3.2. Rheological Behavior
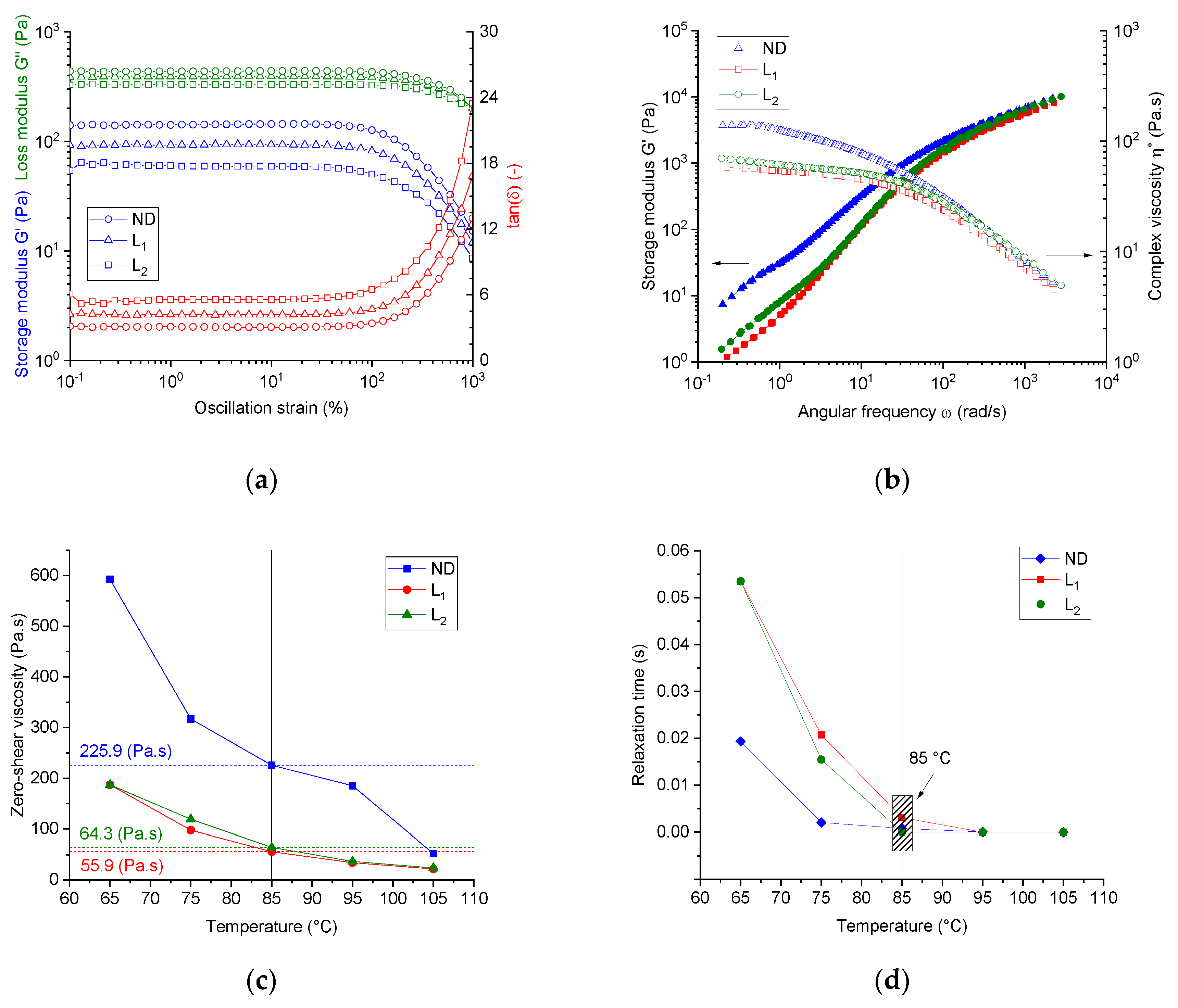
Figure 2a shows the linear viscoelastic (LVE) range of the three spinning dopes at 85 °C. The system ND (PAN/IL) showed the lowest critical deformation strain of 16.6%. Spinning dopes with lignin show higher critical deformation strains, which are enhanced with the increasing lignin content, and the values of the critical deformation strain are 21.4% and 27.7% for the dope L1 and L2, respectively. These results suggest that the presence of lignin promotes the resistance of these dopes to deformation, regardless of the low molecular weight of lignin.
In the LVE region, the loss modulus was greater than the storage modulus for all dopes (PAN/IL and PAN/lignin/IL) in the entire deformation range, indicating a dominant viscoelastic liquid-like behavior of all spinning dopes.
It was also clear that the lignin-free spinning dope (ND) showed a higher storage modulus in the LVE region than the system L1. With any further increase in the lignin content, a decrement of the storage modulus value can be found in the LVE region. The same behavior was observed for the loss modulus. This implies that lignin acts as a softening agent in the dopes owing to the low molecular weight of lignin.
For further oscillatory investigations, a deformation strain of 6% was adjusted, thus this value was high enough concerning the rotational moment resolution of the utilized rheometer and fulfils the criteria of the LVE region for all dopes.
The storage modulus G′ and complex viscosity
versus the angular frequency
as a log–log plot obtained from the frequency sweep tests are illustrated in
Supplementary Data Part II (Figure S1). All spinning dopes show strong dependence of the complex viscosity on the temperature, and a general shear thinning behavior at increasing angular frequency and increasing temperatures was detected.
In the terminal region (at low angular frequencies), linear polymer melts typically show the response of a viscoelastic liquid with the scaling property of approximately G′ ∝ ω2 and G″ ∝ ω, indicating a liquid-like behavior.
In the case of the pseudo-molten spinning dopes, at, for example, 85 °C, however, the PAN/IL (ND) dope showed a slope of G′ versus the angular frequency of 1.04 and a slope of G″ versus of 0.76 at small terminal values of . The dope L1 (PAN/lignin is 2:1) showed corresponding slopes of 1.36 (G′ versus ) and 0.95 (G″ versus ), while the dope L2 (PAN/lignin was 1:1) showed slopes of 1.03 and 0.92, respectively.
