
Discovery of Potential SARS-CoV-2 Papain-like Protease Natural Inhibitors Employing a Multi-Phase In Silico Approach
 ,
,  , ,
, ,  , and
, and 
Abstract
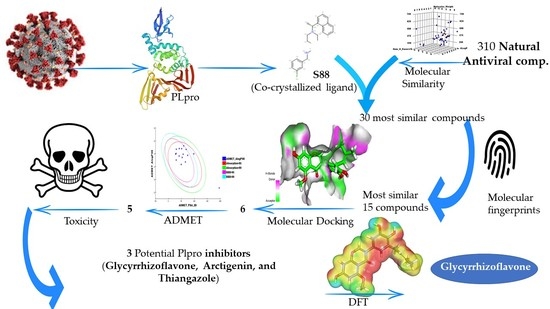
:
1. Introduction
2. Results and Discussion
2.1. Molecular Similarity
2.2. Filter Using Fingerprints
2.3. Docking Studies


2.4. ADMET
2.5. Toxicity Studies
2.6. DFT Studies
2.6.1. Frontier Molecular Orbitals Analysis
2.6.2. Molecular Electrostatic Potential Maps (MEP)
3. Conclusions
4. Method
4.1. Molecular Similarity Detection
4.2. Fingerprint Studies
4.3. Docking Studies
4.4. ADMET Analysis
4.5. Toxicity Studies
4.6. DFT Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 10 September 2021).
- Engel, T. Basic overview of chemoinformatics. J. Chem. Inf. Modeling 2006, 46, 2267–2277. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Hagler, A. Chemoinformatics and drug discovery. Molecules 2002, 7, 566–600. [Google Scholar] [CrossRef]
- Jalmakhanbetova, R.I.; Suleimen, Y.M.; Oyama, M.; Elkaeed, E.B.; Eissa, I.; Suleimen, R.N.; Metwaly, A.M.; Ishmuratova, M.Y. Isolation and In Silico Anti-COVID-19 Main Protease (Mpro) Activities of Flavonoids and a Sesquiterpene Lactone from Artemisia sublessingiana. J. Chem. 2021, 2021, 5547013. [Google Scholar] [CrossRef]
- Koshak, A.E.; Koshak, E.A. Nigella sativa L. as a potential phytotherapy for covid-19: A mini-review of in-silico studies. Curr. Ther. Res. 2020, 93, 100602. [Google Scholar] [CrossRef]
- Basu, S.; Ramaiah, S.; Anbarasu, A. In-silico strategies to combat COVID-19: A comprehensive review. Biotechnol. Genet. Eng. Rev. 2021, 37, 64–81. [Google Scholar] [CrossRef]
- Lo, Y.-C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef]
- Zhang, W.; Pei, J.; Lai, L. Computational multitarget drug design. J. Chem. Inf. Modeling 2017, 57, 403–412. [Google Scholar] [CrossRef]
- Youssef, M.I.; Zhou, Y.; Eissa, I.H.; Wang, Y.; Zhang, J.; Jiang, L.; Hu, W.; Qi, J.; Chen, Z. Tetradecyl 2,3-dihydroxybenzoate alleviates oligodendrocyte damage following chronic cerebral hypoperfusion through IGF-1 receptor. Neurochem. Int. 2020, 138, 104749. [Google Scholar] [CrossRef]
- Kairys, V.; Baranauskiene, L.; Kazlauskiene, M.; Matulis, D.; Kazlauskas, E. Binding affinity in drug design: Experimental and computational techniques. Expert Opin. Drug Discov. 2019, 14, 755–768. [Google Scholar] [CrossRef]
- Al-Warhi, T.; El Kerdawy, A.M.; Aljaeed, N.; Ismael, O.E.; Ayyad, R.R.; Eldehna, W.M.; Abdel-Aziz, H.A.; Al-Ansary, G.H. Synthesis, biological evaluation and in silico studies of certain oxindole–indole conjugates as anticancer CDK inhibitors. Molecules 2020, 25, 2031. [Google Scholar] [CrossRef]
- Sharma, A.K.; Srivastava, G.N.; Roy, A.; Sharma, V.K. ToxiM: A toxicity prediction tool for small molecules developed using machine learning and chemoinformatics approaches. Front. Pharmacol. 2017, 8, 880. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Cordeiro, M.J.D.S. Simultaneous virtual prediction of anti-Escherichia coli activities and ADMET profiles: A chemoinformatic complementary approach for high-throughput screening. ACS Comb. Sci. 2014, 16, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Conceptual DFT as a chemoinformatics tool for the study of the Taltobulin anticancer peptide. BMC Res. Notes 2019, 12, 442. [Google Scholar] [CrossRef] [PubMed]
