Theoretical Study on the Aggregation of Copper Clusters on a Liquid Surface
by
Hong-Ying Mao
1,*, 
Bao-Xing Li
1,*,
Wang-Feng Ding
1,
Yu-Hong Zhu
1,
Xu-Xin Yang
1,
Chao-Yang Li
1 and
Gao-Xiang Ye
2 1
Department of Physics, Hangzhou Normal University, Hangzhou 310036, China
2
Department of Physics, Zhejiang University, Hangzhou 310027, China
*
Authors to whom correspondence should be addressed.
Materials 2019, 12(23), 3877; https://doi.org/10.3390/ma12233877
Submission received: 6 November 2019
/
Revised: 16 November 2019
/
Accepted: 22 November 2019
/
Published: 24 November 2019
(This article belongs to the Section Advanced Nanomaterials and Nanotechnology)
Abstract
:The ground state structures of copper clusters with different sizes along with their aggregation have been systematic investigated using Amsterdam Density Functional (ADF) and Atomistix ToolKit (ATK) programs. On the basis of geometry optimization, some Cu clusters with more stable structures which were not reported previously have been revealed. In most cases, these Cu clusters prefer to adopt icosahedral structures which originate from the 13-atom icosahedron. It has also been demonstrated that the interaction between two Cu clusters is anisotropic, which is attributed to their charge distribution, especially the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of Cu clusters. Moreover, we have carried out the simulation of Cu clusters aggregation on the silicone oil substrate by means of Monte Carlo (MC) method, which shows good consistence with our previous experimental studies.
1. Introduction
There are extensive research efforts for the investigation of metal clusters, which have been regarded as promising candidates for biomedical applications [1,2], chemical and biological testing [3], plasmonic sensitization [4] and optoelectronic applications [5]. Their unique physical and chemical properties mainly originated from their specific geometrical and micro-structures, leading to a large surface area-to-volume ratio. Versatile functionalities of metal clusters have been demonstrated by modulating their compositions. Among all kinds of metal clusters, copper clusters have been intensively studied in the last decade, including the aggregation geometry, electronic structures, physical and chemical properties [6,7,8]. Recently, the conversion efficiency of methanol from carbon dioxide over size-selected Cu4 clusters at low pressures has been investigated [9]. It was indicated that size-selected Cu4 clusters were the most active low-pressure catalyst for catalytic CO2 conversion to CH3OH. The size effect of copper cluster in methanol synthesis has also been investigated using in situ grazing incidence X-ray absorption spectroscopy, catalytic activity measurements and first-principles calculations [10]. It has been demonstrated that the activities of gas-phase Cu clusters increased as the cluster size decreased. Moreover, different theoretic methods, including self-consistent one-electron local-density theory, density functional calculations, Embedded-Atoms Method (EAM) and the Monte Carlo (MC) method, have been employed to investigate the size effect of metal clusters on their structural and electronic properties [11,12,13,14,15,16,17,18,19,20,21,22].
It was generally believed that the growth behavior of metal atoms on liquid substrate differed from that on solid substrate. Comparing with solid substrates, the interaction between metal atoms and liquid substrate is much weaker. The diffusion coefficient is much larger on liquid substrates than that on solid substrates. Moreover, because of the non-lattice characteristic of liquid substrates, liquid substrates can be regarded as an isotropic substrate. In our previous studies, Cu thin film on liquid substrate (silicone oil) has been fabricated using vapor phase deposition method [23]. Compact and ramified Cu clusters can be observed at the initial stage of growth. With the increasing thickness of Cu, the random diffusion of Cu clusters was found and larger ramified aggregates were observed on the silicone oil surface. Other metal nanoparticles and thin films have also been fabricated on liquid substrates, including Au, Ag, Al and Zn [24,25,26,27,28]. In spite of extensive researches for the morphology of metal nanostructures on liquid substrates, the aggregation of metal nanostructures was only discussed and analyzed by classical kinetic method and the growth mechanism of Cu nanostructures on liquid substrates remains unknown. In order to reveal the growth mechanism of Cu nanostructures, especially at the initial stage of growth, the investigation of fundamental structural characteristics of Cu clusters and their aggregation behaviors are highly desirable.
In the present study, first-principles calculations have been carried out to investigate the aggregation of Cu clusters on silicone oil at the initial growth stage using the ADF and ATK program, respectively. Cu clusters with different sizes have been studied. It has been demonstrated that Cu clusters prefer to adopt icosahedral structures deriving from the 13-atom icosahedron. Our calculation results also reveal an anisotropic interaction between two Cu clusters, which is attributed to the charge distribution of Cu clusters. Moreover, the aggregation of Cu clusters on the silicone oil substrate has been simulated by MC method which is in good agreement with our previous experimental studies.
2. Materials and Methods
In the ADF program version (2010.02) [29], molecular orbitals (MOs) were expanded using a large, unconstructed set of Slater-type orbitals (SOs)—Triple-zeta with two polarization functions (TZ2P) [30]. The TZ2P basis is an all-electron basis of triple-z quality, which is augmented by two sets of polarization functions. During the simulation, the frozen-core approximation for inner-core electrons was utilized. The copper orbital which is inner than Cu 3p were kept frozen. We use an auxiliary set of s, p, d, f and g SOs to fit the molecular density and to represent the Coulomb and exchange potentials in each self-consistent field (SCF) cycle. All calculations were made in the framework of the generalized gradient approximation (GGA) using the Becke-Perdew functional, which is based on Becket [31,32] gradient correction to the local expression for the exchange energy and the correlation energy. The self-consistent field was converged to a value of 10−4 in our studies.
Atomism Tool Kit (ATK) is a software package [33,34,35] that offers very fast geometry optimization and molecular dynamics calculations by using such methods ranging from both accurate quantum-mechanical first-principles and fast semi-empirical methods to classical potentials. Therefore, the software can treat large-scale systems with several thousand atoms. In this paper, the optimization of cluster structures has been made by using Perdew-Burke-Ernzerhof (PBE) functional within density functional theory (DFT) in generalized gradient approximation (GGA).
Kohn-Sham density functional theory has been widely used to simulate the ground state structure of metal clusters. In generalized gradient approximation (GGA), the general expression of the exchange-correlation density functional is described as:
It was found that semiempirical GGA’s can be remarkably successful for small molecules. A first-principles numerical GGA can be constructed in the following ways. In a system of slowly varying density, the second-order density-gradient was first expanded for the exchange-correlation hole surrounding the electron. After that, its spurious long-range parts were cut off to satisfy sum rules on the exact hole. Perdew et al. improved the exchange-correlation density functional with the optimized Perdew-Burke-Ernzerhof (PBE) functional which is in the form of [35]:
where rs is the local Seitz radius (), is the relative spin polarization and is a dimensionless density gradient. Here is a spin-scaling factor and is the Thomas- Fermi screening wave number (). They constructed the gradient contribution H from three conditions, which were in the slowly and rapidly varying limits and under uniform scaling to the high-density limit, respectively. The atomization energies of small molecules obtained by PBE functional were in good agreement with the experimental results.
As we know, copper is a non-magnetic material. Except for some special doped structures [36,37,38], the pure copper clusters do not exhibit any nontrivial magnetic moments. ADF and ATK programs had been used to study the magnetic properties of small Cun (n ≤ 10) clusters and no additional magnetic moments were found. Therefore, spin polarization is not considered in the calculations. In order to simulate the aggregation of the Cu clusters on a liquid surface, a Cu cluster can be regarded as a Brownian particle and the forces exerting on it by liquid substrates are sum up as a random force. For a free cluster floating on the liquid substrate, its motion is mainly driven by Brownian dynamics. In this case, MC methods with small trial steps have been employed to simulate random Brownian motions. The simulations are initiated with 2000 clusters randomly distributed in the 2-dimensional system and are carried out in 3 steps. Firstly, MC steps are performed until the energy of system is optimized to a minimum. Then, another 2000 clusters are dispersed to the free space of the system one by one, following a random walk until it is attached firmly to aggregations of early clusters. Finally, the optimization of the total amount of clusters is performed to reach an energy minimum of the whole system.
