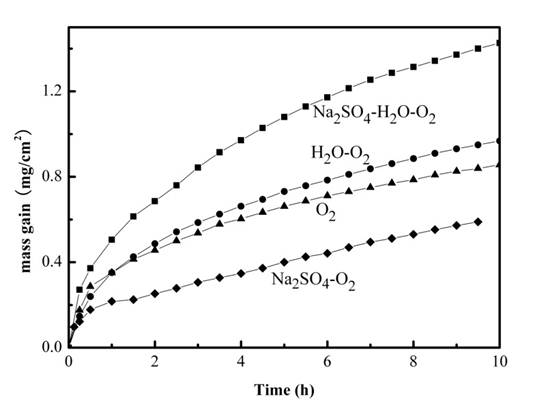
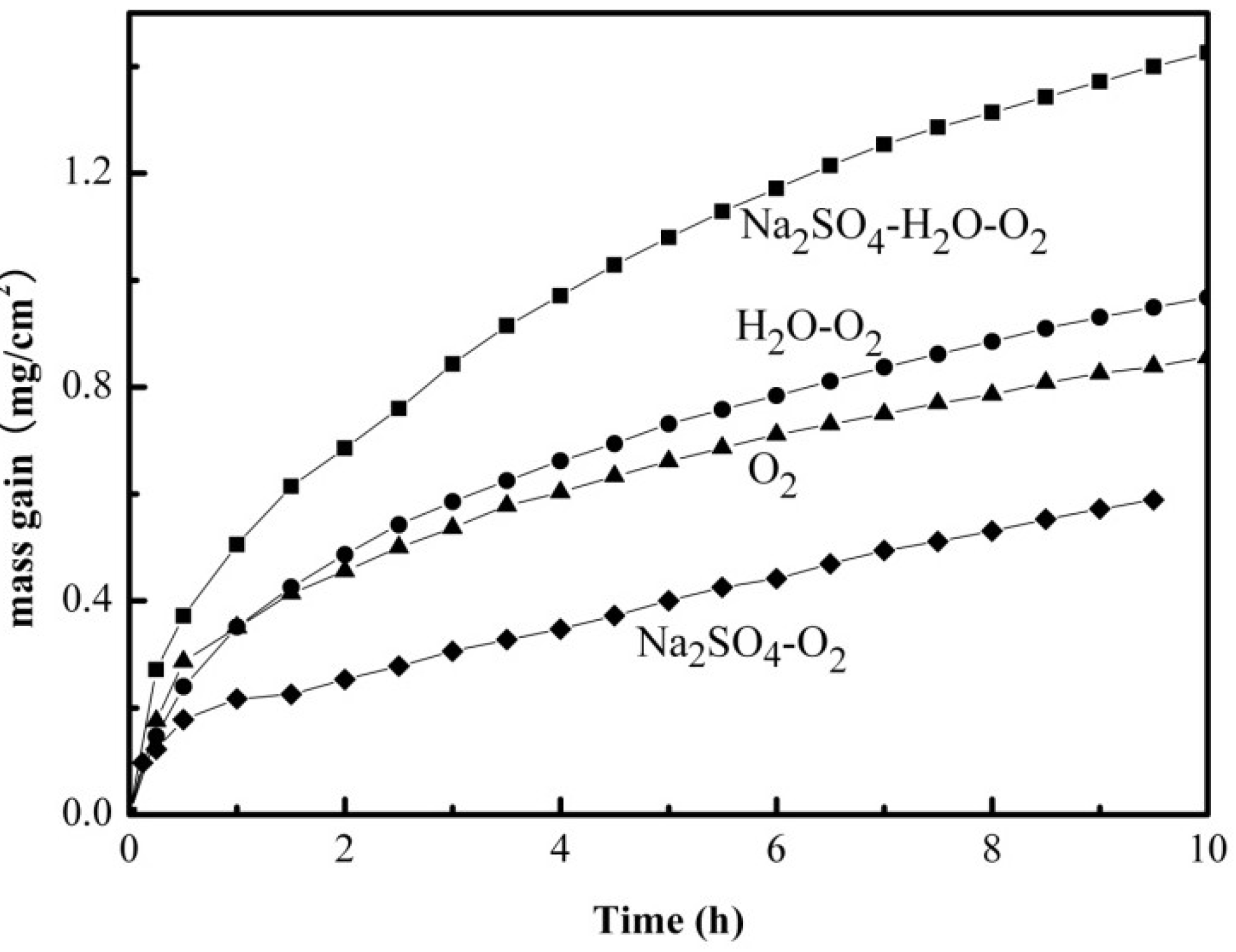
2.1. Mass Gain Measurements

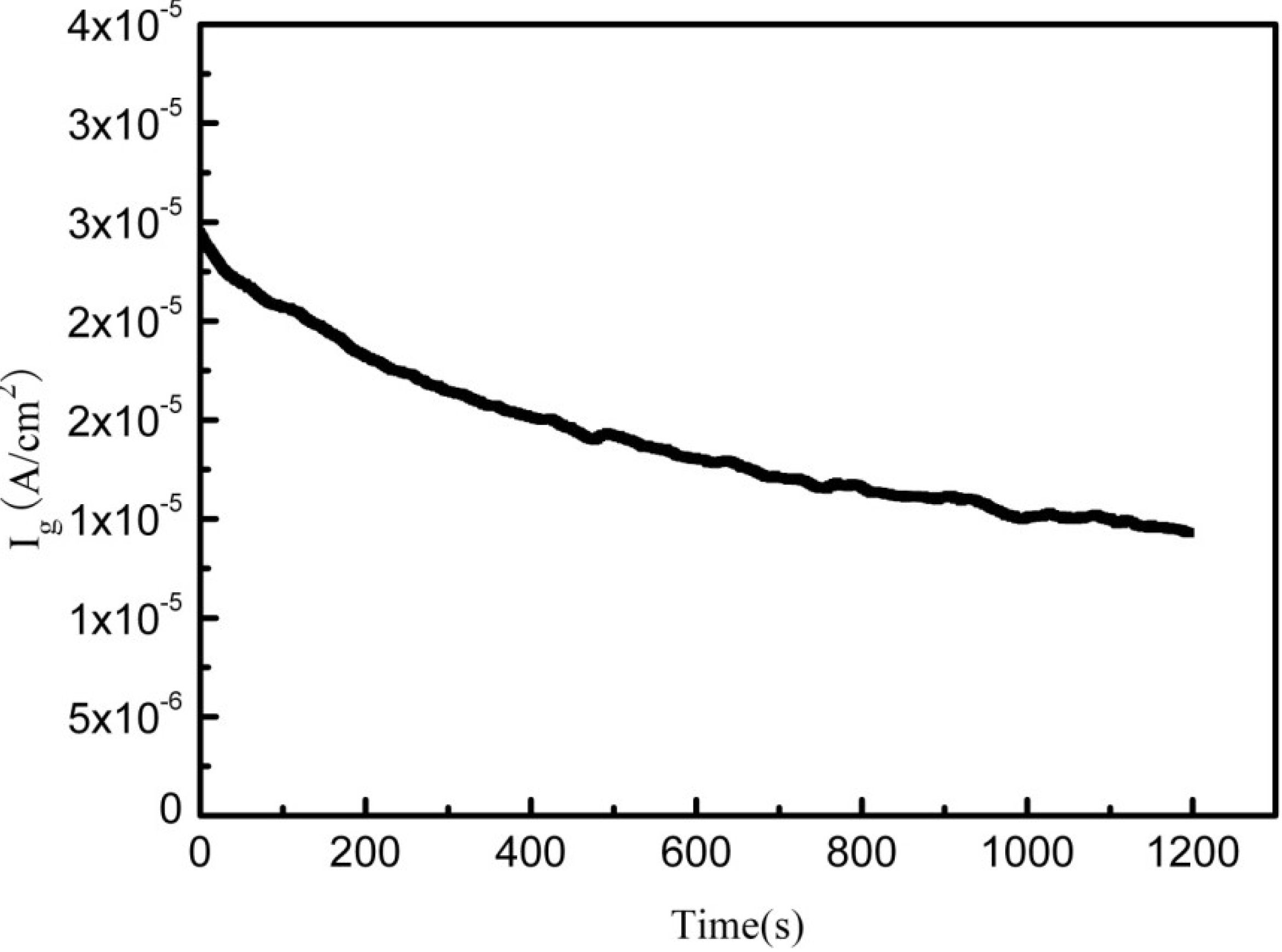
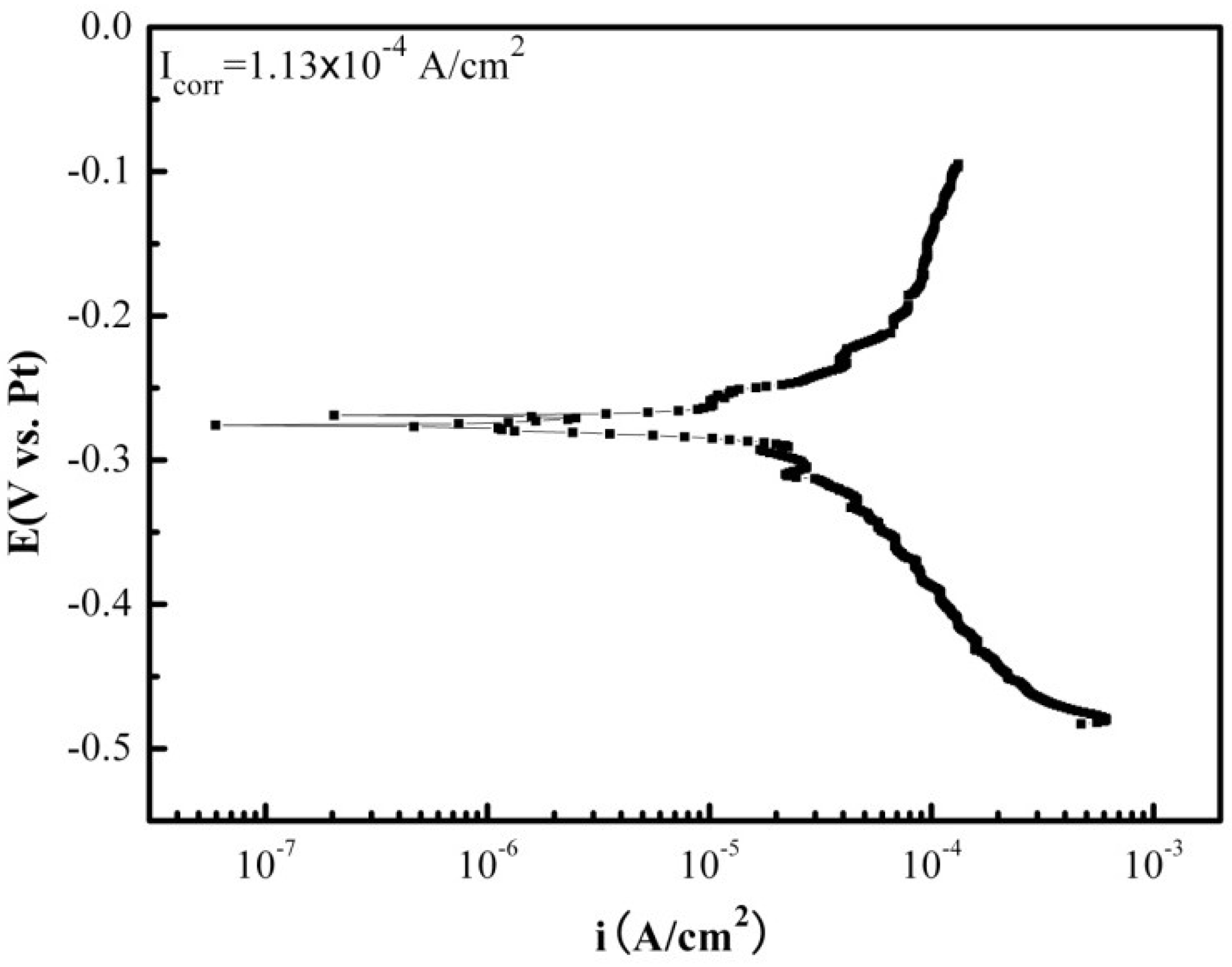
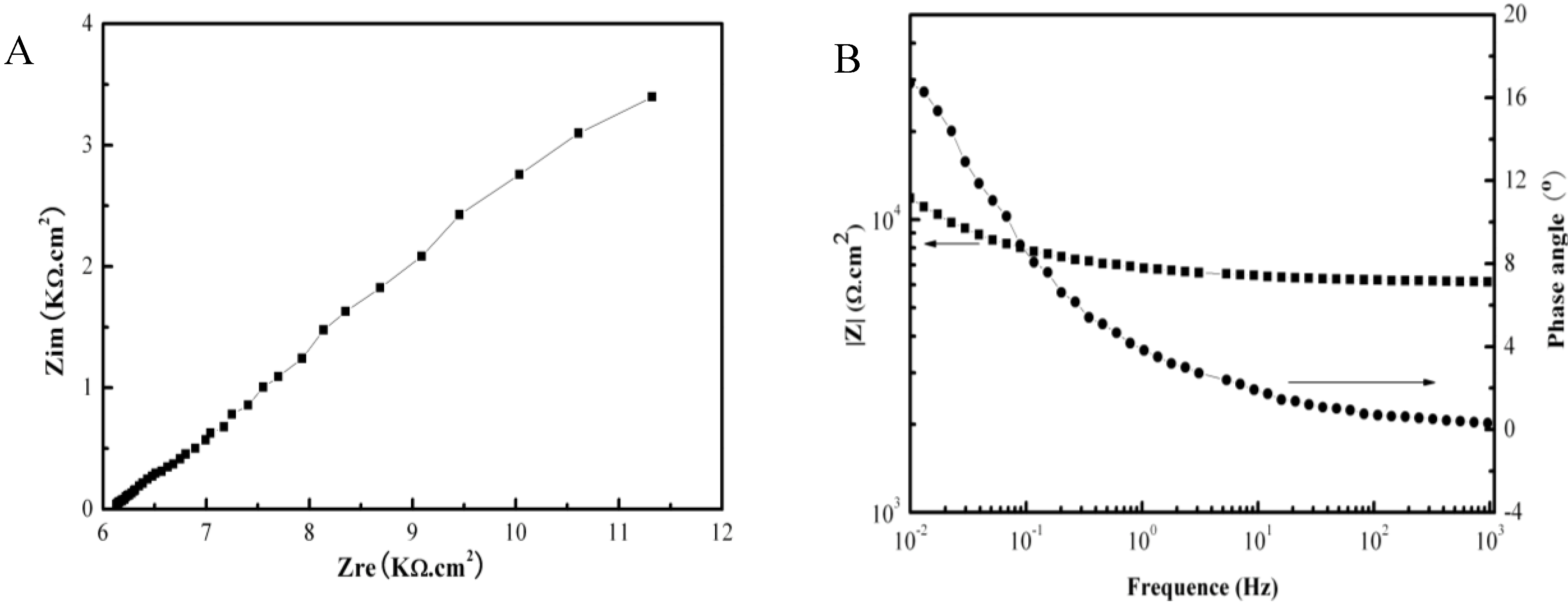
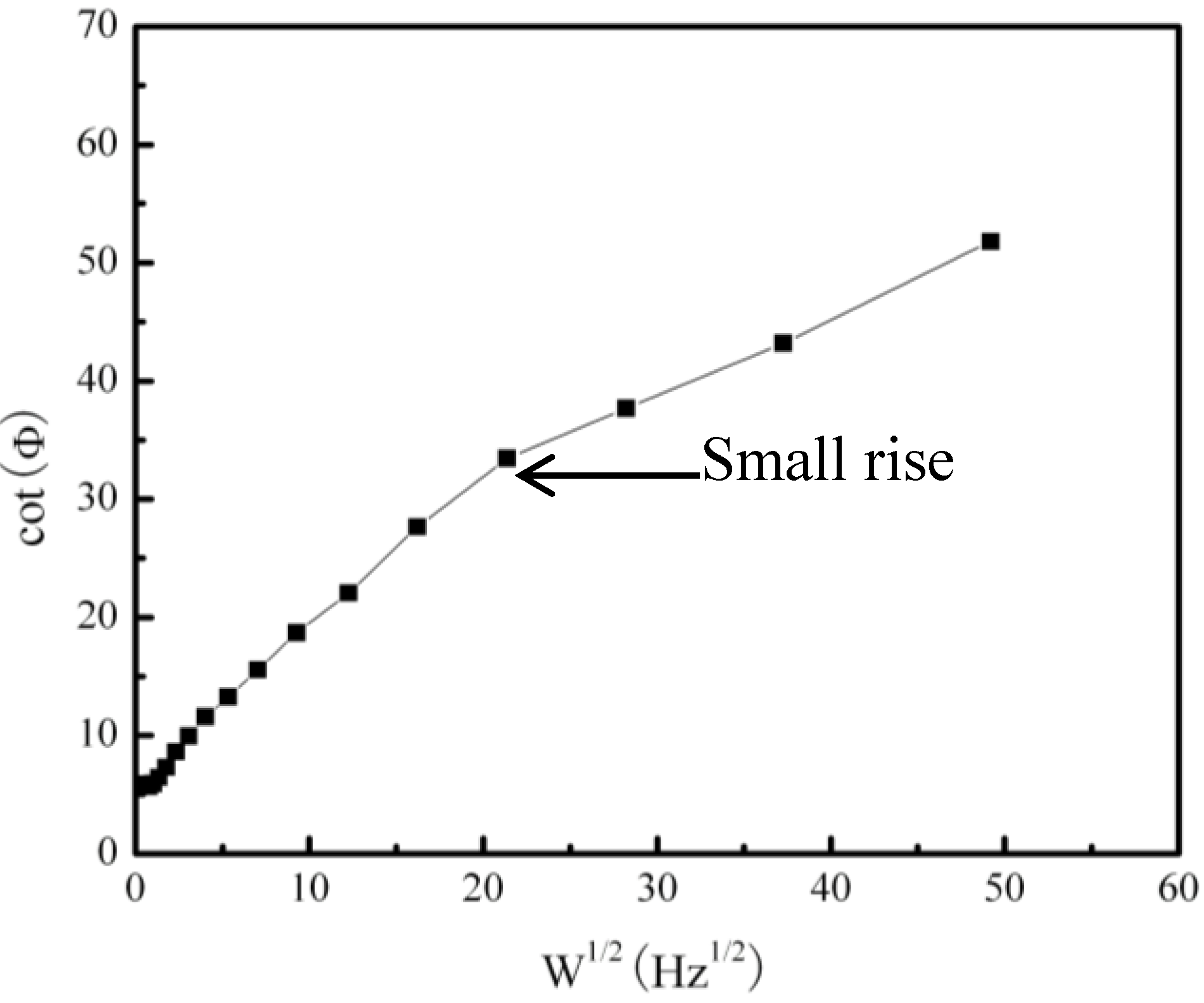
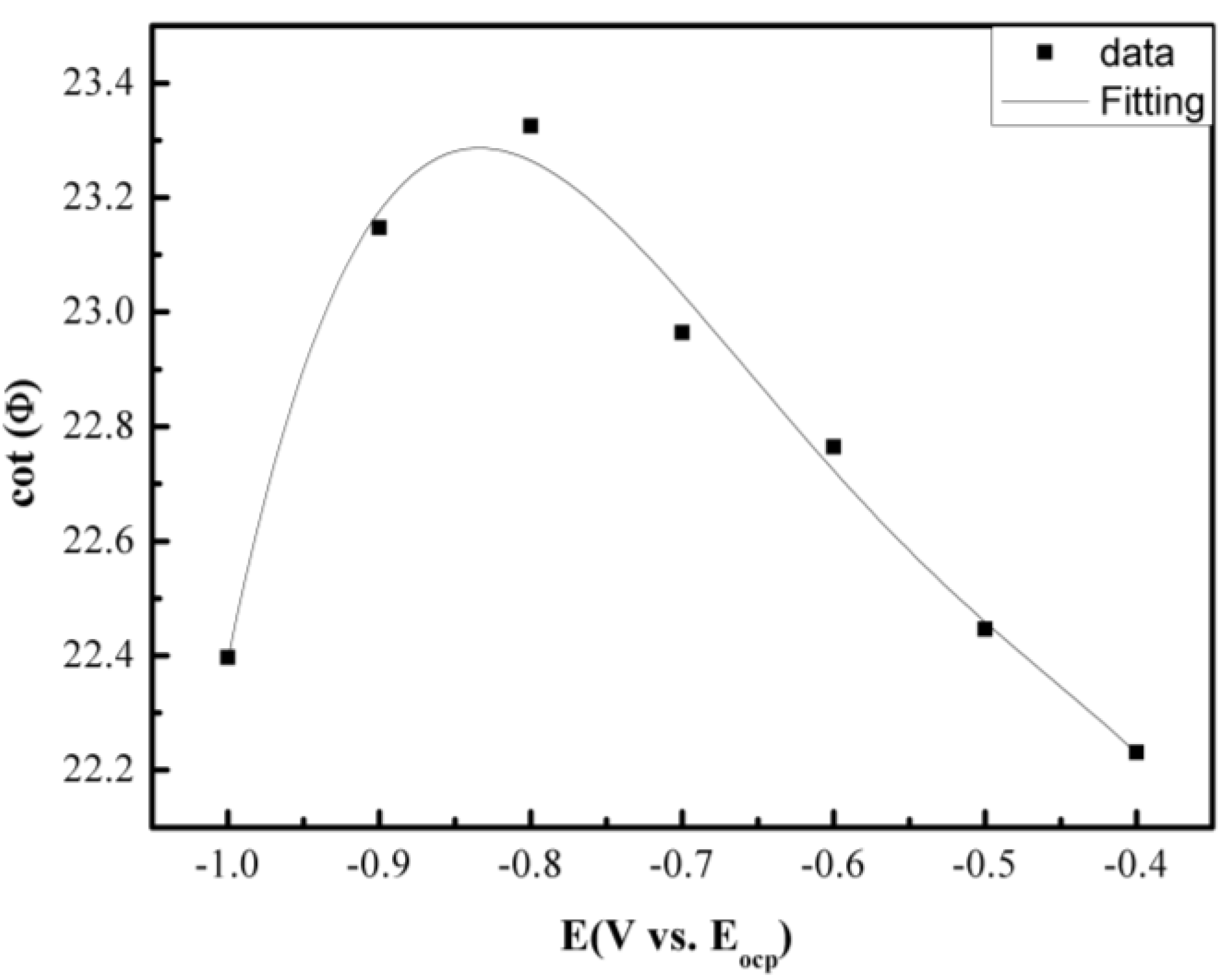
2.2. Electrochemical Corrosion





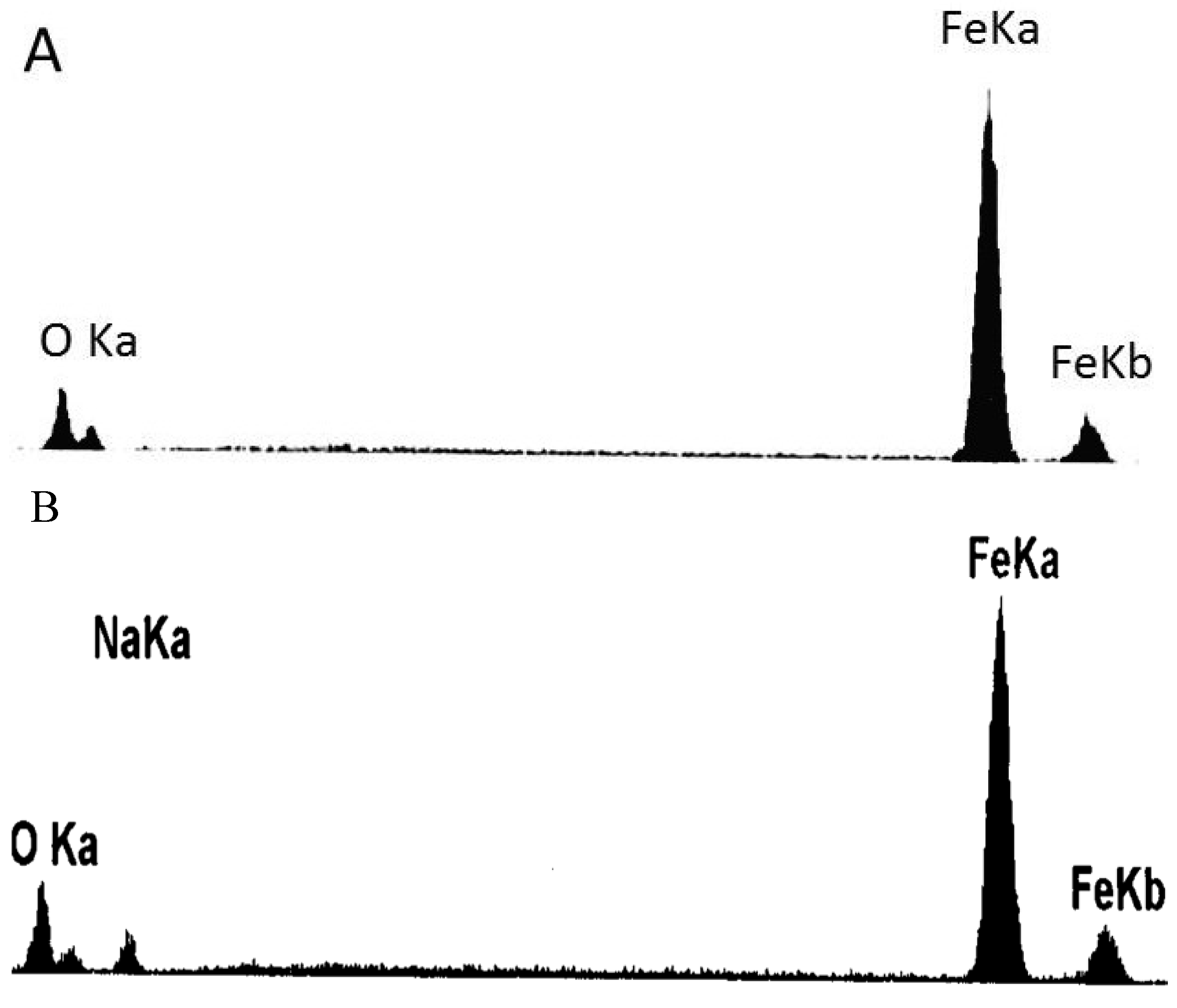
2.3. Corrosion Products and Morphologies




3. Discussion
3.1. The Interaction between Electrochemical Corrosion Reaction and Chemical Corrosion Reaction
3.2. The Corrosion Behavior of the Pure Fe under a Na2SO4 Deposit in an Atmosphere of H2O + O2

3.3. Thermodynamic Calculation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactions | Conditions |
|---|---|