- Metwaly, A.M.; Ghoneim, M.M.; Eissa, I.H.; Elsehemy, I.A.; Mostafa, A.E.; Hegazy, M.M.; Afifi, W.M.; Dou, D. Traditional ancient Egyptian medicine: A review. Saudi J. Biol. Sci. 2021, 28, 5823–5832. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, Y.; Metwaly, A.M.; Xue, Y.; Shi, Y.; Dou, D. The Chinese herbal formulae (Yitangkang) exerts an antidiabetic effect through the regulation of substance metabolism and energy metabolism in type 2 diabetic rats. J. Ethnopharmacol. 2019, 239, 111942. [Google Scholar] [CrossRef]
- Ghildiyal, R.; Prakash, V.; Chaudhary, V.; Gupta, V.; Gabrani, R. Phytochemicals as antiviral agents: Recent updates. In Plant-Derived Bioactives; Springer: Berlin/Heidelberg, Germany, 2020; pp. 279–295. [Google Scholar]
- El Sayed, K.A. Natural products as antiviral agents. Stud. Nat. Prod. Chem. 2000, 24, 473–572. [Google Scholar]
- Uzair, B.; Mahmood, Z.; Tabassum, S. Antiviral activity of natural products extracted from marine organisms. BioImpacts 2011, 1, 203. [Google Scholar]
- Owen, L.; Laird, K.; Shivkumar, M. Antiviral plant-derived natural products to combat RNA viruses: Targets throughout the viral life cycle. Lett. Appl. Microbiol. 2021. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Báez-Santos, Y.M.; John, S.E.S.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef]
- Alesawy, M.S.; Abdallah, A.E.; Taghour, M.S.; Elkaeed, E.B.; Eissa, I.H.; Metwaly, A.M. In Silico Studies of Some Isoflavonoids as Potential Candidates against COVID-19 Targeting Human ACE2 (hACE2) and Viral Main Protease (Mpro). Molecules 2021, 26, 2806. [Google Scholar] [CrossRef]
- El-Demerdash, A.; Metwaly, A.M.; Hassan, A.; El-Aziz, A.; Mohamed, T.; Elkaeed, E.B.; Eissa, I.H.; Arafa, R.K.; Stockand, J.D. Comprehensive virtual screening of the antiviral potentialities of marine polycyclic guanidine alkaloids against SARS-CoV-2 (COVID-19). Biomolecules 2021, 11, 460. [Google Scholar] [CrossRef] [PubMed]
- Eissa, I.H.; Khalifa, M.M.; Elkaeed, E.B.; Hafez, E.E.; Alsfouk, A.A.; Metwaly, A.M. In Silico Exploration of Potential Natural Inhibitors against SARS-Cov-2 nsp10. Molecules 2021, 26, 6151. [Google Scholar] [CrossRef] [PubMed]
- Alesawy, M.S.; Elkaeed, E.B.; Alsfouk, A.A.; Metwaly, A.M.; Eissa, I. In Silico Screening of Semi-Synthesized Compounds as Potential Inhibitors for SARS-CoV-2 Papain-Like Protease: Pharmacophoric Features, Molecular Docking, ADMET, Toxicity and DFT Studies. Molecules 2021, 26, 6593. [Google Scholar] [CrossRef] [PubMed]
- Eissa, I.H.; Alesawy, M.S.; Saleh, A.M.; Elkaeed, E.B.; Alsfouk, B.A.; El-Attar, A.-A.M.; Metwaly, A.M. Ligand and structure-based in silico determination of the most promising SARS-CoV-2 nsp16-nsp10 2′-o-Methyltransferase complex inhibitors among 3009 FDA approved drugs. Molecules 2022, 27, 2287. [Google Scholar] [CrossRef] [PubMed]