3. Results
3.1. Copper Clusters Cun (n = 2–80)
We have investigated copper clusters with different sizes using the ADF program and the ATK software package, respectively. Initial atomic configurations are randomly placed within a real three dimensional box or cage or sphere. To avoid over-crowded or over-relaxed among atoms, their separations are confined to be 2.2–2.8 Å. For each Cu cluster with less than 32 Cu atoms (n ≤ 32), about one hundred thousand initial geometrical configurations have been constructed by this method. After that, the initial configurations are optimized using the ADF program and the ATK software package, respectively. Our results show that most initial structures are convergent after geometry optimization. In many cases, the same convergent structure is obtained after the geometry optimization of different initial configurations. At last, optimized structures with larger binding energies are optimized further with higher calculation accuracy. During the optimization, some isomers with different structures have almost the same binding energy. The cutoff energy is selected as 0.1 eV. Their energy order will not be reversed under different calculation accuracy when the energy difference between two isomers is larger than 0.1 eV. During structural optimization, the symmetries are not restricted. The atom vibration frequencies for each structure have also been calculated and structures with imaginary frequencies have been eliminated. As a result, if the energy difference between the structures is less than 0.1 eV, they are considered to be degenerated
Our calculations results of Cun (n = 2–80) constructed by Ref. [13,14] are presented in Table 1. The estimated binding energies of Cu clusters are different when the ADF or ATK program is employed but the trend of binding energies is similar. Although Jahn–Teller effect was mostly encountered in octahedral complexes of the transition metals and this phenomenon was commonly observed in six-coordinate copper(II) complexes. Since we are mainly focused on the formation of Cu clusters and their aggregations, Jahn-Teller effect is not considered in the present study.
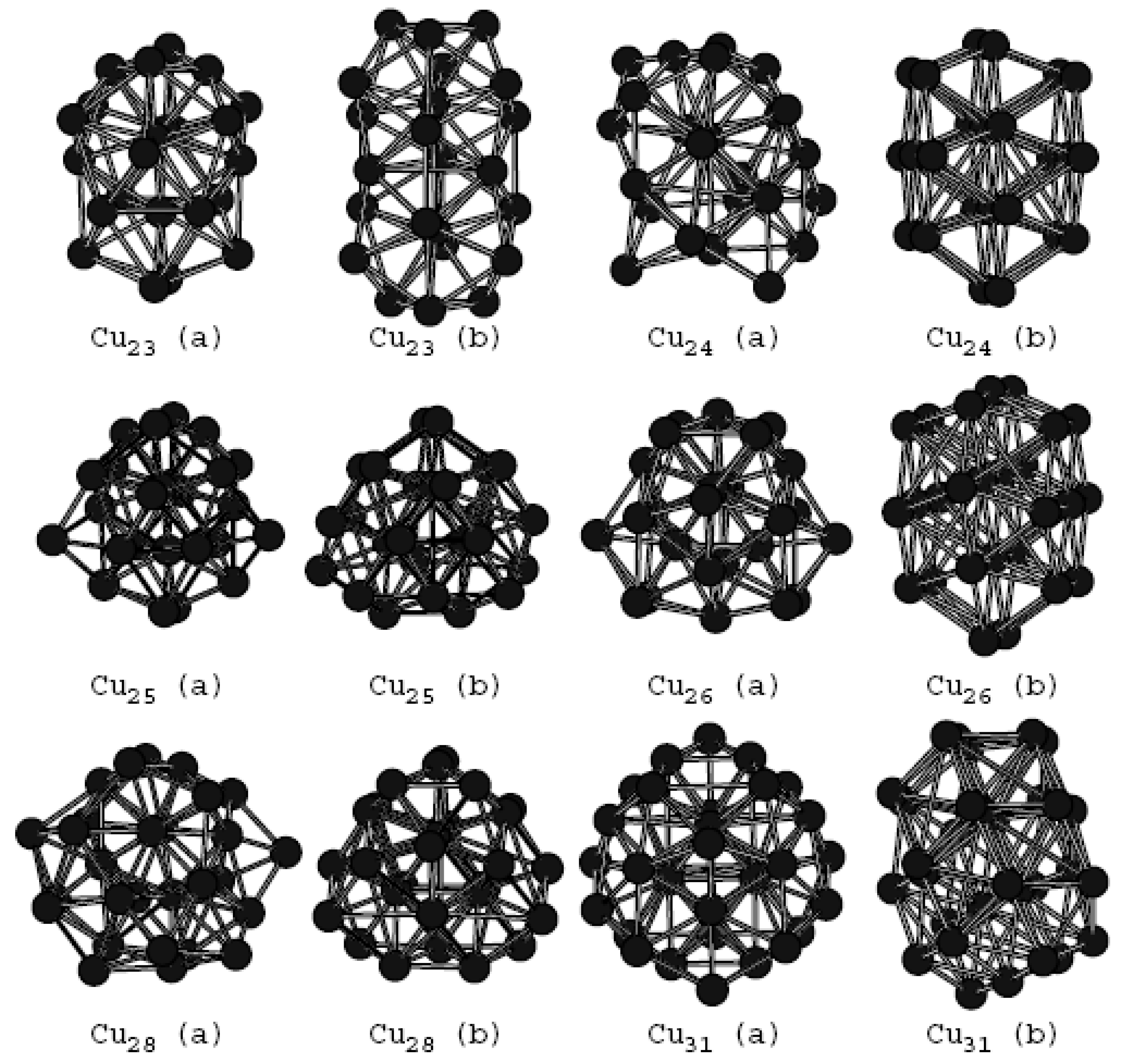
In Table 1, the data marked with * in the range of n from 3 to 8 are calculated from experimental data [19,39]. The binding energy of Cu clusters calculated using ADF shows good consistence with experimental data. There is a discrepancy between the calculated binding energy using the ATK program and experimental data, which is attributed to the fact that DFT-GGA (density functional theory and generalized gradient approximation) does not capture long-range type electrostatic effects [40,41,42]. Choosing different methods or correlation functions based on first-principles calculations, the binding energies are different. But their change trends are nearly the same, which will be discussed later. Our calculation results indicate that the constructed Cu clusters are stable because they do not have imaginary frequencies. Furthermore, the global minima of Cu clusters ranging from 2 to 32 atoms have been investigated. Some Cu clusters with more stable structures, which have been not reported previously, are revealed. Geometric structures and binding energies of these Cu clusters are presented in Table 1 and Figure 1, respectively. The newly revealed structures are marked by the letter “a”. The letter “b” is used to refer to the structures in the previous study [14].
The Cu31(a) and Cu31(b) structure shown in Figure 1 are different but they are almost degenerated. Other structures marked with “a” are more stable than those marked with “b”. Cu23(a) and Cu28(a) structures are the same as those reported in Ref [19]. The most stable structure of the Cu23 cluster is the polyicosahedral structure in the form of a “triple icosahedron”. In the case of Cu28 cluster, it is a disordered structure with polycapped atoms by removing two surface atoms from the 19- atom triple icosahedron. All other structures marked with “a” are newly revealed but they are also based on the icosahedral growth. Although the energy difference between “a” and “b” structures calculated by the ADF program is different from those calculated by the ATK software package, the energy order between them does not change. In Figure 1, structure “a” is more stable than structure “b”.
In order to have a better understanding about the structural evolution, we have also calculated the second order difference (Δ2E) of the binding energies for ground state structures of Cun (n = 2–32) clusters. The Δ2E function is defined as Δ2E = E(Cun − 1) + E(Cun + 1) − 2E(Cun). The larger positive Δ2E’s demonstrated that the corresponding clusters (n = 5, 8, 18, 20, 26, 28) are more stable than their neighboring Cu clusters. The calculated results are shown in Figure 2a,b. Figure 2c presents the change of the binding energy per atom with the increasing number of Cu atoms. Comparisons between the ADF and ATK results show that they have similar trends.