| 4Fe + 3O2 + 2H2O + 2Na2SO4 → NaFeO2 + 2H2SO4 | PH2SO4 < 19,090.3 Pa |
| H2SO4 + Fe → FeSO4 + H2 | PH2 < 1.02 PH2SO4 |
| 6FeSO4 + O2 + 6H2O → 2Fe3O4 + 6H2SO4 | PH2SO4 < 42,116.9 Pa |
| 5FeSO4 + O2 + 5H2O → Fe2O3 + Fe3O4 + 5H2SO4 | PH2SO4 < 41,040.8 Pa |
| 4Fe + 3O2 + 2Na2SO4 → 4NaFeO2 + 2SO3 | PSO3 < 42,491.1 Pa |
4. Experimental Section
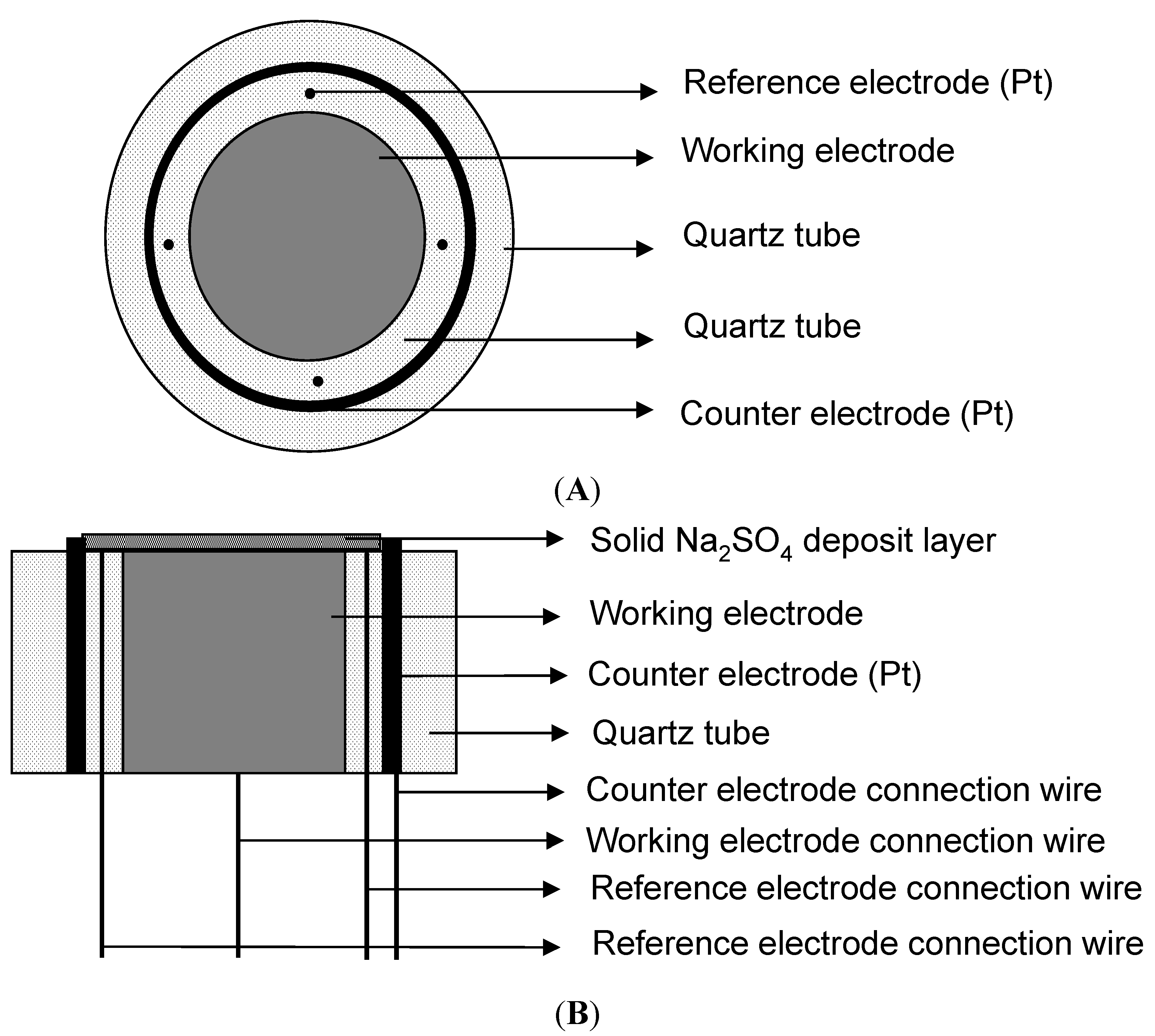
4.1. Materials and Experimental Conditions
4.2. Mass Gain Measurements
4.3. Electrochemical Experiments

5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shu, Y. Corrosion Behavior of Same Metals and Coating under the Synergistic Effect of Solid NaCl and H2O at 500–700 °C. Ph.D. Dissertation, Shenyang, China, June 1999. [Google Scholar]