- Elkaeed, E.B.; Eissa, I.H.; Elkady, H.; Abdelalim, A.; Alqaisi, A.M.; Alsfouk, A.A.; Elwan, A.; Metwaly, A.M. A Multistage In Silico Study of Natural Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Int. J. Mol. Sci. 2022, 23, 8407. [Google Scholar] [CrossRef] [PubMed]
- Elkaeed, E.B.; Youssef, F.S.; Eissa, I.H.; Elkady, H.; Alsfouk, A.A.; Ashour, M.L.; El Hassab, M.A.; Abou-Seri, S.M.; Metwaly, A.M. Multi-step in silico discovery of natural drugs against COVID-19 targeting main protease. Int. J. Mol. Sci. 2022, 23, 6912. [Google Scholar] [CrossRef]
- Elkaeed, E.B.; Elkady, H.; Belal, A.; Alsfouk, B.A.; Ibrahim, T.H.; Abdelmoaty, M.; Arafa, R.K.; Metwaly, A.M.; Eissa, I.H. Multi-Phase In Silico Discovery of Potential SARS-CoV-2 RNA-Dependent RNA Polymerase Inhibitors among 3009 Clinical and FDA-Approved Related Drugs. Processes 2022, 10, 530. [Google Scholar] [CrossRef]
- Altamash, T.; Amhamed, A.; Aparicio, S.; Atilhan, M. Effect of hydrogen bond donors and acceptors on CO2 absorption by deep eutectic solvents. Processes 2020, 8, 1533. [Google Scholar]
- Wan, Y.; Tian, Y.; Wang, W.; Gu, S.; Ju, X.; Liu, G. In silico studies of diarylpyridine derivatives as novel HIV-1 NNRTIs using docking-based 3D-QSAR, molecular dynamics, and pharmacophore modeling approaches. RSC Adv. 2018, 8, 40529–40543. [Google Scholar] [CrossRef]
- Turchi, M.; Cai, Q.; Lian, G. An evaluation of in-silico methods for predicting solute partition in multiphase complex fluids–A case study of octanol/water partition coefficient. Chem. Eng. Sci. 2019, 197, 150–158. [Google Scholar] [CrossRef]
- Sullivan, K.M.; Enoch, S.J.; Ezendam, J.; Sewald, K.; Roggen, E.L.; Cochrane, S. An adverse outcome pathway for sensitization of the respiratory tract by low-molecular-weight chemicals: Building evidence to support the utility of in vitro and in silico methods in a regulatory context. Appl. Vitr. Toxicol. 2017, 3, 213–226. [Google Scholar] [CrossRef]
- Escamilla-Gutiérrez, A.; Ribas-Aparicio, R.M.; Córdova-Espinoza, M.G.; Castelán-Vega, J.A. In silico strategies for modeling RNA aptamers and predicting binding sites of their molecular targets. Nucleosides Nucleotides Nucleic Acids 2021, 40, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.C.; Kumar, A.; Bharadwaj, S.; Chaudhary, R.; Sahi, S. Ligand-Based Approach for In-silico Drug Designing. In Bioinformatics Techniques for Drug Discovery; Springer: Berlin/Heidelberg, Germany, 2018; pp. 11–19. [Google Scholar]
- Zhang, H.; Ren, J.-X.; Ma, J.-X.; Ding, L. Development of an in silico prediction model for chemical-induced urinary tract toxicity by using naïve Bayes classifier. Mol. Divers. 2019, 23, 381–392. [Google Scholar] [CrossRef]
- Burke, B.J. Developments in Molecular Shape Analysis to Establish Spatial Similarity among Flexible Molecules. Ph.D. Thesis, University of Illinois at Chicago, Health Sciences Center, Chicago, IL, USA, 1993. [Google Scholar]
- Briem, H.; Kuntz, I.D. Molecular similarity based on DOCK-generated fingerprints. J. Med. Chem. 1996, 39, 3401–3408. [Google Scholar] [CrossRef]