The storage and loss moduli of the dopes show, in general, frequency dependency, and are thus considered to be viscoelastic, although the scaling properties are different from polymer melt, where G′ ∝ ω2 and G″ ∝ ω.
Adding lignin in small concentrations, that is, dope L1, leads to an increase of both the slope of G′ versus ω and the slope of G″ versus ω in comparison with the case of neat PAN dopes (ND). For dope L1, the increase of the slope of G′ versus ω was higher than the increase of G″ versus ω, implying that lignin oligomers promote more elasticity to the dopes. This suggests, furthermore, some molecular interaction between the PAN and lignin oligomers. Further increase of the share of lignin in the blend, that is, the case of dope L2, however, reduces the slope to values comparable of that of PAN. This suggests the softening effect of lignin at high lignin shares.
From the frequency sweep tests, the master curves of the complex viscosity versus the angular frequency with a reference temperature of 85 °C, for all dope systems, can be seen from
Figure 2b. By assuming the applicability of the Cox–Merz rule (|η* (ω)|→η(
)) for the developed dopes, the steady shear viscosity as a function of the shear rate
can be calculated. All dopes showed a temperature-dependent shear-thinning behavior.
Low molecular lignin reduced the zero-rate viscosity, that is, in the case of dope L1. However, when increasing the lignin share, that is, dope L2, a slight increase of the zero-shear viscosity over that of dope L1 was observed, obviously owing to the increased overall polymer content in the dope. From the transformed curves of the steady shear viscosity versus the shear rate and applying the Carreau–Yasuda fit function, the specific temperature-dependent and composition-dependent characteristics of all the dopes, that is, the zero-rate viscosity and relaxation time, were calculated.
Figure 2c shows the dependence of the zero-shear viscosity on temperature for all dopes. In general, the dopes with lignin shares of 33.5 wt. % in the PAN matrix showed lowered zero-shear viscosity over the studied temperature range than that of the neat PAN dopes. Higher lignin shares, that is, dope L2, slightly increased the zero-shear viscosity compared with that of dope L1, owing to the higher overall polymer content in the dopes (please refer to
Table 1).
Figure 2d illustrates the influence of the temperature and the composition of the dopes on the calculated relaxation time (the consistency parameter). A higher temperature reduced the relaxation time of all dopes and the neat PAN dopes showed the lowest relaxation times. Introducing lignin to the dopes, that is, dope L1, increases the longest relaxation time of the dopes. This effect became strongly visible at lower softening temperatures <85 °C. This implies that the low molecular weight lignin oligomers considerably decelerate the relaxation process of the PAN chains. This effect is very important to be taken into consideration, as the coagulation process will be performed at much lower temperatures.
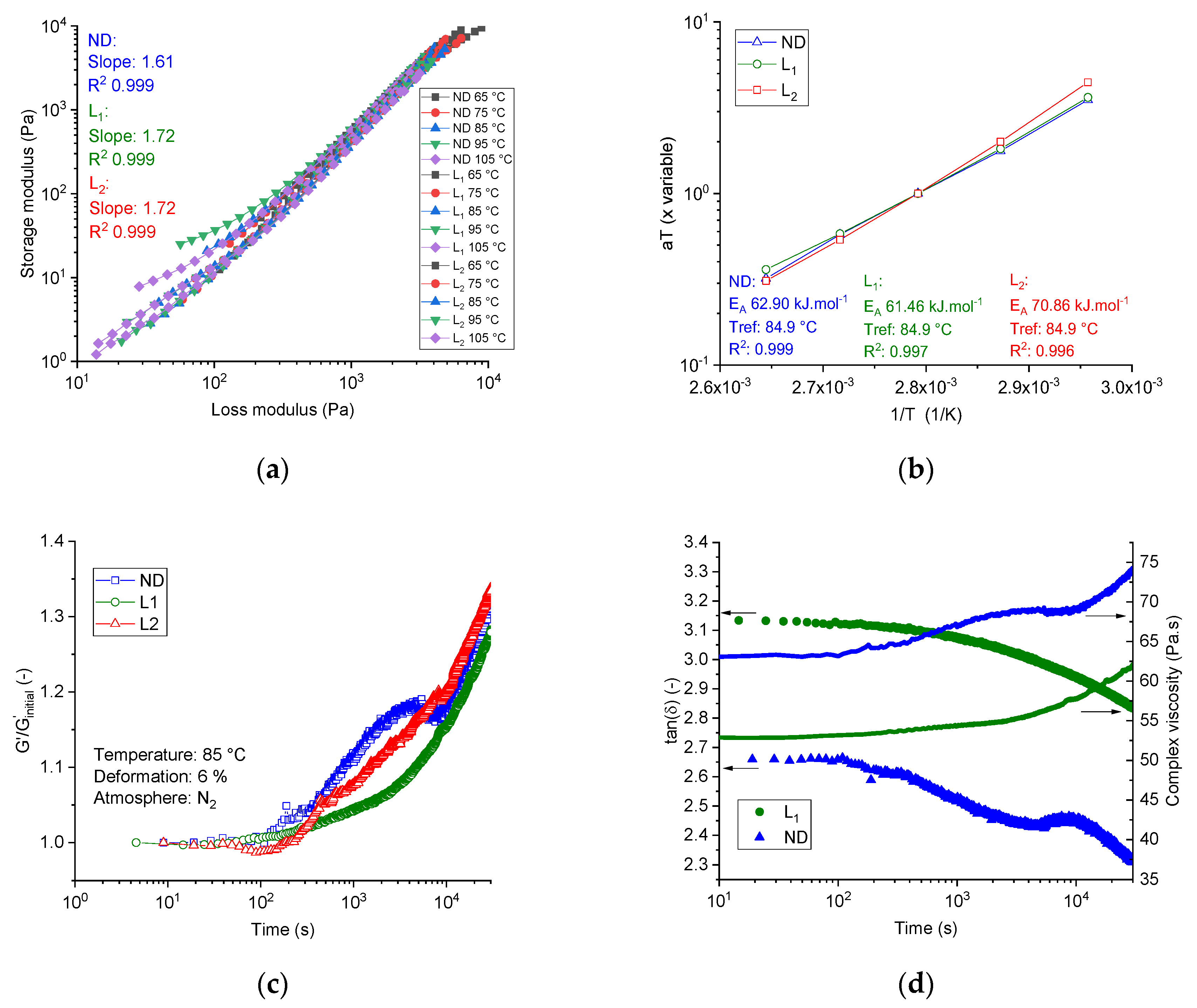
The compatibility of the dopes was determined using the Han plot, which is temperature invariant and showed a linear correlation in the plot log G′ versus log G″. The system can be considered as compatible if nearly the same slope of the pure matrix can be observed at different lignin shares in the blend; otherwise, the system was considered to be phase-separated [
41].