3.2. Aggregation of the Cun Clusters
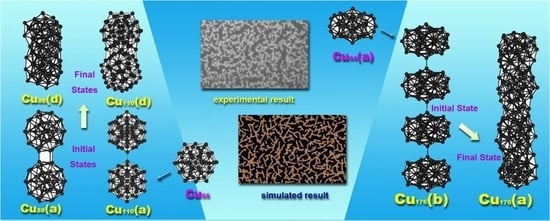
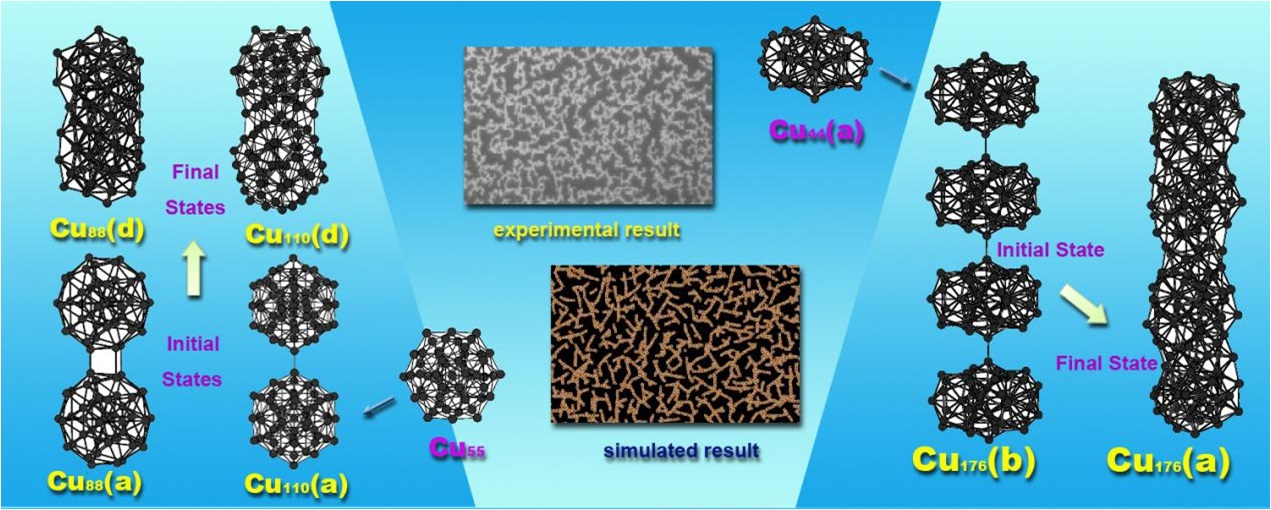
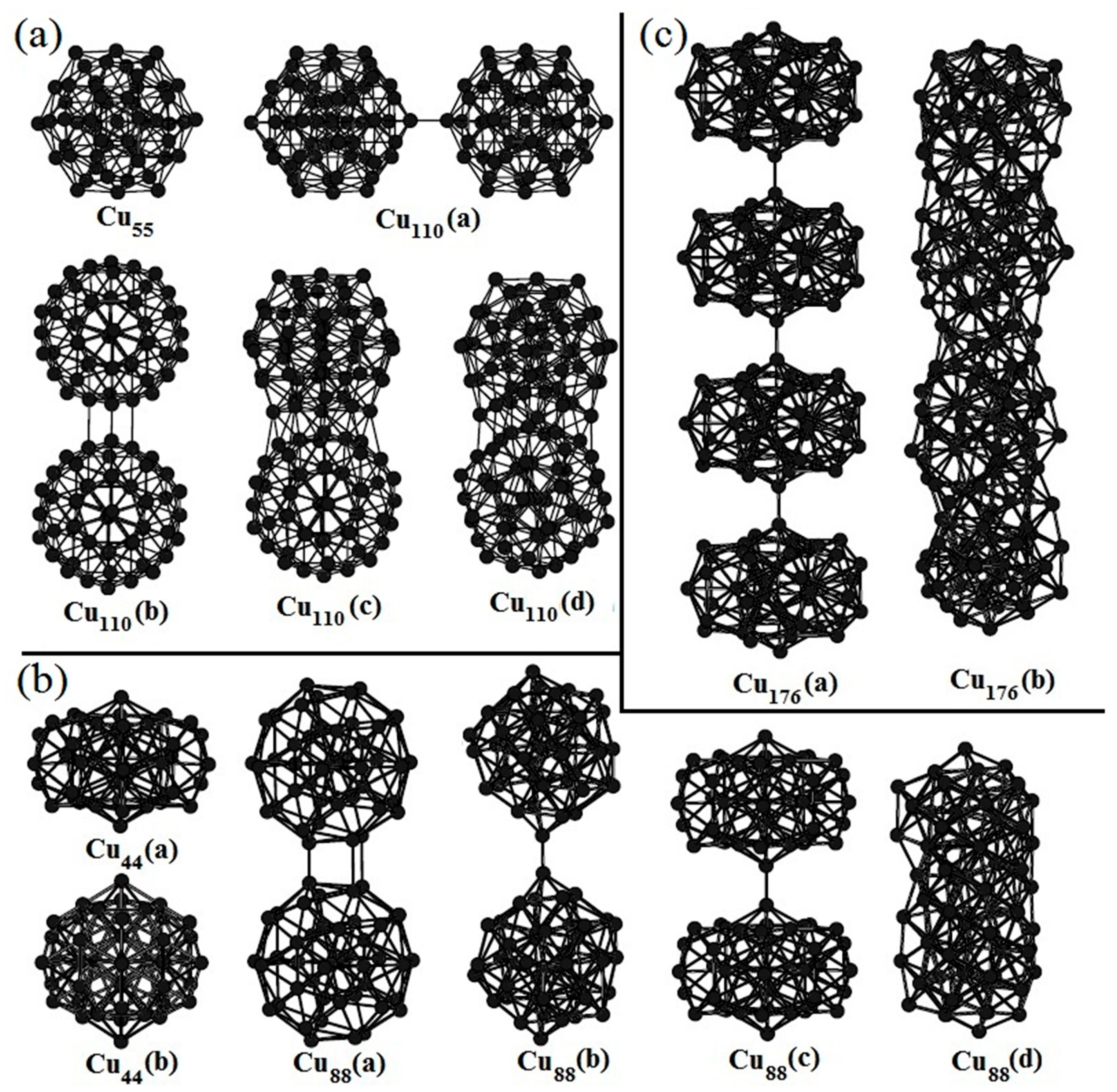
To date, there have been many reports about copper clusters but there is little investigation about the aggregation of Cu clusters. By means of the ATK software, we have studied the aggregation of medium-sized copper clusters. The optimized compact Cu55 in Figure 3a has an icosahedral structure which is its ground state structure. If two Cu55 structures approach to each other in different directions, it is found that the interaction between them is anisotropic, which means that their interaction is rather weak in some directions, while quite strong in other directions. Cu110(a) is the aggregation of two Cu55 clusters in the direction with relatively weak interaction. The binding energy between two Cu55 structures is only 0.96 eV. If they are approached in the direction with strong interaction, structural optimization reveals that they interact strongly with each other and reconstruct to form a new stable structure. The relaxed Cu110(b) in Figure 3a is its initial structure. Cu110(c) is its intermediate structure which is optimized to its final stable structure of Cu110(d). The energy released from the Cu110(b) to the Cu110(d) is up to 7.5 eV, which is much larger than that of Cu110(a).
The initial structure of Cu44(a) in Figure 3b is also a compact structure. After structural optimization, it becomes more compact which is denoted by Cu44(b). But the binding energy is 2.44 eV smaller than the ground state structure with 44 copper atoms (not shown here). Similar to the aggregation of two Cu55 clusters, the aggregation of two or more Cu44 is also highly dependent on the direction along which they are approached. After the geometry optimization of the initial Cu88(a), one stable structure Cu88(b) is obtained. The binding energy of the Cu88(b) is only 1.00 eV. If two Cu44(a) structures are approached in another direction (Cu88(c) in Figure 3b), a stable structure of Cu88(d) with larger binding energy of 14.89 eV is obtained. It is worth noting that the Cu88(d) is even more stable than the aggregated structures from the ground state structure with 44 copper atoms. A large number of calculations indicate that the maximum ratio of the binding energies between two medium-sized ground state structures in different directions is ranging from 7 to 15. In addition, the initial structure of Cu176(a) shown in Figure 3c is constructed from four Cu44(a) clusters. After geometry optimization, a completely different structure, Cu176(b), has been obtained. The reconstructed structure is highly stable with the binding energy of 32.56 eV. We need to mention that a large number of initial structures can be constructed using four Cu44(a) structures. The optimized structures are also different with each other. The aggregation of these clusters depends on the initial structures but quasi-linear stacking seems to be more favorable.