- Chu, H.; He, Q.-X.; Wang, J.; Hu, Y.; Wang, Y.-Q.; Lin, Z.-H. In silico design of novel benzohydroxamate-based compounds as inhibitors of histone deacetylase 6 based on 3D-QSAR, molecular docking, and molecular dynamics simulations. New J. Chem. 2020, 44, 21201–21210. [Google Scholar] [CrossRef]
- Ieritano, C.; Campbell, J.L.; Hopkins, W.S. Predicting differential ion mobility behaviour in silico using machine learning. Analyst 2021, 146, 4737–4743. [Google Scholar] [CrossRef]
- Taha, M.; Ismail, N.H.; Ali, M.; Rashid, U.; Imran, S.; Uddin, N.; Khan, K.M. Molecular hybridization conceded exceptionally potent quinolinyl-oxadiazole hybrids through phenyl linked thiosemicarbazide antileishmanial scaffolds: In silico validation and SAR studies. Bioorganic Chem. 2017, 71, 192–200. [Google Scholar] [CrossRef]
- Opo, F.A.D.M.; Rahman, M.M.; Ahammad, F.; Ahmed, I.; Bhuiyan, M.A.; Asiri, A.M. Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein. Sci. Rep. 2021, 11, 4049. [Google Scholar] [CrossRef]
- Wang, J.; Morin, P.; Wang, W.; Kollman, P.A. Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J. Am. Chem. Soc. 2001, 123, 5221–5230. [Google Scholar] [CrossRef] [PubMed]
- Houston, D.R.; Walkinshaw, M.D. Consensus docking: Improving the reliability of docking in a virtual screening context. J. Chem. Inf. Modeling 2013, 53, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.; Cox, M. G protein-coupled receptors and second messengers. In Lehninger Principles of Biochemistry, 5th ed.; WH Freeman and Company: New York, NY, USA, 2008; pp. 423–439. [Google Scholar]
- Malau, N.D.; Azzahra, S.F. Molecular Docking Studies of Potential Quercetin 3,4′-dimethyl ether 7-alpha-LArabinofuranosyl-(1-6)-glucoside as Inhibitor antimalaria. In Journal of Physics: Conference Series; IOP Publishing: Bristol, UK, 2020; p. 012057. [Google Scholar]
- Patel, H.; Dhangar, K.; Sonawane, Y.; Surana, S.; Karpoormath, R.; Thapliyal, N.; Shaikh, M.; Noolvi, M.; Jagtap, R. In search of selective 11β-HSD type 1 inhibitors without nephrotoxicity: An approach to resolve the metabolic syndrome by virtual based screening. Arab. J. Chem. 2018, 11, 221–232. [Google Scholar] [CrossRef]
- Mannhold, R.; Kubinyi, H.; Folkers, G. Pharmacokinetics and Metabolism in Drug Design; John Wiley & Sons: Hoboken, NJ, USA, 2012; Volume 51. [Google Scholar]
- Klopman, G.; Stefan, L.R.; Saiakhov, R.D. ADME evaluation: 2. A computer model for the prediction of intestinal absorption in humans. Eur. J. Pharm. Sci. 2002, 17, 253–263. [Google Scholar] [CrossRef]
- Roy, P.P.; Roy, K. QSAR studies of CYP2D6 inhibitor aryloxypropanolamines using 2D and 3D descriptors. Chem. Biol. Drug Des. 2009, 73, 442–455. [Google Scholar] [CrossRef]
- Ghafourian, T.; Amin, Z. QSAR models for the prediction of plasma protein binding. BioImpacts BI 2013, 3, 21. [Google Scholar]
- Xia, X.; Maliski, E.G.; Gallant, P.; Rogers, D. Classification of kinase inhibitors using a Bayesian model. J. Med. Chem. 2004, 47, 4463–4470. [Google Scholar] [CrossRef]
- BIOVIA. QSAR, ADMET and Predictive Toxicology. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/qsar-admet-and-predictive-toxicology.html (accessed on 1 May 2020).