Figure 3a shows the Han plot of the three dopes at different temperatures (over the frequency range of 0.1 to 100 Hz). The neat PAN dope (ND) showed a slope of 1.61. Introducing lignin to the dope systems at different lignin shares, that is, dope L1 and L2, slightly increased the slope to 1.72 (+ 6.2%) in both cases. Although lignin consists of low molecular oligomers and showed no repeated monomer sequences, the spinning dopes with different lignin content showed temperature-independent and almost composition-independent slope values of 1.61–1.72, indicating the compatible composition and single-phase consistency between the PAN and low molecular weight purified softwood Kraft lignin BioChoice
TM.
Additionally, the miscibility/immiscibility of the PAN, lignin, and ionic liquids was also investigated using the time–temperature–superposition plot. The G′
curves for thermo-rheologically simple pseudo-melts were superimposed by horizontal shifts (αT) along the angular frequency axis. The vertical shifts (
bT) were set to unity.
Figure 3b showed the TTS-Arrhenius plot of the three investigated dopes at a reference temperature of 84.9 °C. Regardless of how strong or weak the intra-molecular interactions between lignin oligomers and PAN chains were, the TTS principle was valid for the three spinning dopes. Certainly, lignin oligomers caused no break to the TTS plot at shares up to 50 wt. %, hence the compositions can be miscible.
Isothermal time sweep oscillation tests under nitrogen were used to characterize the thermal stability by observing the development of the nominal value of the storage modulus (G′/G′Initial) at 85 °C and 6% deformation over a pre-defined period of time of 30,000 s.
The initial value of the G′
Initial was set to 200 ± 10 Pa. The values of the angular frequencies, at which the initial value of G′ maintained the aforementioned values, can be attained from the intercept of the line (G′ = 200 Pa) with the function G′
(
Figure 2b). In general, if no molecular changes in the dopes under isothermal conditions take place, the storage modulus should remain constant, and thus time-independent. The system can be, hence, termed as thermally stable. Any decrease in the storage modulus over time will be caused by the molecular degradation affecting the relaxation process, whereas the increase of the storage modulus over the time signifies many molecular changes, for example, cross-linking reaction or gelation. In the case of the filled system, this would mean development in the structural rigidity or elasticity.
Figure 3c illustrates the time sweep test of the dopes at 85 °C and a deformation of 6%. After 30,000 s, the neat PAN dopes show an increase of the storage modulus of 32.6% relative to the initial value. Lignin-containing dopes L1 and L2 showed comparatively similar behavior and an increase of the storage modulus by 27.9% and 33.3%, respectively.
Figure 3d shows the development of the loss factor in the time sweep tests at 85 °C over the pre-defined period. Obviously, the neat PAN and lignin-containing dopes show a similar tendency. The loss factors decreased by 11.5% for the neat PAN dope system and by 9% for lignin-based dopes over time, where the increasing rate of the storage modulus was higher than the increasing rate of the loss modulus. This means that the material became, under the applied isothermal conditions over the time, more elastic than viscous. The complex viscosity of the neat PAN system showed an increase of 15%. The complex viscosity of the lignin-containing dopes L1 and L2 increased by 14.6% and 18.7%, respectively, as illustrated in
Figure 3d.
3.4. Evaluation of the Drawing Dependent Fineness and Diameter
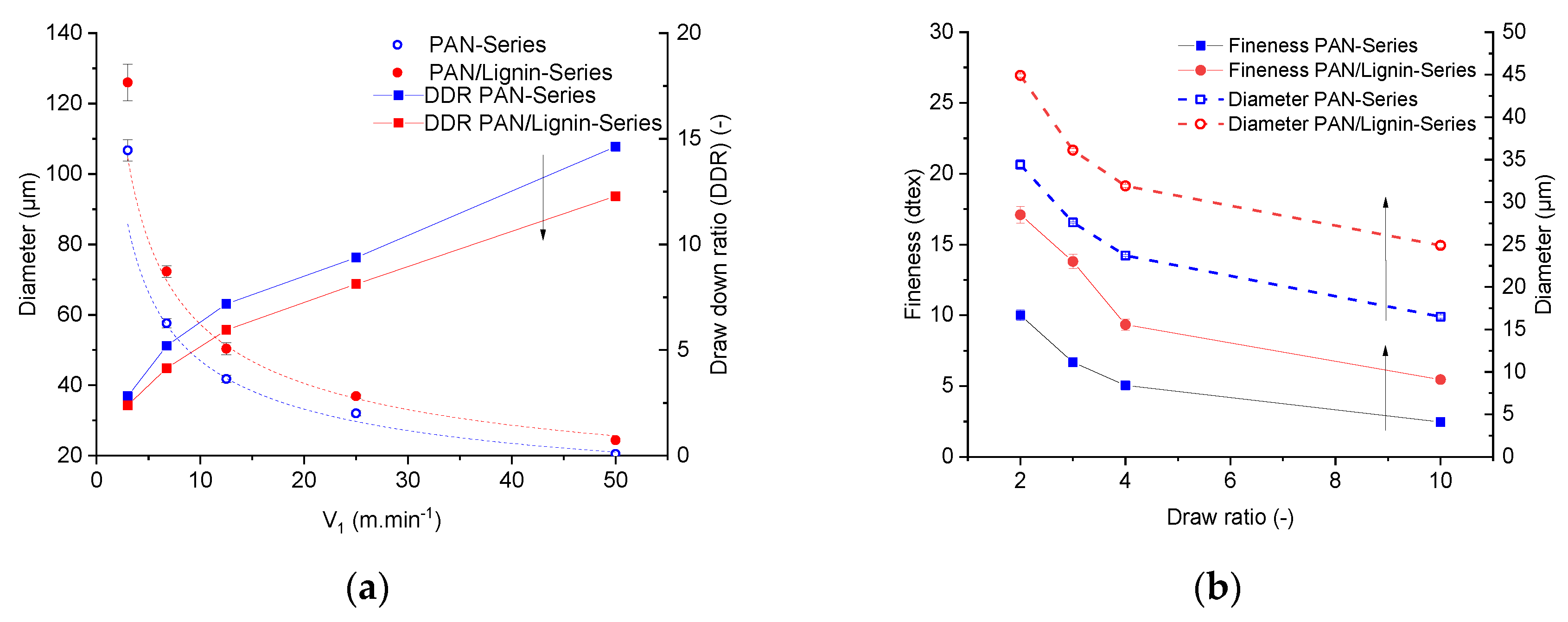
Figure 5a showed the influence of the take-up speed of the first godet (V1) on the diameter of the as-spun fibers and on the draw down ratio. Obviously, thicker filaments were obtained from lignin-containing dope in comparison with the neat PAN dope under similar processing conditions (spinning temperature 85 °C; T
C_bath = 6 °C).