3.3. Anisotropic Interaction Between Two Cu Clusters
As mentioned above, we have demonstrated that the interaction between two Cu clusters is anisotropic. In order to reveal the origin of the anisotropic interaction between two Cu clusters, the aggregation of two separated Cu55 clusters to Cu110(a) and Cu110(d) have been investigated. For the convenience of discussion, the Cu atoms in the Cu55 cluster are divided into two types. The equivalent atoms which can exchange positions by symmetry operation are represented with white in Figure 4, the other atoms are represented with black. Our results indicate that other atoms or clusters are hard to adsorb on the tops (top sites) of the white atoms. In some cases, other atoms can be adsorbed; however its adsorption energy is relatively low, leading to unstable structures. For example, in the case of Cu110(a) shown in Figure 3a, two Cu55 clusters are adsorbed on the top of each white atom but the binding energy is less than 1.0 eV. On the other hand, it is more favorable for other atoms or clusters to adsorbed on the hollow site consisting of three adjacent black atoms. In the initial structure of Cu110(b) shown in Figure 3a, a Cu55 cluster is adsorbed on the hollow site of another Cu55 cluster. Consequently, the binding energy of Cu110(d) is 7.5 eV higher that of Cu110(b), indicating that Cu110(d) is more stable than Cu110(b).
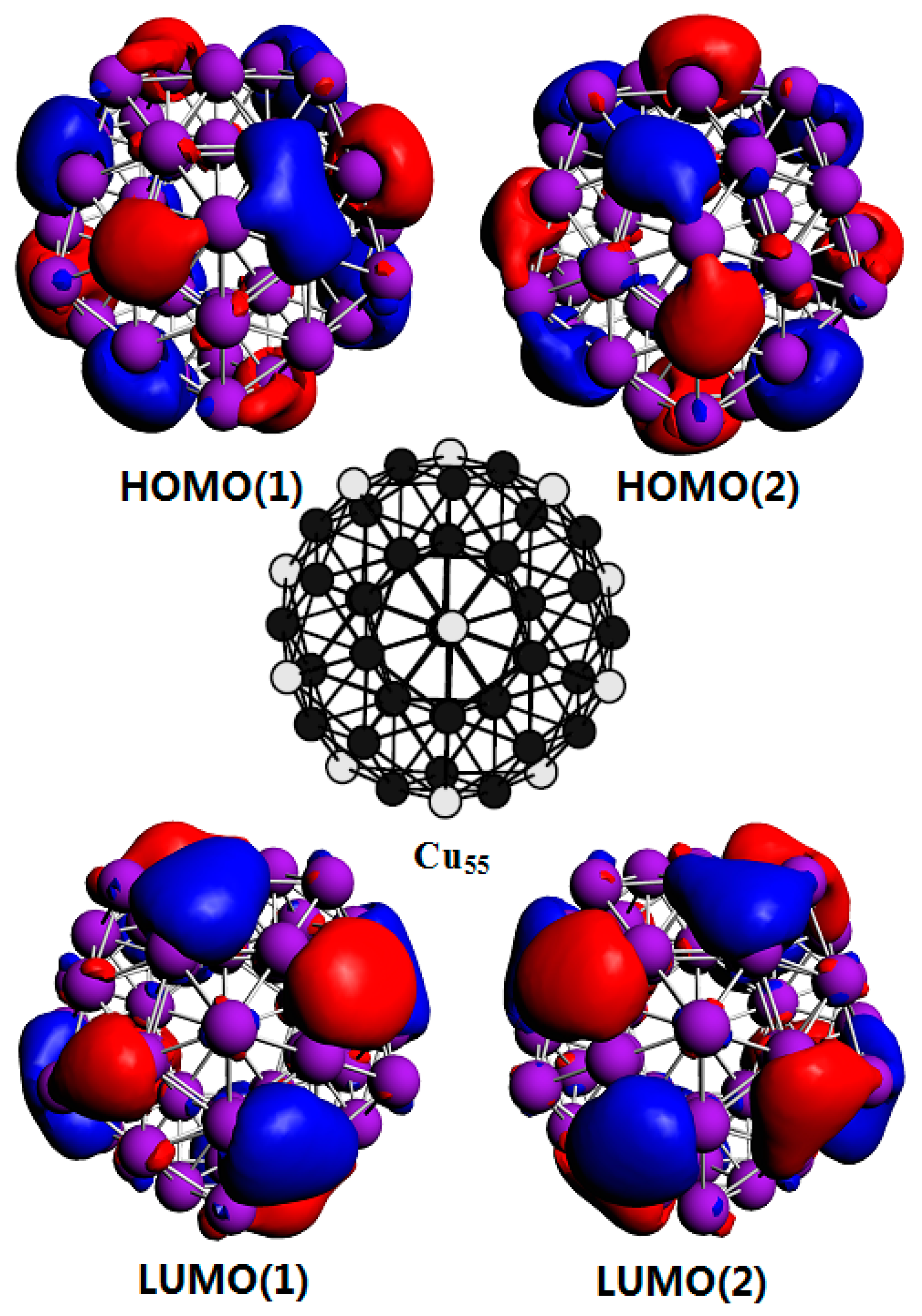
Figure 4 shows the highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO) of the Cu55 cluster. Both of them are degenerate orbitals. It is found that the electron density distribution of the HOMO is mainly concentrated on the hollow sites, while the charge density distribution on the top sites is very low. This feature is more pronounced for the LUMO of Cu55 cluster. This is the main reason why Cu clusters are easily adsorbed on hollow sites. The most stable adsorption position that can attract other clusters differs from cluster to cluster. The compact Cu44(b) shown in Figure 3b is still a meta-stable structure. Our calculation results show that some of hollow sites are not energy favorable for the adsorption of other clusters, as shown in Cu88(a). On the other hand, some of its top sites can strongly attract other clusters as shown in Cu88(c). This further complicates the aggregation of Cu clusters. However, by checking the HOMO and LUMO (not shown here) of Cu44(b) clusters, it is clear that the charge density distribution of the HOMO and LUMO of Cu44(b) are mainly concentrated on top sites, which indicates that the charge distribution of Cu clusters is of vital importance in determining the aggregation of Cu clusters. Therefore, the strong interaction positions depend mainly on the charge distribution rather than on geometry positions.