- Shu, Y.; Wang, F.; Wu, W. Synergistic effect of NaCl and water vapor on the corrosion of 1Cr11Ni2W2MoV steel at 500–700 °C. Oxid. Met. 1999, 51, 97–110. [Google Scholar] [CrossRef]
- Shu, Y.; Wang, F.; Wu, W. Corrosion behavior of Ti60 alloy coated with a solid NaCl deposit in O2 plus water vapor at 500–700 °C. Oxid. Met. 1999, 52, 463–473. [Google Scholar] [CrossRef]
- Shu, Y.; Wang, F.; Wu, W. Corrosion behavior of pure Cr with a solid NaCl deposit in O2 plus water vapor. Oxid. Met. 2000, 54, 457–471. [Google Scholar] [CrossRef]
- Wang, F.; Geng, S.; Zhu, S. Corrosion behavior of a sputtered K38G nanocrystalline coating with a solid NaCl deposit in wet oxygen at 600 to 700 °C. Oxid. Met. 2002, 58, 185–195. [Google Scholar] [CrossRef]
- Wang, F.; Shu, Y. Influence of Cr content on the corrosion of Fe-Cr alloys: The synergistic effect of NaCl and water vapor. Oxid. Met. 2003, 59, 201–214. [Google Scholar] [CrossRef]
- Wang, C.; Jiang, F.; Wang, F. Corrosion inhibition of 304 stainless steel by nano-sized Ti/Silicone coatings in an environment containing NaCl and H2O at 400–600 °C. Oxid. Met. 2004, 62, 1–13. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Zeng, C.; Wang, F. Electrochemical impedance spectroscopy (EIS) studies of the corrosion of pure Fe and Cr at 600 °C under solid NaCl deposit in H2O. Electrochem. Acta 2006, 51, 4763–4743. [Google Scholar]
- Tang, Y.; Liu, L.; Li, Y.; Wang, F. Evidence for the occurrence of electrochemical reactions and their interaction with chemical reactions during the corrosion of pure Fe with solid NaCl deposit in water vapor at 600 °C. Electrochem. Commun. 2010, 12, 191–193. [Google Scholar] [CrossRef]