In the Dry-Jet Wet spinning process, the flow of the extruded mass in the air gap can be considered as a non-isothermal viscoelastic extensional flow assuming a simplified cylindrical symmetry along the spinning line. Ionic liquids have in general low-volatility [
42]. Taking the mass-transfer or continuity equation, the momentum equation, the energy equation, and the constitutive equation into consideration, the diameter of the fibers at the first godet is related to the following parameters [
43]:
where
is the diameter of the fiber in µm collected on the first godet,
Q is the mass throughput in g·min−1,
is the density of the dope in g·cm−3,
is the polymer fraction in the spinning dope in wt. %,
is the take-up speed of the first godet in m·min−1.
At a constant take-up speed set-up and the spinning nozzle (D0 = 300 µm), the ratio of the final fiber diameter of lignin-containing fibers D2 to the diameter of the neat PAN fibers D1 was related to the following parameters:
Figure 5a illustrates higher diameter values of the lignin-containing fibers compared with the diameter values of the neat PAN fibers for a given series of the take-up speed.
The mass throughput of the lignin-containing dopes and the neat PAN was 0.09 g·min
−1 and 0.11 g·min
−1, respectively; the polymer content ratio c2/c1 was equal to 18.37/13.04 = 1.41. Taking these parameters into account, the density ratio of the spinning dopes
must be greater than 1 in order to analyze the experimental data of the fiber diameter. The dynamic simulation of diameter formation in the Dry-Jet wet spinning of lignin-containing dopes will be conducted in another paper and was not within the scope of the present paper. A discrepancy in the draw down ratio between the neat PAN fibers and PAN/lignin fibers was detected and the discrepancy was increased by increasing the take-up speed. This effect became intense by in-line stretching and solid state post drawing the filaments, as shown in the
Figure 5a.
At a low in-line draw ratio (DR = 2), the discrepancy was ca. 30%. At the highest solid-state drawing (DR = 10), the difference was roughly 51%. This means that the draw ratio-dependent decrement of the diameter (lignin-containing fiber) is much lower than that of neat PAN fibers. This effect can also be seen from the values of the linear density.
The fineness of the developed fibers can be seen in
Figure 5b. At a total draw ratio of 10, neat PAN fibers showed linear density of 2.46 ± 0.14 dtex and diameter values of 16.5 ± 0.16 µm. Neat PAN fibers were thinner than the lignin-containing fibers 5.46 ± 0.37 dtex with diameter values of 24.9 ± 0.27 µm. From the linear density and the fiber diameter, the theoretically determined density
of the fibers can be calculated from the following equation:
where
is the fineness of the fibers in tex,
is the density of the fibers in g·cm
−3, and D is the diameter of the fiber in µm.
The theoretically determined density of highly drawn (DR = 10) neat PAN fibers and highly drawn (DR = 10) lignin-containing fibers was 1.15 g·cm−3 and 1.12 g·cm−3, respectively.