3.4. Simulation of the Aggregation of Copper Clusters on a Liquid Surface
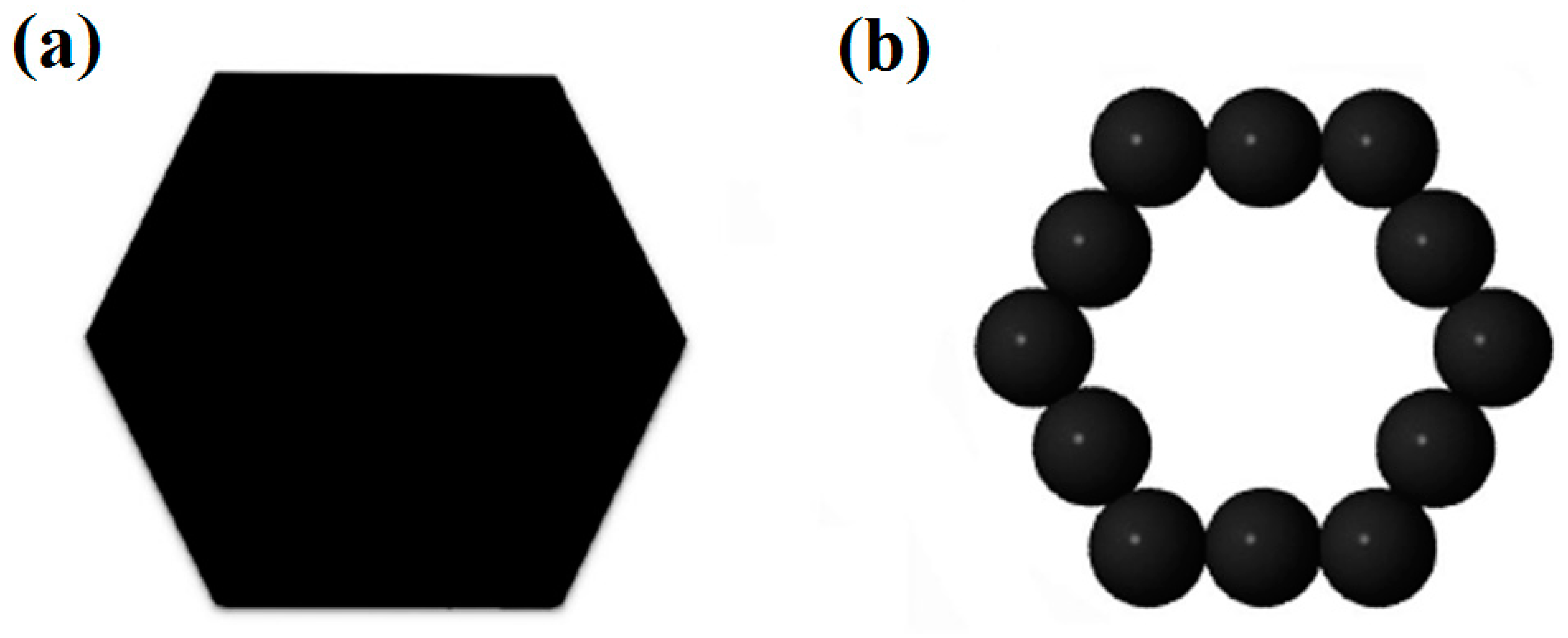
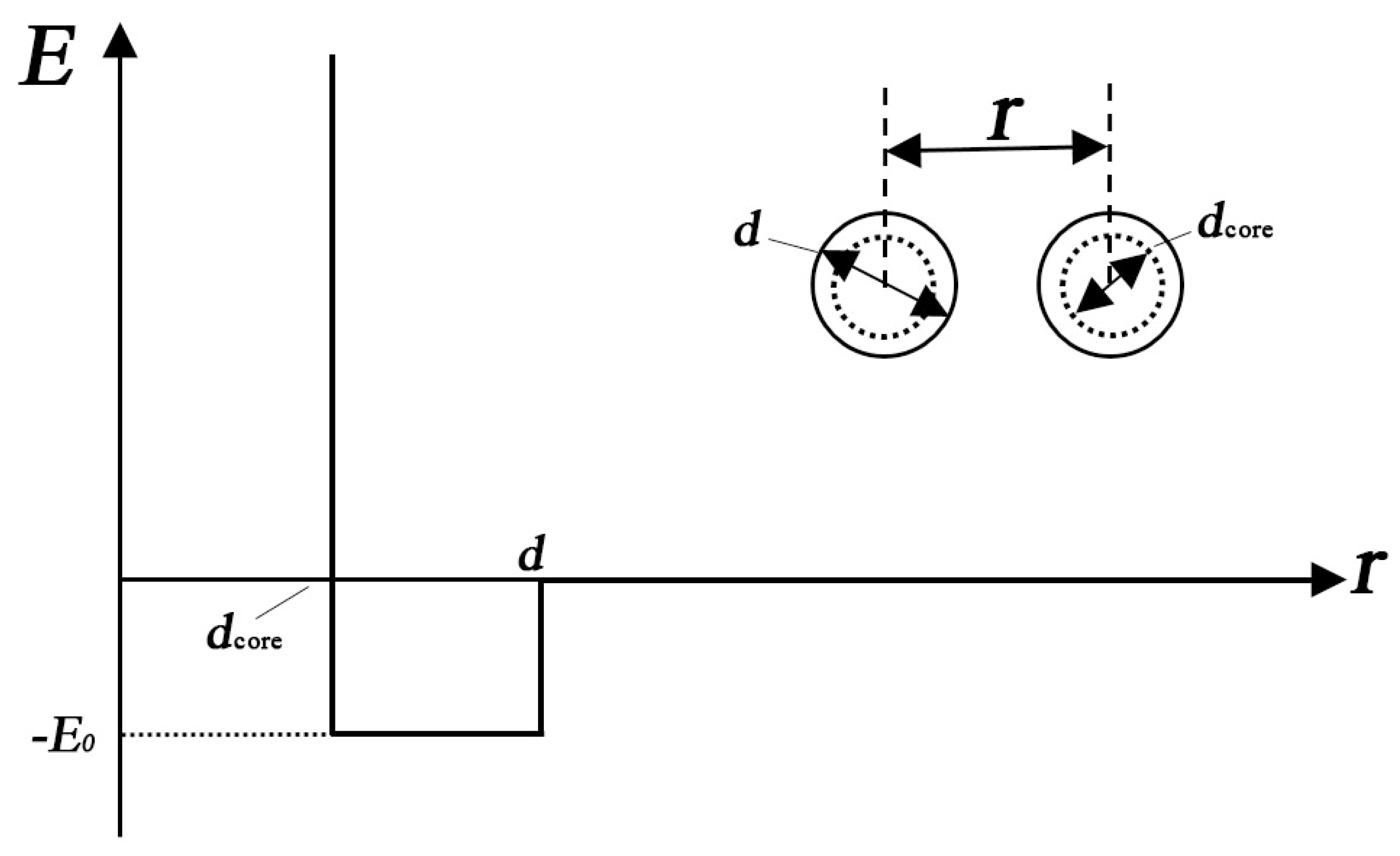
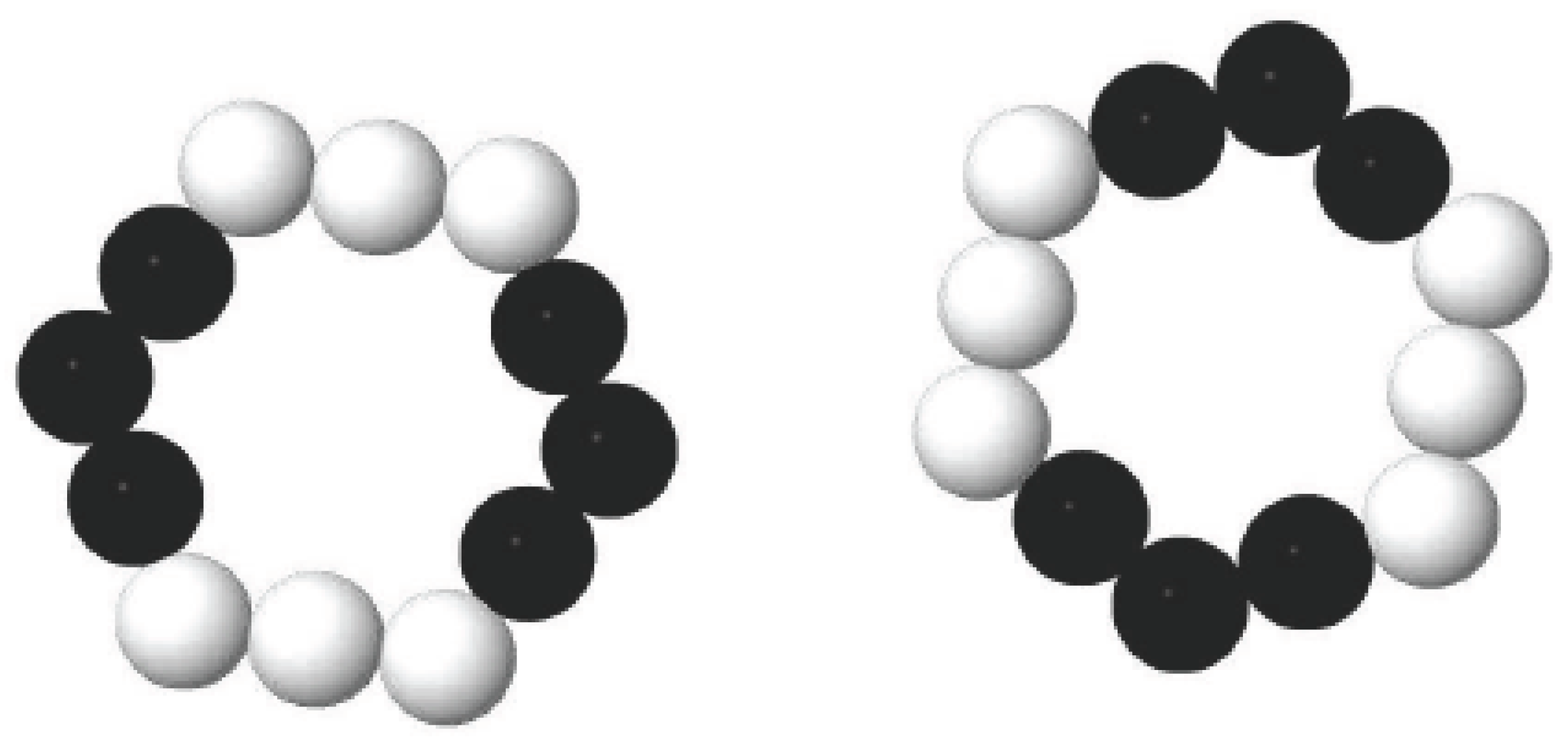
For geometric configurations of copper clusters deposited on a liquid surface, a two-dimensional polygons system has been constructed based on our simulation results. As we have discussed above, most stable Cu clusters are constructed by hexagons and pentagons structures as shown in Figure 1. In some cases, triangles, squares and heptagons can also be observed. During MC simulations, the simulation results are nearly the same when triangles, squares, pentagons, hexagons or heptagons are chosen to model Cu clusters. Therefore, in the present study, hexagons (Figure 5a) have been chosen during the MC simulation. However, the hexagons cannot be treated as the basic units in simulations. The common strategy is to treat each side of a hexagon as n linearly-connected spheres [39]. Figure 5b shows a small hexagon with n = 3 and a total number of N =12 for spheres. Larger hexagons with more spheres on each side have been also used to simulate the aggregation of Cu clusters but the results do not change substantially. By assuming that there is no interaction between two clusters until they touch each other, the interaction between two spheres from surrounded clusters can be simplified as a potential well as shown in Figure 6. When the center-center distance r between two spheres is within the range from dcore to d, they attract each other due to the potential well. However, when the hard-core of spheres overlap (the center-center distance r is smaller than dcore), the infinitely repulsive force dominates. Conversely, when the distance is larger than d, the interaction between them can be neglected. Moreover, on the basis of anisotropic interaction between two Cu clusters which have been discussed above, the binding energy of each side is set to be different when these hexagons combine with each other during the simulation. The cluster is assigned to a hexagonal shape with each side has 3 spheres. In order to study the anisotropic interactions between two clusters, we assign two diagonal sides of a hexagon (the white spheres in Figure 7) to be different from the other sides (black spheres).
Considering that the maximum ratio of binding energies between two clusters in different directions is within the range from 7 to 15, the binding energy ratio of 9 is chosen during our simulations. It is worth noting that choosing different binding energy ratio from 7 to 15 only affects the lengths of cluster aggregations but the morphology of Cu clusters aggregation does not change. Therefore, when the distance between two white spheres lies in the attraction range, there is a drop of −0.9 E0 (a binding energy between two clusters.) and the value is −0.1 E0 for the interaction between a white sphere and a black sphere. The attraction between two black spheres is assumed to be zero.
Figure 8a,c present the typical experimental images of the Cu atomic aggregates on silicone oil surfaces obtained by our group previously [23]. Their nominal film thickness is 1.0 and 5.0 nm, respectively. In the experiment, the deposition rate is f = 0.08 nm/s. Each image has a size of 56 × 42 µm2. We run the simulation on a 2-dimensional scale of 400 d × 400 d by means of MC method. Figure 8b,d show the simulation results obtained by the above method. They correspond to the condensation of 2000 and 4000 clusters on the liquid surface, respectively. The calculation shows that the fractal dimensions of Figure 8c,d are 1.87 and 1.83, respectively. This further indicates that the simulation results are in good agreement with the experimental results.
4. Conclusions
Using the ADF and ATK programs based on first-principles calculations, we have investigated the structures and aggregates of the small or medium-sized Cu clusters. Their aggregation on silicone oil surfaces is simulated by our self-programming and some conclusions are drawn.
1. Most of the Cu clusters adopt icosahedral structures, which can be derived from the 13-atom icosahedron. Some clusters with n = 5, 8, 18, 20, 26, 28 are more stable than their neighboring clusters within the scope of our global search.