- Rapp, R.A. Chemistry and electrochemistry of the hot corrosion of metals. Corrosion 1986, 42, 568–576. [Google Scholar] [CrossRef]
- Simons, E.L.; Browning, G.V.; Liehafsky, H.A. Sodium sulfate in gas turbines. Corrosion 1955, 11, 505–513. [Google Scholar]
- Bornstein, N.S.; Decrescente, M.A. The role of sodium and sulfur in the accelerated oxidation phenomena-sulfidation. Corrosion 1970, 26, 209–214. [Google Scholar]
- Bornstein, N.S.; Decrescente, M.A. The role of sodium in the accelerated oxidation phenomenon termed sulfidation. Metall. Mater.Trans. 1971, 2, 2875–2883. [Google Scholar] [CrossRef]
- Goebel, J.A.; Pettit, F.S. The influence of sulfides on the oxidation behavior of nickel-base alloys. Metall. Mater. Trans. 1970, 1, 3421–3429. [Google Scholar]
- Goebel, J.A.; Pettit, F.S. Na2SO4-induced accelerated oxidation hot corrosion of nickel. Metall. Mater. Trans. 1970, 1, 1943–1954. [Google Scholar]
- Li, M. High Temperature Corrosion of Metals; Metallurgical Industry Press: Beijing, China, 2001; pp. 398–403. [Google Scholar]
- Monyoncho, E.; Bissessur, R. Unique properties of α-NaFeO2: De-intercalation of sodium via hydrolysis and the intercalation of guest molecules into the extract solution. Mater. Res. Bull. 2013, 48, 2678–2686. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, L.; Li, Y.; Wang, F. The electrochemical corrosion mechanisms of pure Cr with NaCl deposit in water vapor at 600 °C. J. Electrochem. Soc. 2011, 158, C237–C241. [Google Scholar] [CrossRef]