3.5. Mechanical Properties
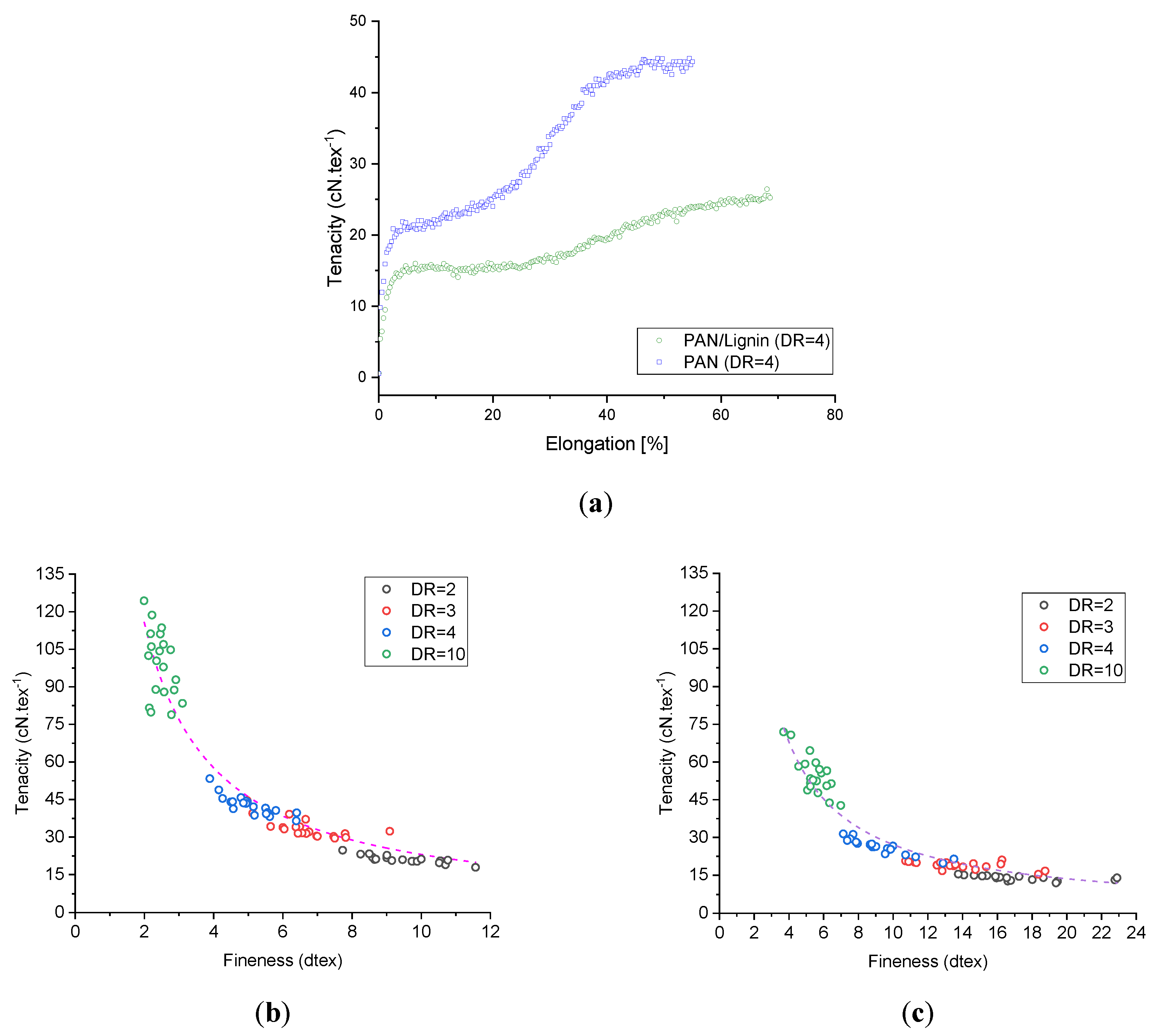
The PAN and PAN/lignin fibers show typical tenacity–elongation curves, as shown in
Figure 6a. Lignin-containing fibers show lower tenacity values under the same manufacturing conditions. This general tenacity effect is owing to (a) the higher fineness values (lower diameters) of the neat PAN fibers and (b) the disturbed fine structure of the lignin-containing fibers by the low molecular weight lignin oligomers. Nonetheless, lignin-containing fibers in general show higher elongation at break values.
The tenacity of the neat PAN fibers and lignin-containing fibers increases with higher draw ratio, as seen in
Figure 6b,c. As it was possible to produce roughly two times finer neat PAN fibers than the lignin-containing fibers under the same conditions (owing to the higher polymer content and lower density of the lignin-containing dopes), better mechanical properties of the neat PAN fibers were reached.
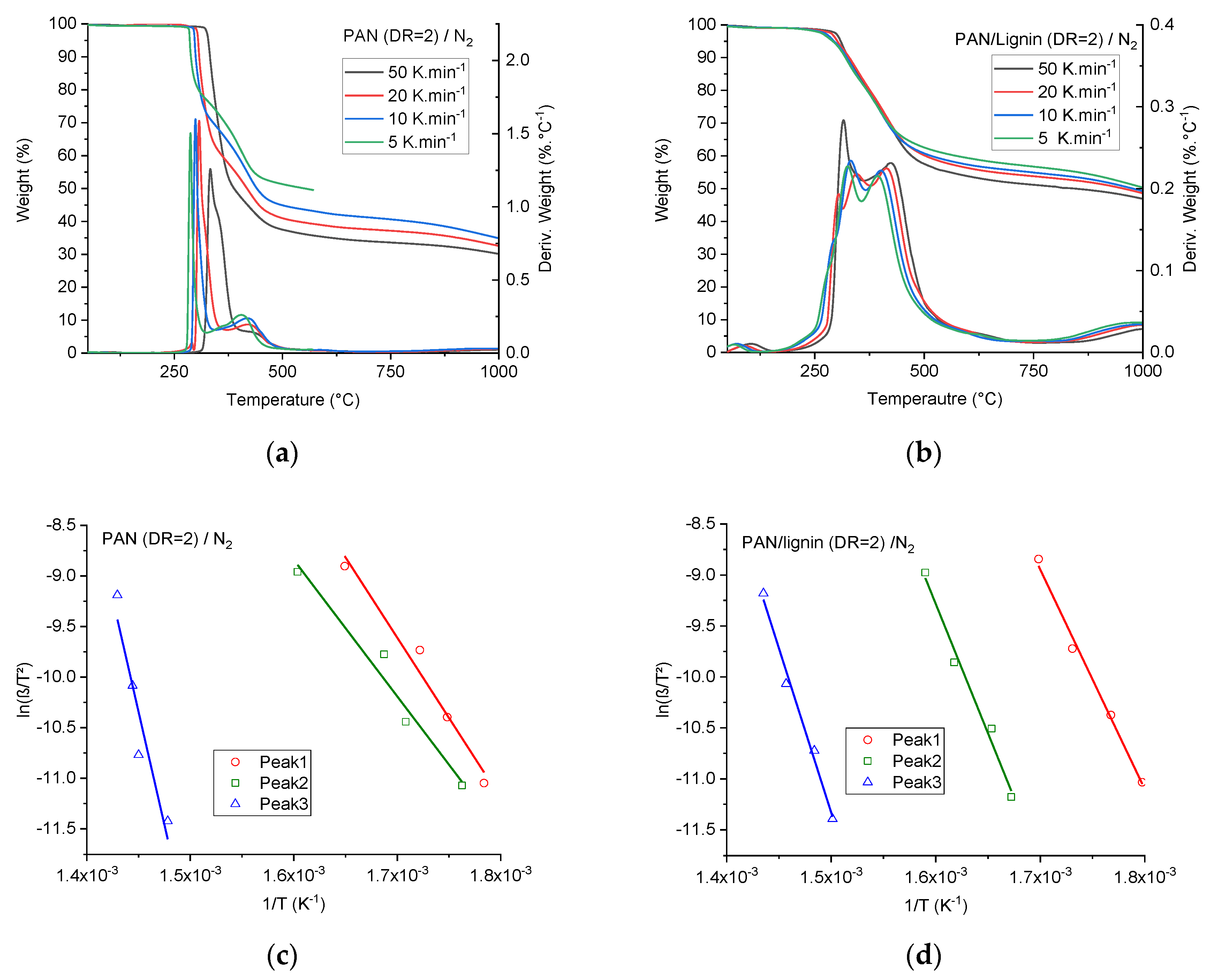
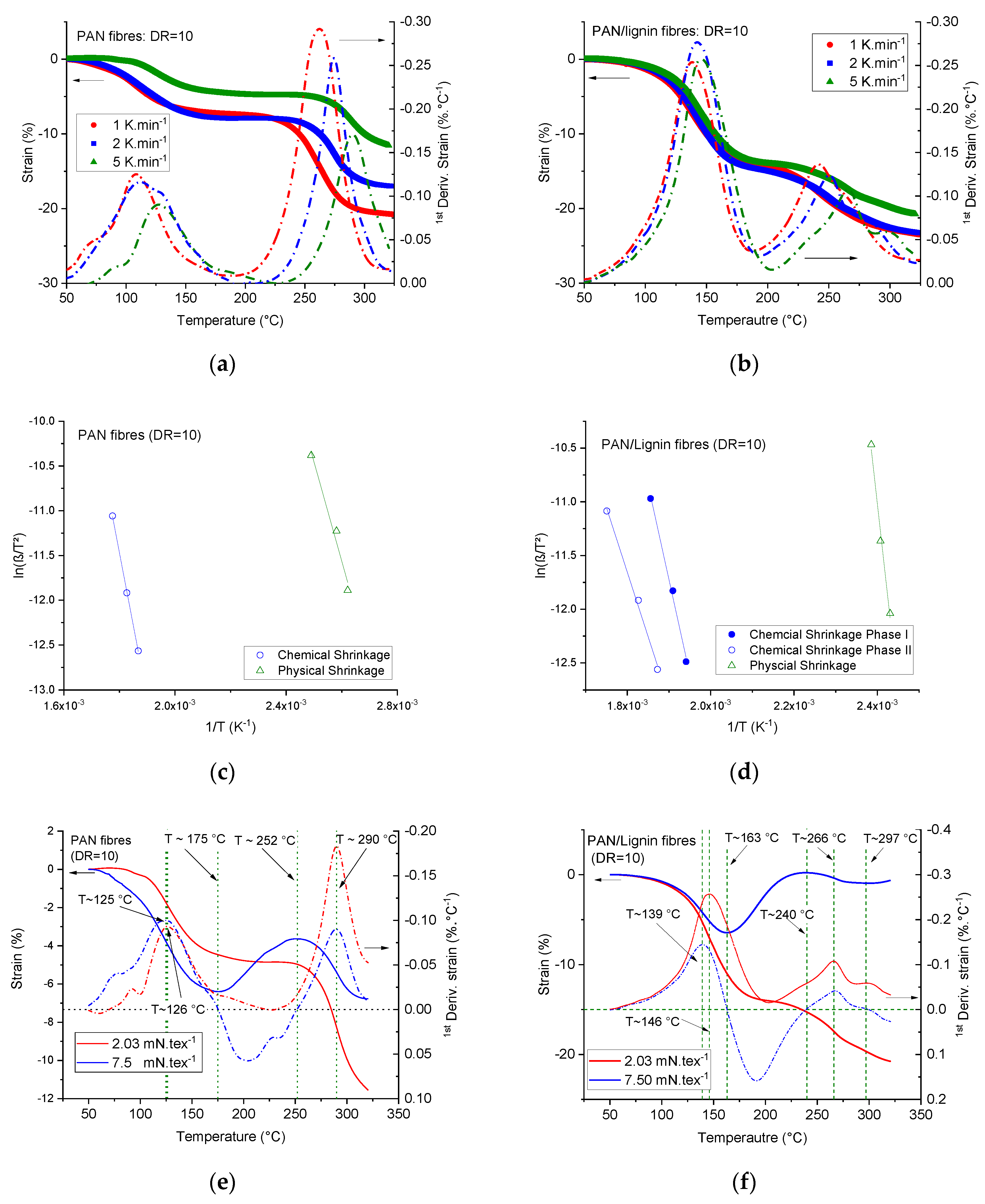
Table 3 illustrates in detail the impact of two important processing parameters, that is, the draw ratio and the temperature of the coagulation bath, on the fineness and mechanical performance of the neat PAN and lignin-containing fibers. Obviously, in both cases of neat PAN fibers and lignin-containing fibers coagulated at 6 °C, the tenacity increases with increased drawing. Generally, lignin-containing fibers show lower tenacities and tensile moduli. The highest tenacity of the neat PAN fibers reached an average value of 99.18 ± 2.99 cN·tex
−1 at a total draw ratio of 10. On the basis of the diameter calculations (D = 16.5 ± 0.16 µm), the tensile strength and the tensile modulus were 1141.04 ± 34.38 MPa and 66.86 ± 3.19 GPa, respectively. These tenacity values obtained from a textile grade PAN by this method were higher than those of many high-performance PAN fibers for CF described in the available literature [
44,
45,
46,