2. The aggregation of the Cu clusters depends on their structures and directions. In some directions, they converge tightly but in other directions, the attractions between them are very weak. The HOMO and LUMO analyses reveal that this anisotropic interaction between Cu clusters is determined by the charge distribution of the clusters.
3. The aggregation of the copper clusters on a liquid surface has been simulated by means of MC method. The fractal dimension with 4000 clusters is 1.83, which is close to the experimental value 1.87 with nominal film thickness df = 5 nm. The simulation results of the morphologies are in good agreement with the experimental results.
Author Contributions
Conceptualization, H.-Y.M. and B.-X.L.; methodology, H.-Y.M. and B.-X.L.; software, W.-F.D., Y.-H.Z. and B.-X.L.; formal analysis, X.-X.Y. and C.-Y.L.; resources, B.-X.L. and G.-X.Y.; writing—original draft preparation, H.-Y.M. and B.-X.L.; writing—review and editing, H.-Y.M., B.-X.L. and G.-X.Y.; supervision, B.-X.L.; funding acquisition, H.-Y.M. and B.-X.L.
Funding
This research was funded by the Natural Science Foundation of Zhejiang Province (Grant No. LY16A040011, LY16A040009 and LY18F010019), the National Natural Science Foundation of China (Grant No. 11374082 and No. 11547012) and the Innovation Project in Hangzhou For Returned Scholar.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ling, D.S.; Lee, N.; Hyeon, T. Chemical synthesis and assembly of uniformly sized iron oxide nanoparticles for medical application. Acc. Chem. Res. 2015, 48, 1276–1285. [Google Scholar] [CrossRef]
- Dou, X.Y.; Chen, X.Y.; Zhu, H.G.; Liu, Y.; Chen, D.Y.; Yuan, X.; Yao, Q.F.; Xie, J.P. Water-soluble metal nanoculsters: Recent advances in molecular-level exploration and biomedical applications. Dalton Trans. 2019, 48, 10385–10392. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Agasti, S.S.; Kim, C.; Li, X.N.; Rotello, V.M. Gold Nanoparticles in Chemical and Biological Sensing. Chem. Rev. 2012, 112, 2739. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.X.; Zeng, Z.P.; Liu, B. Bridging the gap: Electron relay and plasmonic sentization of metal nanocrystals for metal clusters. J. Am. Chem. Soc. 2015, 137, 10735–10744. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Chen, R.H.; Zhang, X.M.; Chen, F.J.; Yan, J.Z.; Sun, C.F.; Ou, D.H.; Peng, J.; Lin, S.C.; Tang, Z.C.; et al. Ether-soluble Cu53 nanoclusters as an effective precursor of high-quality cul films for optoelectronic applications. Angew. Chem. Int. Ed. 2019, 58, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Die, D.; Zheng, B.X.; Kuang, X.Y.; Zhao, Z.Q.; Guo, J.H.; Du, Q. Exploration of the structural, electronic and tunable magnetic properties of Cu4M (M = Sc-Ni) Clusters. Materials 2017, 10, 946. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Li, Q.J.; Si, R.; Li, G.D.; Li, W.; Liu, D.P.; Wang, D.J.; Sun, L.; Zhang, Y.; Zou, X.X. Coupling sub-nanometric copper clusters with quasi-amorphous cobalt sulfide yields efficient and robust electrocatalysts for water splitting reaction. Adv. Mater. 2017, 29, 1606200. [Google Scholar] [CrossRef]
- Li, C.G.; Yuan, Y.Q.; Hu, Y.F.; Zhang, J.; Tang, Y.N.; Ren, B.Z. Density function theory study of the structures and electronic properties of copper and sulfur doped copper clusters. Comput. Theor. Chem. 2016, 1080, 47–55. [Google Scholar] [CrossRef]
- Liu, C.; Yang, B.; Tyo, E.; Seifert, S.; DeBartolo, J.; Issendorff, B.V.; Zapol, P.; Vajda, S.; Curtiss, L.A. Carbon dioxide conversion to methanol over size-selected Cu4 clusters at low pressures. J. Am. Chem. Soc. 2015, 137, 8676–8679. [Google Scholar] [CrossRef]
- Yang, B.; Liu, C.; Halder, A.; Tyo, E.C.; Martinson, A.B.F.; Seifert, S.; Zapol, P.; Curtiss, L.A.; Vajda, S. Copper cluster size effect in methanol synthesis from CO2. J. Phys. Chem. C 2017, 121, 10406–10412. [Google Scholar] [CrossRef]
- Delley, B.; Ellis, D.E.; Freeman, A.J.; Baerends, E.J.; Post, D. Binding energy and electronic structure of small copper particles. Phys. Rev. B 1983, 27, 2132. [Google Scholar] [CrossRef]
- Christensen, O.B.; Jacobsen, K.W.; Nørskov, J.K.; Manninen, M. Cu cluster shell structure at elevated temperatures. Phys. Rev. Lett. 1991, 66, 2219. [Google Scholar] [CrossRef] [PubMed]
- Doye, J.P.K.; Wales, D.J. Global minima for transition metal clusters described by Sutton-Chen potentials. New J. Chem. 1998, 22, 733. [Google Scholar] [CrossRef]
- Sutton-Chen Clusters. Available online: http://doye.chem.ox.ac.uk/jon/structures/SC.html (accessed on 24 November 2019).