- Venkatapathy, R.; Wang, N.C.Y.; Martin, T.M.; Harten, P.F.; Young, D. Structure–Activity Relationships for Carcinogenic Potential. Gen. Appl. Syst. Toxicol. 2009. [Google Scholar] [CrossRef]
- Goodrnan, G.; Wilson, R. Comparison of the dependence of the TD50 on maximum tolerated dose for mutagens and nonmutagens. Risk Anal. 1992, 12, 525–533. [Google Scholar] [CrossRef]
- Council, N.R. Correlation between Carcinogenic Potency and the Maximum Tolerated Dose: Implications for Risk Assessment. In Issues in Risk Assessment; National Academies Press (US): Washington, DC, USA, 1993. [Google Scholar]
- Gonella Diaza, R.; Manganelli, S.; Esposito, A.; Roncaglioni, A.; Manganaro, A.; Benfenati, E. Comparison of in silico tools for evaluating rat oral acute toxicity. SAR QSAR Environ. Res. 2015, 26, 1–27. [Google Scholar] [CrossRef]
- Pizzo, F.; Benfenati, E. In silico models for repeated-dose toxicity (RDT): Prediction of the no observed adverse effect level (NOAEL) and lowest observed adverse effect level (LOAEL) for drugs. In In Silico Methods for Predicting Drug Toxicity; Springer: Berlin/Heidelberg, Germany, 2016; pp. 163–176. [Google Scholar]
- Venkatapathy, R.; Moudgal, C.J.; Bruce, R.M. Assessment of the oral rat chronic lowest observed adverse effect level model in TOPKAT, a QSAR software package for toxicity prediction. J. Chem. Inf. Comput. Sci. 2004, 44, 1623–1629. [Google Scholar] [CrossRef]
- Wilhelmus, K.R. The Draize eye test. Surv. Ophthalmol. 2001, 45, 493–515. [Google Scholar] [CrossRef]
- Abdallah, A.E.; Alesawy, M.S.; Eissa, S.I.; El-Fakharany, E.M.; Kalaba, M.H.; Sharaf, M.H.; Abo Shama, N.M.; Mahmoud, S.H.; Mostafa, A.; Al-Karmalawy, A.A.; et al. Design and synthesis of new 4-(2-nitrophenoxy)benzamide derivatives as potential antiviral agents: Molecular modeling and in vitro antiviral screening. New J. Chem. 2021, 45, 16557–16571. [Google Scholar] [CrossRef]
- Subashchandrabose, S.; Saleem, H.; Erdogdu, Y.; Rajarajan, G.; Thanikachalam, V. FT-Raman, FT-IR spectra and total energy distribution of 3-pentyl-2,6-diphenylpiperidin-4-one: DFT method. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 82, 260–269. [Google Scholar] [CrossRef]
- Bazeera, A.Z.; Selvaraj, S.; Mohamed, A.S. Spectroscopic analysis (Raman, FT-IR, UV, NMR), HUMO, LUMO and first order hyper polarizability calculations of Nor Leucine Maleate (DLNM) using DFT methods. Wutan Huatan Jisuan Jishu 2020, 16, 266–277. [Google Scholar]
- Mohammed, H.S.; Tripathi, V.D.; Darghouth, A.A. Synthesis, Characterization, DFT calculation and Antimicrobial Activity of Co (II) and Cu (II) complexes with azo dye. In Journal of Physics: Conference Series; IOP Publishing: Bristol, UK, 2019; p. 052051. [Google Scholar]
- Fleming, I. Frontier Orbitals and Organic Chemical Reactions; Wiley: New York, NY, USA, 1977. [Google Scholar]
- El-Nahass, M.; Kamel, M.; El-Deeb, A.; Atta, A.; Huthaily, S. Ab initio HF, DFT and experimental (FT-IR) investigation of vibrational spectroscopy of PN, N-dimethylaminobenzylidenemalononitrile (DBM). Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 79, 443–450. [Google Scholar] [CrossRef]
- Parambil, S.H.K.; Parambil, H.A.T.; Hamza, S.P.; Parameswaran, A.T.; Thayyil, M.S.; Karuvanthodi, M. DFT and Molecular Docking Studies of a Set of Non-Steroidal Anti-Inflammatory Drugs: Propionic Acid Derivatives. In Density Functional Theory Calculations; IntechOpen: London, UK, 2020. [Google Scholar]
- Discovery Studio. Accelrys, BIOVIA: San Diego, CA, USA, 2008.