47]. For lignin-containing fibers stretched 10 times, the tenacity was 54.96 ± 1.73 cN·tex
−1. On the basis of the diameter mean value (D = 24.90 ± 0.27 µm) of this fiber series, the tensile strength was 616.24 ± 19.50 MPa and the tensile modulus was 29.89 ± 1.25 GPa. The elongation at break of the neat PAN fibers decreases from 89.34 ± 3.83% for fibers with DR = 2 to 12.73 ± 0.46% for the fibers drawn ten times. Comparatively, lignin-containing fibers showed similar behavior, where the elongation at break reduced from 88.56 ± 4.30% to 13.27 ± 0.41% by increasing the draw ratio from 2 to 10. This implies that low molecular lignin oligomers do not generally affect the elongation at break of the fibers. Additional work regarding the critical fiber radius and its dependency on the fineness will be conducted in the future to understand in-depth the impact of lignin on the brittleness of the fiber structure. Furthermore,
Table 3 showed the impact of the coagulation bath temperature on the mechanical performance of the developed neat PAN and lignin-containing fibers. Clearly, the higher the coagulation temperature, the thicker the developed fibers offered lower the mechanical properties of the fibers. This tendency was observed for neat PAN fibers as well as for lignin-containing fibers. For instance, in the case of neat PAN fibers drawn four times in-line, the tenacity was reduced from 41.38 ± 1.88 cN·tex
−1 to 24.57 ± 0.96 cN·tex
−1 (reduction by ca. 68.4%). For the lignin-containing fibers, the reduction was roughly 38.1% from 26.13 ± 0.68 cN·tex
−1 to 18.92 ± 0.55 cN·tex
−1. This important effect can be explained by the influence of the diffusion rate of the ionic liquids from the high concentration region (the attenuated fiber being drawn in the coagulation zone) to the low-concentration region (the deionized water in the coagulation bath) being higher at higher temperatures. This shortens in turn the available period to draw and thus orient the polymer chains. This was also obvious in terms of higher fineness values and lower orientation