- Luo, Z.X.; Castleman, A.W.; Khanna, S.N. Reactivity of metal clusters. Chem. Rev. 2016, 116, 14456–14492. [Google Scholar] [CrossRef] [PubMed]
- Jaque, P.; Toro-Labbé, A. Characterization of copper clusters through the use of density functional theory reactivity descriptors. J. Chem. Phys. 2002, 117, 3208. [Google Scholar] [CrossRef]
- Darby, S.; Mortimer-Jones, T.V.; Johnston, R.L.; Roberts, C. Theoretical study of Cu-Au nanoalloy clusters using a genetic algorithm. J. Chem. Phys. 2002, 116, 1536. [Google Scholar] [CrossRef]
- Tang, Q.; Lee, Y.J.; Li, D.Y.; Choi, W.J.; Lee, D.; Jiang, D.E. Lattice-hydride mechanism in electrocatalytic CO2 reduction by structurally precise copper-hydride nanoclusters. J. Am. Chem. Soc. 2017, 139, 9728–9736. [Google Scholar] [CrossRef]
- Kabir, M.; Mookerjee, A.; Bhattacharya, A.K. Structure and stability of copper clusters: A tight-binding molecular dynamics study. Phys. Rev. A 2004, 69, 043203. [Google Scholar] [CrossRef]
- Grigoryan, V.G.; Alamanova, D.; Springborg, M. Structure and energetics of CuN clusters with (2 ≤ N ≤ 150): An embedded-atom-method study. Phys. Rev. B 2006, 73, 115415. [Google Scholar] [CrossRef]
- Zhang, M.L.; Li, G.P. Energy and Structure of Copper Clusters (n = 2–70,147,500) Studied by the Monte Carlo Method. Solid State Phenom. 2007, 121, 607–610. [Google Scholar] [CrossRef]
- Pan, X.D.; Gai, Z.G.; Gong, P.L. Atomic and molecular physics: Energy and structure of copper clusters (n = 70–150) studied by the Monte Carlo computer simulation. Chin. Phys. B 2008, 17, 3329. [Google Scholar]
- Chen, M.G.; Xie, J.P.; Ye, G.X. Formation mechanism and ordered patterns in Cu films deposited on silicone oil surfaces. Phys. Lett. A 2006, 360, 323–326. [Google Scholar] [CrossRef]
- Ye, G.X.; Zhang, Q.R.; Feng, C.M.; Ge, H.L.; Jiao, Z.K. Structural and electrical properties of a metallic rough thin film system deposited on liquid substrates. Phys. Rev. B 1996, 54, 14754. [Google Scholar] [CrossRef] [PubMed]
- Wender, H.; De Oliveira, L.F.; Feil, A.F.; Lissner, E.; Migowski, P.; Meneghetti, M.R.; Teixeira, S.R.; Dupont, J. Synthesis of gold nanoparticles in a biocompatible fluid from sputtering deposition onto castor oil. Chem. Commun. 2010, 46, 7019–7021. [Google Scholar] [CrossRef]
- Yoshida, H.; Kawamoto, K.; Kubo, H.; Tsuda, T.; Fujii, A.; Kuwabata, S.; Ozaki, M. Nanoparticle-Dispersed Liquid Crystals Fabricated by Sputter Doping. Adv. Mater. 2010, 22, 622–626. [Google Scholar] [CrossRef]
- Pan, Q.F.; Cheng, Y.; Tao, X.M.; Yang, B.; Li, B.X.; Ye, G.X. Temperature Dependence of the Aggregation Behavior of Aluminum Nanoparticles on Liquid Substrate. J. Nanopart. Res. 2015, 17, 161. [Google Scholar] [CrossRef]
- Lu, C.X.; Jin, Y.; Tao, X.M.; Yang, B.; Ye, G.X. Nucleation and growth of zinc crystals on a liquid surface. CrystEngComm 2018, 20, 122–127. [Google Scholar] [CrossRef]
- ADF2010.02, SCF. Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Lenthe, E.V.; Baerends, E.J. Optimized slater-type basis sets for the elements 1–118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822. [Google Scholar] [CrossRef]
- Brandbyge, M.; Mozos, J.L.; Ordejon, P.; Taylor, J.; Stokbro, K. Density-functional method for nonequilibrium electron transport. Phys. Rev. B 2002, 65, 165401. [Google Scholar] [CrossRef]
- Taylor, J.; Guo, H.; Wang, J. Ab initio modeling of quantum transport properties of molecular electronic devices. Phys. Rev. B 2001, 63, 245407. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Kawazoe, Y. Hund’s rule in metal clusters: Prediction of high magnetic moment state of Al12Cu from first-principles calculations. Phys. Rev. B 2001, 64, 115405. [Google Scholar] [CrossRef]
- Die, D.; Zheng, B.X.; Zhao, L.Q.; Zhu, Q.W.; Zhao, Z.Q. Insights into the structural, electronic and magnetic properties of V-doped copper clusters: Comparison with pure copper clusters. Sci. Rep. 2016, 6, 31978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulbazine, M.; Boudjahem, A.G. Stability, Electronic and Magnetic Properties of Mn-Doped Copper Clusters: A Meta-GGA Functional Investigation. J. Clust. Sci. 2019, 30, 31–44. [Google Scholar] [CrossRef]
- Spasov, V.A.; Lee, T.H.; Ervin, K.M. Threshold collision-induced dissociation of anionic copper clusters and copper cluster monocarbonyls. J. Chem. Phys. 2000, 112, 1713–1720. [Google Scholar] [CrossRef]
- Tamijani, A.A.; Salam, A.; Lara-Castells, M.P. Adsorption of Noble-Gas Atoms on the TiO2(110) Surface: An Ab Initio-Assisted Study with van der Waals-Corrected DFT. J. Phys. Chem. C 2016, 120, 18126. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Berland, K.; Cooper, V.R.; Lee, K.; Schröder, E.; Thonhauser, T.; Hyldgaard, P.; Lundqvist, B.I. van der Waals forces in density functional theory: A review of the vdW-DF method. Rep. Prog. Phys. 2015, 78, 066501. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
The newly obtained ground state structure (marked with “a”) except for n = 23 and n = 28 and the original ground state structure (marked with “b”).
Figure 1.
The newly obtained ground state structure (marked with “a”) except for n = 23 and n = 28 and the original ground state structure (marked with “b”).

Figure 2.
The second order difference (Δ2E) of the binding energies for the ground state structures of Cun (n = 2–32) clusters obtained by the (a) ADF and (b) ATK software, respectively; (c) The binding energy per atom as a function of Cu clusters with different sizes.
Figure 2.
The second order difference (Δ2E) of the binding energies for the ground state structures of Cun (n = 2–32) clusters obtained by the (a) ADF and (b) ATK software, respectively; (c) The binding energy per atom as a function of Cu clusters with different sizes.