- Pegu, D.; Deb, J.; Van Alsenoy, C.; Sarkar, U. Theoretical investigation of electronic, vibrational, and nonlinear optical properties of 4-fluoro-4-hydroxybenzophenone. Spectrosc. Lett. 2017, 50, 232–243. [Google Scholar] [CrossRef]
- Matin, M.M.; Hasan, M.S.; Uzzaman, M.; Bhuiyan, M.M.H.; Kibria, S.M.; Hossain, M.E.; Roshid, M.H. Synthesis, spectroscopic characterization, molecular docking, and ADMET studies of mannopyranoside esters as antimicrobial agents. J. Mol. Struct. 2020, 1222, 128821. [Google Scholar] [CrossRef]
- Hatano, T.; Eerdunbayaer, C.; Kuroda, T.; Shimozu, Y. Licorice as a resource for pharmacologically active phenolic substances: Antioxidant and antimicrobial effects. In Biological Activities and Action Mechanisms of Licorice Ingredients; InTech: Rijeka, Croatia, 2017; pp. 59–75. [Google Scholar]
- Uchiumi, F.; Hatano, T.; Ito, H.; Yoshida, T.; Tanuma, S.-I. Transcriptional suppression of the HIV promoter by natural compounds. Antivir. Res. 2003, 58, 89–98. [Google Scholar] [CrossRef]
- Vlietinck, A.; De Bruyne, T.; Apers, S.; Pieters, L. Plant-derived leading compounds for chemotherapy of human immunodeficiency virus (HIV) infection. Planta Med. 1998, 64, 97–109. [Google Scholar] [CrossRef] [Green Version]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Molecular Formula | ALog p | M. Wt | HBA | HBD | Rotatable Bonds | Rings | Aromatic Rings | MFPSA | Minimum Distance |
|---|---|---|---|---|---|---|---|---|---|---|
| 4 | C24H27NO4 | 2.658 | 394.483 | 4 | 1 | 4 | 5 | 3 | 0.102 | 0.654 |
| 28 | C17H19N3 | 1.457 | 266.361 | 1 | 2 | 1 | 4 | 3 | 0.119 | 0.693 |
| 41 | C24H27NO3 | 3.131 | 378.484 | 3 | 1 | 3 | 5 | 3 | 0.083 | 0.546 |
| 46 | C20H20NO4 | 3.936 | 338.377 | 4 | 1 | 3 | 4 | 3 | 0.149 | 0.709 |
| 47 | C21H22NO4 | 4.161 | 352.404 | 4 | 0 | 4 | 4 | 3 | 0.11 | 0.714 |
| 76 | C21H20O6 | 3.98 | 368.38 | 6 | 3 | 4 | 3 | 2 | 0.257 | 1.101 |
| 87 | C21H22O4 | 4.667 | 338.397 | 4 | 2 | 6 | 2 | 2 | 0.178 | 1.102 |
| 94 | C21H24O6 | 3.743 | 372.412 | 6 | 1 | 7 | 3 | 2 | 0.192 | 1.057 |
| 98 | C19H20O3 | 4.784 | 296.36 | 3 | 2 | 6 | 2 | 2 | 0.153 | 1.100 |
| 99 | C18H18O2 | 4.8 | 266.334 | 2 | 2 | 5 | 2 | 2 | 0.14 | 1.108 |
| 101 | C22H18O7 | 3.584 | 394.374 | 7 | 0 | 4 | 5 | 3 | 0.192 | 0.356 |
| 111 | C23H30O5 | 4.65 | 386.481 | 5 | 2 | 4 | 5 | 1 | 0.209 | 0.486 |
| 127 | C14H12O4 | 2.466 | 244.243 | 4 | 1 | 1 | 3 | 2 | 0.235 | 0.539 |
| 146 | C21H30O2 | 6.109 | 314.462 | 2 | 1 | 4 | 3 | 1 | 0.084 | 0.493 |
| 147 | C27H34O5 | 3.325 | 437.548 | 5 | 0 | 4 | 5 | 1 | 0.182 | 0.412 |
| 188 | C14H16BrN3OS | 1.287 | 355.273 | 3 | 2 | 0 | 4 | 2 | 0.282 | 0.789 |