parameters (please refer to the crystallographic fine structure section). The orientation is furthermore only possible if the ionic liquids can act as long as possible as a “lubricant-agent” in the fiber structure while orienting the fibers via the extensional forces being applied in the coagulation zone. Another important parameter, which influences the orientation, is the temperature-dependent relaxation time of the spinning dope. At higher temperatures, the dopes, in general, show shorter relaxation times than at lower temperatures. This was of rather importance in the entry zone (after the air-gap) of the attenuated fibers in the coagulation bath. This effect together with the influence of the thermodynamic diffusion rate can be pursued in detail by the orientation values of the fibers, that is, the relative orientation degree and Herman’s orientation factor, as described in the following section. A similar effect of the relaxation time and temperature-dependent diffusion rate of the ionic liquids on the mechanical properties was found in the study of the Dry-Jet Wet spinning of cellulose fibers from ionic liquid dopes [
48].
3.6. Crystallographic Fine Structure
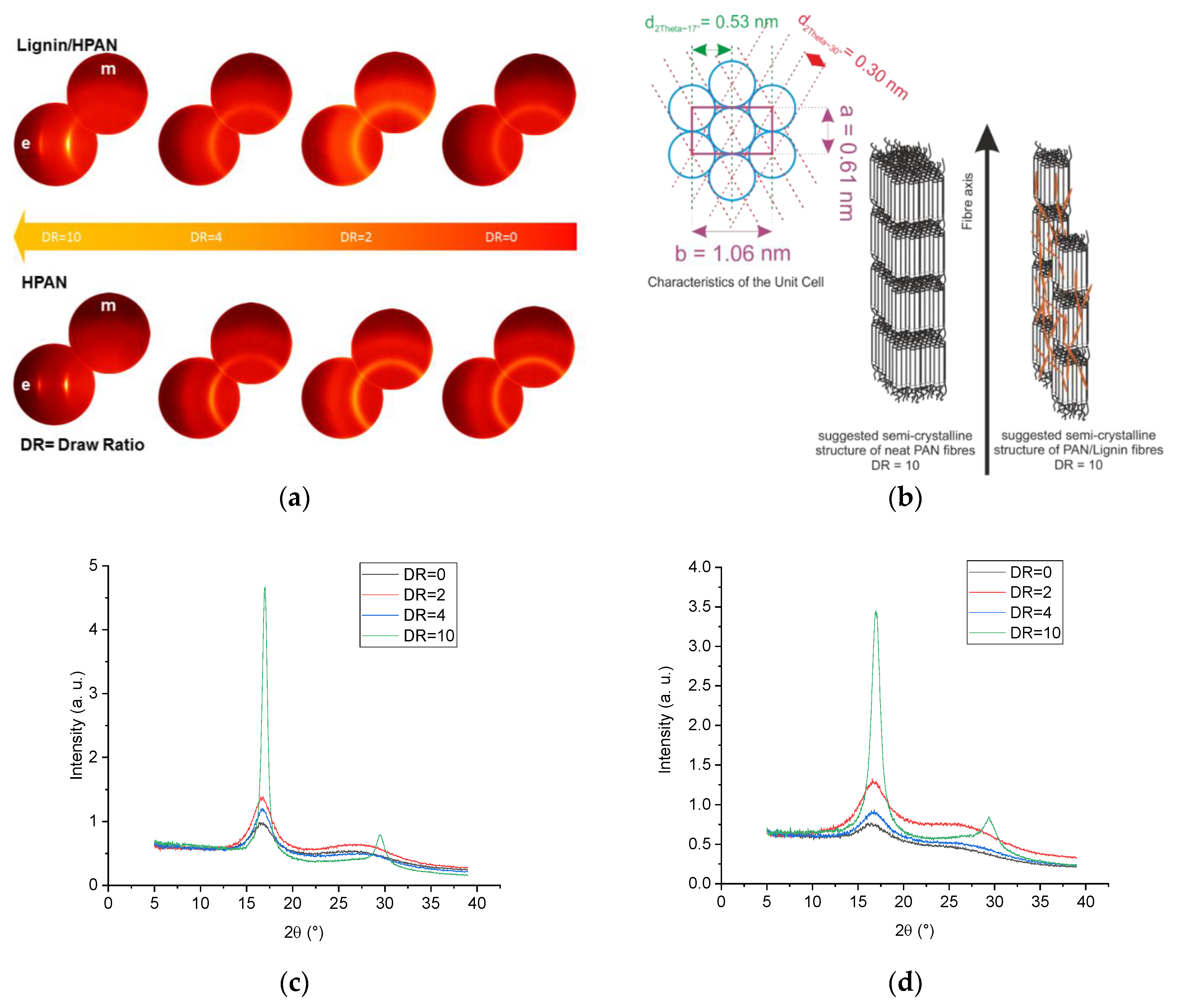
The structure–process–property relations of the developed fibers were influenced to a wide extent by (a) the material composition, that is, low molecular lignin oligomers, on the deformation behavior of the fibers; and (b) the processing parameters, that is, the coagulation bath temperature, and in-line and solid state stretching. It is well known that the mechanical performance and deformation behavior of the fibers are influenced to a wide extent by the fine structure of the fibers, that is, the crystal structure and the orientation parameters of the crystalline domains. For each type of fiber, equatorial and meridional scans were performed at different drawing ratios and at different coagulation bath temperatures. The azimuthal scans of the intensity of the PAN reflection at 2θ~17° were recorded.