Figure 3.
The aggregation of two typical (a) Cu44, (b) Cu55 and (c) four Cu44 clusters.

Figure 4.
The top part shows the HOMO(1) and HOMO(2) corresponding to two degenerate highest occupied molecular orbitals (HOMO). The bottom part shows the LUMO(1) and LUMO(2) referring to two degenerate lowest unoccupied molecular orbitals (LUMO). The middle structure diagram is the ground state structure of Cu55 cluster, in which white balls represent equivalent atoms.
Figure 4.
The top part shows the HOMO(1) and HOMO(2) corresponding to two degenerate highest occupied molecular orbitals (HOMO). The bottom part shows the LUMO(1) and LUMO(2) referring to two degenerate lowest unoccupied molecular orbitals (LUMO). The middle structure diagram is the ground state structure of Cu55 cluster, in which white balls represent equivalent atoms.

Figure 5.
In a two-dimension system: A (a) hexagon-shaped cluster is treated as a (b) sphere-connected model.
Figure 5.
In a two-dimension system: A (a) hexagon-shaped cluster is treated as a (b) sphere-connected model.

Figure 6.
The potential well between two spheres belonging to two different clusters.

Figure 7.
Sides of the hexagons are assigned to be different in order to study their anisotropic properties.
Figure 7.
Sides of the hexagons are assigned to be different in order to study their anisotropic properties.

Figure 8.
(a,c) are the typical experimental images of the Cu atomic aggregates on silicone oil surfaces [23]. (b,d) are the simulation results obtained by the MC method.
Figure 8.
(a,c) are the typical experimental images of the Cu atomic aggregates on silicone oil surfaces [23]. (b,d) are the simulation results obtained by the MC method.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The binding energies (BE, in eV) of Cun (n =2–80) clusters obtained by the Amsterdam Density Functional (ADF) and Atomistix ToolKit (ATK) programs, respectively. The data marked with * in the upper right corner is calculated from the experimental data [19,39]. The newly revealed structures are marked by the letter “a”. The letter “b” is used to refer to the structures in the previous study. The first and second lines of the table correspond to the data calculated by ADF and ATK respectively.
Table 1.
The binding energies (BE, in eV) of Cun (n =2–80) clusters obtained by the Amsterdam Density Functional (ADF) and Atomistix ToolKit (ATK) programs, respectively. The data marked with * in the upper right corner is calculated from the experimental data [19,39]. The newly revealed structures are marked by the letter “a”. The letter “b” is used to refer to the structures in the previous study. The first and second lines of the table correspond to the data calculated by ADF and ATK respectively.
| Cun | BE | Cun | BE | Cun | BE | Cun | BE | Cun | BE |
|---|---|---|---|---|---|---|---|---|---|
| Cu2 | 2.47 | Cu16 | 35.17 | Cu29 | 68.08 | Cu45 | 111.02 | Cu63 | 160.52 |
| 2.58 | 41.79 | 81.32 | 133.22 | 193.04 | |||||
| Cu3 | 3.69 | Cu17 | 38.18 | Cu30 | 71.09 | Cu46 | 114.81 | Cu64 | 163.45 |
| 4.00 | 45.23 | 85.05 | 137.89 | 196.63 | |||||
| 3.21 * | Cu18 | 40.98 | Cu31(a) | 74.11 | Cu47 | 117.46 | Cu65 | 165.98 | |
| Cu4 | 4.88 | 48.47 | 88.58 | 141.03 | 199.82 | ||||
| 5.68 | Cu19 | 43.14 | Cu31(b) | 74.14 | Cu48 | 119.50 | Cu66 | 169.07 | |
| 5.92 * | 51.21 | 88.52 | 143.53 | 203.19 | |||||
| Cu5 | 7.89 | Cu20 | 45.89 | Cu32 | 76.82 | Cu49 | 123.33 | Cu67 | 171.52 |
| 9.00 | 54.40 | 91.83 | 148.18 | 206.57 | |||||
| 7.80 * | Cu21 | 48.26 | Cu33 | 79.67 | Cu50 | 125.15 | Cu68 | 174.76 | |
| Cu6 | 9.91 | 57.29 | 95.14 | 150.33 | 209.99 | ||||
| 11.80 | Cu22 | 50.32 | Cu34 | 82.36 | Cu51 | 127.93 | Cu69 | 176.76 | |
| 10.38 * | 59.91 | 98.26 | 154.00 | 212.87 | |||||
| Cu7 | 13.40 | Cu23(a) | 53.14 | Cu35 | 85.26 | Cu52 | 131.33 | Cu70 | 179.57 |
| 15.33 | 63.30 | 101.83 | 157.99 | 216.22 | |||||
| 13.02 * | Cu23(b) | 52.80 | Cu36 | 87.58 | Cu53 | 134.57 | Cu71 | 182.38 | |
| Cu8 | 16.06 | 63.04 | 104.81 | 161.90 | 219.77 | ||||
| 18.47 | Cu24(a) | 55.62 | Cu37 | 90.26 | Cu54 | 137.97 | Cu72 | 185.20 | |
| 16.00 * | 66.07 | 108.04 | 165.85 | 223.11 | |||||
| Cu9 | 17.89 | Cu24(b) | 55.00 | Cu38 | 93.08 | Cu55 | 141.24 | Cu73 | 187.97 |
| 20.77 | 65.63 | 111.53 | 169.80 | 226.41 | |||||
| Cu10 | 19.83 | Cu25(a) | 57.77 | Cu39 | 95.66 | Cu56 | 143.81 | Cu74 | 190.55 |
| 23.45 | 69.01 | 114.50 | 172.75 | 229.72 | |||||
| Cu11 | 22.02 | Cu25(b) | 57.35 | Cu40 | 98.53 | Cu57 | 146.41 | Cu75 | 193.34 |
| 26.05 | 68.56 | 118.01 | 175.73 | 233.20 | |||||
| Cu12 | 24.36 | Cu26(a) | 60.66 | Cu41 | 100.45 | Cu58 | 149.39 | Cu76 | 196.20 |
| 28.76 | 72.33 | 120.31 | 179.14 | 236.56 | |||||
| Cu12 | 24.38 | Cu26(b) | 59.68 | Cu42 | 104.06 | Cu59 | 150.69 | Cu77 | 199.21 |
| 28.78 | 71.64 | 124.59 | 180.85 | 240.04 | |||||
| Cu13 | 26.32 | Cu27 | 62.62 | Cu43(a) | 106.50 | Cu60 | 154.45 | Cu78 | 202.35 |
| 31.58 | 74.90 | 127.66 | 185.30 | 243.34 | |||||
| Cu14 | 29.19 | Cu28(a) | 65.83 | Cu43(b) | 104.87 | Cu61 | 157.05 | Cu79 | 204.59 |
| 34.88 | 78.41 | 127.66 | 188.65 | 246.44 | |||||
| Cu15 | 32.21 | Cu28(b) | 65.42 | Cu44 | 109.00 | Cu62 | 159.56 | Cu80 | 207.37 |
| 38.38 | 78.24 | 130.74 | 191.59 | 249.72 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mao, H.-Y.; Li, B.-X.; Ding, W.-F.; Zhu, Y.-H.; Yang, X.-X.; Li, C.-Y.; Ye, G.-X. Theoretical Study on the Aggregation of Copper Clusters on a Liquid Surface. Materials 2019, 12, 3877. https://doi.org/10.3390/ma12233877
AMA Style
Mao H-Y, Li B-X, Ding W-F, Zhu Y-H, Yang X-X, Li C-Y, Ye G-X. Theoretical Study on the Aggregation of Copper Clusters on a Liquid Surface. Materials. 2019; 12(23):3877. https://doi.org/10.3390/ma12233877
Chicago/Turabian StyleMao, Hong-Ying, Bao-Xing Li, Wang-Feng Ding, Yu-Hong Zhu, Xu-Xin Yang, Chao-Yang Li, and Gao-Xiang Ye. 2019. "Theoretical Study on the Aggregation of Copper Clusters on a Liquid Surface" Materials 12, no. 23: 3877. https://doi.org/10.3390/ma12233877
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.