| 189 | C14H16BrN3OS | 1.287 | 355.273 | 3 | 2 | 0 | 4 | 2 | 0.282 | 0.789 |
| 192 | C21H18BrN3O | 3.919 | 408.291 | 3 | 2 | 3 | 5 | 3 | 0.168 | 0.418 |
| 193 | C21H20N4S | 2.122 | 361.483 | 3 | 1 | 3 | 5 | 4 | 0.167 | 0.509 |
| 200 | C15H8N2O2 | 2.331 | 248.236 | 3 | 0 | 0 | 4 | 2 | 0.222 | 0.670 |
| 211 | C21H17N3O | 4.078 | 327.379 | 2 | 1 | 1 | 5 | 3 | 0.176 | 0.529 |
| 215 | C12H9ClN2 | 3.043 | 216.666 | 1 | 0 | 0 | 3 | 3 | 0.084 | 0.582 |
| 216 | C12H8Cl2N2 | 3.707 | 251.111 | 1 | 0 | 0 | 3 | 3 | 0.076 | 0.558 |
| 217 | C12H8Cl2N2O | 2.846 | 267.111 | 1 | 1 | 0 | 3 | 2 | 0.142 | 0.578 |
| 227 | C22H32O3 | 5.507 | 344.488 | 3 | 1 | 1 | 4 | 1 | 0.101 | 0.798 |
| 287 | C29H37NO5 | 4.1 | 479.608 | 5 | 3 | 2 | 4 | 1 | 0.196 | 0.679 |
| 291 | C15H19NO2 | 2.932 | 245.317 | 2 | 2 | 5 | 2 | 2 | 0.198 | 0.524 |
| 298 | C18H21N3O2S | 2.716 | 343.443 | 4 | 1 | 5 | 3 | 2 | 0.252 | 0.600 |
| 300 | C23H25NO4 | 5.22 | 379.449 | 4 | 0 | 7 | 3 | 2 | 0.149 | 0.473 |
| 303 | C19H26ClNO3 | 3.006 | 350.86 | 4 | 1 | 6 | 2 | 1 | 0.158 | 0.650 |
| S88 | C25H27FN2O | 3.098 | 391.501 | 1 | 2 | 5 | 4 | 3 | 0.083 |
| Comp. | Similarity | SA | SB | SC |
|---|---|---|---|---|
| S88 | 1.000 | 565 | 0 | 0 |
| Brevicollin (28) | 0.614 | 304 | −70 | 261 |
| Cryptopleurine (41) | 0.642 | 401 | 60 | 164 |
| Columbamine (46) | 0.605 | 353 | 18 | 212 |
| Palmatine (47) | 0.584 | 363 | 57 | 202 |
| Glycyrrhizoflavone (76) | 0.561 | 329 | 21 | 236 |
| Licochalcone A (87) | 0.645 | 354 | −16 | 211 |
| Arctigenin (94) | 0.591 | 355 | 36 | 210 |
| Termilignan (98) | 0.635 | 343 | −25 | 222 |
| Anolignan B (99) | 0.615 | 346 | −2 | 219 |
| 4,5-dihydroxy-6″-deoxybromotopsentin (192) | 0.720 | 394 | −18 | 171 |
| Dercitin (193) | 0.621 | 357 | 10 | 208 |
| Tryptanthrin (200) | 0.633 | 337 | −33 | 228 |
| 6-Cyano-5-methoxy-12-methylindolo [2, 3A] carbazole (211) | 0.594 | 329 | −11 | 236 |
| Thiangazole (298) | 0.580 | 307 | −36 | 258 |
| Phenoxan (300) | 0.574 | 354 | 52 | 211 |
| Comp. | ΔG [Kcal/mol] | Comp. | ΔG [Kcal/mol] |
|---|---|---|---|
| 28 | −40.44 | 99 | −39.43 |
| 41 | −47.34 | 192 | −30.85 |
| 46 | −44.13 | 193 | −44.02 |
| 47 | −46.06 | 200 | −41.31 |
| 76 | −51.63 | 211 | −37.33 |
| 87 | −35.48 | 298 | −48.46 |
| 94 | −50.82 | 300 | −33.61 |
| 98 | −52.21 | S88 | −59.13 |
| Comp. | BBB a | HIA b | Aq c | CYP2D6 d | Hepatotoxicity Probability e | PPB f |
|---|---|---|---|---|---|---|
| 28 | c | a | d | n | 0.298 | c |
| 41 | b | a | c | i | 0.39 | b |
| 46 | b | a | c | i | 0.907 | a |
| 47 | b | a | c | i | 0.966 | c |
| 76 | e | a | c | i | 0.894 | b |
| 87 | b | a | c | n | 0.735 | b |
| 94 | c | a | c | n | 0.774 | c |