- Smith, D.E. Alternating current polarography of electrode processes with coupled homogeneous chemical reactions 1. Theory for systems with first-order preceding, following, and catalytic chemical reactions. Anal. Chem. 1963, 35, 602–609. [Google Scholar] [CrossRef]
- Shen, J.; Zhou, L.; Li, T. High-temperature oxidation of Fe-Cr alloys in wet oxygen. Oxid. Met. 1997, 48, 347–356. [Google Scholar] [CrossRef]
- Öijerholm, J.; Pan, J.; Jönsson, B. Influence of grain-size on ionic conductivity of pure and dense alpha-Al2O3 in the temperature range 400–1000 °C. Mater. Sci. Forum 2004, 461, 865–872. [Google Scholar]
- Öijerholm, J.; Pan, J.; Leygraf, C. In situ measurements by impedance spectroscopy of highly resistive α-alumina. Corros. Sci. 2006, 48, 243–257. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tang, Y.; Liu, L.; Fan, L.; Li, Y.; Wang, F. The Corrosion Behavior of Pure Iron under Solid Na2SO4 Deposit in Wet Oxygen Flow at 500 °C. Materials 2014, 7, 6144-6157. https://doi.org/10.3390/ma7096144
Tang Y, Liu L, Fan L, Li Y, Wang F. The Corrosion Behavior of Pure Iron under Solid Na2SO4 Deposit in Wet Oxygen Flow at 500 °C. Materials. 2014; 7(9):6144-6157. https://doi.org/10.3390/ma7096144
Chicago/Turabian StyleTang, Yanbing, Li Liu, Lei Fan, Ying Li, and Fuhui Wang. 2014. "The Corrosion Behavior of Pure Iron under Solid Na2SO4 Deposit in Wet Oxygen Flow at 500 °C" Materials 7, no. 9: 6144-6157. https://doi.org/10.3390/ma7096144