Figure 7a shows the equatorial (e) and meridional (m) wide-angle X-ray diffraction patterns of the as-spun fibers, in-line drawn fibers, and solid-state post drawn fibers. As-spun neat PAN and lignin-containing fibers (DR = 0) and in-line drawn fibers (DR to 4) show diffuse reflection at 2θ~17° as well as a broad diffuse scattering maximum 2θ values between 22° and 30°. The peak intensity of the reflection at 2θ~17° in the equatorial mode increased and the full width at half maximum decreased with the increased stretching, as shown in
Figure 7c,d. Only at high stretching values (DR = 10), neat PAN and lignin-containing fibers showed the highest intensity of the equatorial reflection at 2θ~17° and an equatorial peak at 2θ values ~30° became clear in both cases (
Figure 7a,c,d).
Table 4 summarizes the crystallographic fine structural data of the neat PAN fibers and lignin-containing fibers. Additional information about the impact of some processing parameters was also listed. As lignin consists of low molecular oligomers with no defined semi crystalline structure, the semi-crystalline structure of the lignin-containing fibers can only be originally given by the PAN chains. At low temperatures (6 °C) of the coagulation bath, neat PAN fibers show increased equatorial cryptal size L
2θ~17°(e) with increasing in-line drawing. In contrast, the meridional crystal size L
2θ~17°(m) decreased with increasing drawing. The crystal size ratio L
2θ~17°(e)/L
2θ~17°(m) increased around 39% at in-line draw ratio of 4. The interlayer spacing d
2θ~17° = 5.3 Å of the PAN chains was found to be similar to that of the original powder 5.26 Å at all drawing ratios. At a total drawing ratio of 10, the equatorial crystal size L
2θ~17°(e) was 12.83 nm. The equatorial crystal size of the reflection at 2θ~30° L
2θ~30°(e) was 7.70 nm and the associated interlayer spacing d
2θ~30° was 3.0 Å. The aspect ratio of the unit cell b/a (
Figure 7b) was equal to 1.732 and the crystal unit is thus hexagonal [
49].
Lignin deteriorated to a wide extent the size of the crystal domains, while the characteristics of the unit cell by PAN were still preserved (
Figure 7b). In other words, the interlayer spacing d
2θ~17° was 5.3 Å and the d
2θ~30° was 3.0 Å, hence the aspect ratio of the unit cell b/a was 1.732. The crystal size of the highly oriented lignin-containing fibers was less than that of PAN at the same draw ratio. At 10 times drawing, the crystal size L
2θ~17°(e) was 7.16 nm and L
2θ~30°(e) was 4.92 nm. This gave a lower number of reflection plains N
2θ~17° = 15 and N
2θ~30° = 17 than that of neat PAN fibers (N
2θ~17° = 26 and N
2θ~30° = 26). This implies that lignin did not affect the unit cell characteristics, but disturbed the number of PAN chains contributed in the crystal domain, as seen illustratively in
Figure 7b. By increasing the temperature of the coagulation bath, the crystal size was reduced in both cases of neat PAN and lignin-containing fibers as a result of the fast diffusion of ionic liquids from the extruded fibers in the coagulation bath, as seen in
Table 4 in both PAN and PAN/lignin-series. The aspect ratio of the L
2θ~17°(e)/L
2θ~17°(m) at a defined drawing was reduced by increasing the coagulation temperature. For instance, this ratio was reduced from 1.53 for neat PAN fibers drawn at DR = 4 to 1.28 for neat PAN fibers coagulated at 20 °C. This suggests that, at higher diffusion rates of the ionic liquids at the equivalent extensional deformation in the coagulation bath (V1 was 10 m·min
−1 for both cases), the ionic liquids left the fiber at a very early stage, and the chains became unsoftened and thus less able to be oriented. Lowered crystal size was a result of the less orientation, as seen in the
Table 4. The similar effect was found in the case of lignin-containing materials. In summary, it was obvious that lignin does not affect the increasing rate of the ratio L
2θ~17°(e)/L
2θ~17°(m), but deteriorates to a wide extent the crystal size L
2θ~17°. From the azimuthal distribution of the reflection at 2θ~17°, the relative orientation degree (OD) and the Herman’s orientation factor of all developed fibers were calculated.
As shown in
Table 4, OD increased by increasing the drawing ratio. For the fiber series coagulated at 6 °C, the highest relative OD was 94.7% for neat PAN fibers and 88.9 for lignin-containing fibers. This implies that lignin reduced and blocked the ability of the PAN chains to be oriented during stretching. The same effect was also visible in terms of Herman’s orientation effect, which was 0.73 and 0.69 for neat PAN and lignin-containing fibers at DR of 10, respectively.
Increasing the coagulation temperature to 20 °C reduced the orientation degree for both series of the developed fibers compared with those fibers coagulated at 6 °C and at the same in-line draw ratio of 2 and 4, as seen in
Table 4. These findings support the explanations of the generally lowered mechanical properties at a higher coagulation temperature discussed in the section of the mechanical properties.
Additional research work was also required to investigate the influence of lignin on the periodical structure length (ordered domain length) of the crystal units parallel to the fibers axis as well as the true void length and tilt angle of the voids with respect to the fiber axis. This can be done using the small angle X-ray scattering and will be discussed in another extended research work.

 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}