| 98 | b | a | c | n | 0.834 | c |
| 99 | b | a | c | n | 0.847 | c |
| 192 | b | a | c | n | 0.152 | c |
| 193 | b | a | c | i | 0.814 | c |
| 200 | c | a | c | n | 0.98 | c |
| 211 | b | a | b | i | 0.874 | c |
| 298 | c | a | c | n | 0.549 | c |
| 300 | b | a | c | n | 0.622 | c |
| Remdesivir | e | d | d | n | 1.777 | b |
| Comp. | FDA * Rat Carcinogenicity | TD50 (Rat) mg/kg Body Weight/Day | MTD * | LD50 * | LOAEL * | Ocular Irritancy *** | Skin Irritancy *** |
|---|---|---|---|---|---|---|---|
| 28 | s | 9.571 | 0.050 | 0.939 | 0.077 | m | m |
| 41 | n | 0.219 | 0.042 | 0.202 | 0.018 | m | n |
| 46 | n | 0.730 | 0.081 | 1.248 | 0.009 | m | n |
| 47 | n | 0.169 | 0.035 | 1.446 | 0.008 | m | n |
| 76 | n | 19.216 | 0.153 | 0.362 | 0.150 | m | n |
| 87 | n | 48.173 | 0.113 | 0.364 | 0.030 | n | n |
| 94 | n | 8.907 | 0.091 | 9.209 | 0.107 | m | m |
| 98 | n | 35.370 | 0.103 | 1.133 | 0.398 | n | n |
| 99 | m | 69.077 | 0.240 | 2.040 | 0.301 | m | n |
| 192 | n | 0.857 | 1.099 | 0.348 | 0.016 | m | n |
| 193 | s | 1.587 | 0.012 | 0.352 | 0.048 | m | m |
| 200 | s | 7.568 | 0.055 | 0.689 | 0.277 | m | n |
| 211 | s | 0.604 | 0.013 | 0.245 | 0.001 | m | m |
| 298 | n | 65.542 | 0.018 | 0.118 | 0.019 | m | n |
| 300 | s | 13.502 | 0.029 | 0.405 | 0.029 | n | m |
| Remdesivir | n | 1.012 | 0.235 | 0.309 | 0.003 | m | m |
| Comp. | Total Energy (Ha) | Binding Energy (Ha) | HOMO Energy (Ha) | LUMO Energy (Ha) | Dipole Mag | Band Gap Energy (Ha) |
|---|---|---|---|---|---|---|
| 76 | −1252.956 | −9.601 | −0.166 | −0.070 | 1.700 | 0.096 |
| 94 | −1255.298 | −10.037 | −0.177 | −0.036 | 3.582 | 0.141 |
| 298 | −1401.286 | −8.702 | −0.195 | −0.064 | 1.094 | 0.131 |
| S88 | −1242.952 | −11.181 | −0.292 | −0.192 | 3.621 | 0.101 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elkaeed, E.B.; Metwaly, A.M.; Alesawy, M.S.; Saleh, A.M.; Alsfouk, A.A.; Eissa, I.H. Discovery of Potential SARS-CoV-2 Papain-like Protease Natural Inhibitors Employing a Multi-Phase In Silico Approach. Life 2022, 12, 1407. https://doi.org/10.3390/life12091407
Elkaeed EB, Metwaly AM, Alesawy MS, Saleh AM, Alsfouk AA, Eissa IH. Discovery of Potential SARS-CoV-2 Papain-like Protease Natural Inhibitors Employing a Multi-Phase In Silico Approach. Life. 2022; 12(9):1407. https://doi.org/10.3390/life12091407
Chicago/Turabian StyleElkaeed, Eslam B., Ahmed M. Metwaly, Mohamed S. Alesawy, Abdulrahman M. Saleh, Aisha A. Alsfouk, and Ibrahim H. Eissa. 2022. "Discovery of Potential SARS-CoV-2 Papain-like Protease Natural Inhibitors Employing a Multi-Phase In Silico Approach" Life 12, no. 9: 1407. https://doi.org/10.3390/life12091407