
Genome Organization and Comparative Evolutionary Mitochondriomics of Brown Planthopper, Nilaparvata lugens Biotype 4 Using Next Generation Sequencing (NGS)

 ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mitogenome Sequencing and Assembly
2.2. N. lugens Mitogenome Annotation
2.3. Phylogenetic Analysis
3. Results and Discussion
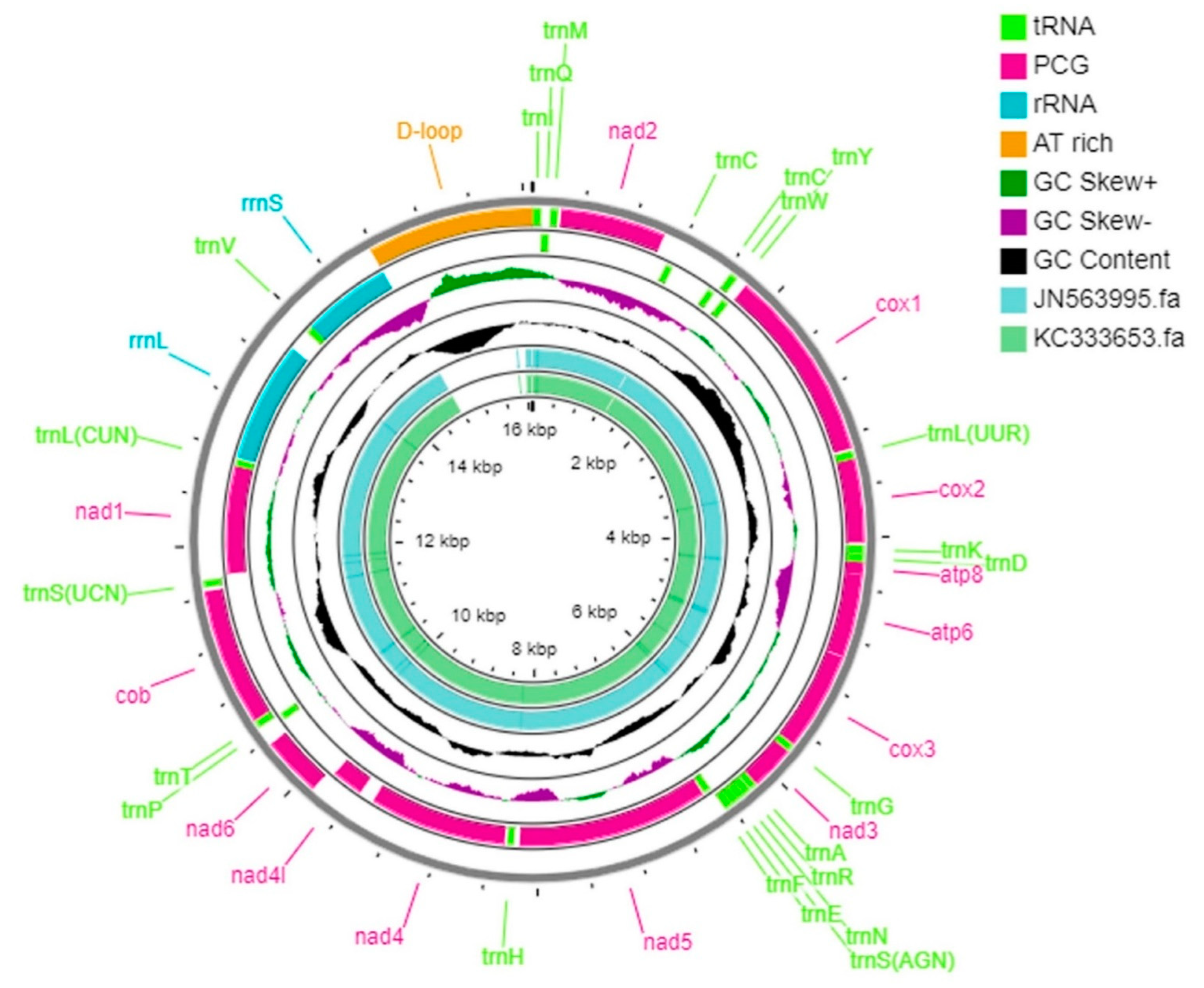
3.1. Sequencing and Assembly of N. lugens Mitogenome
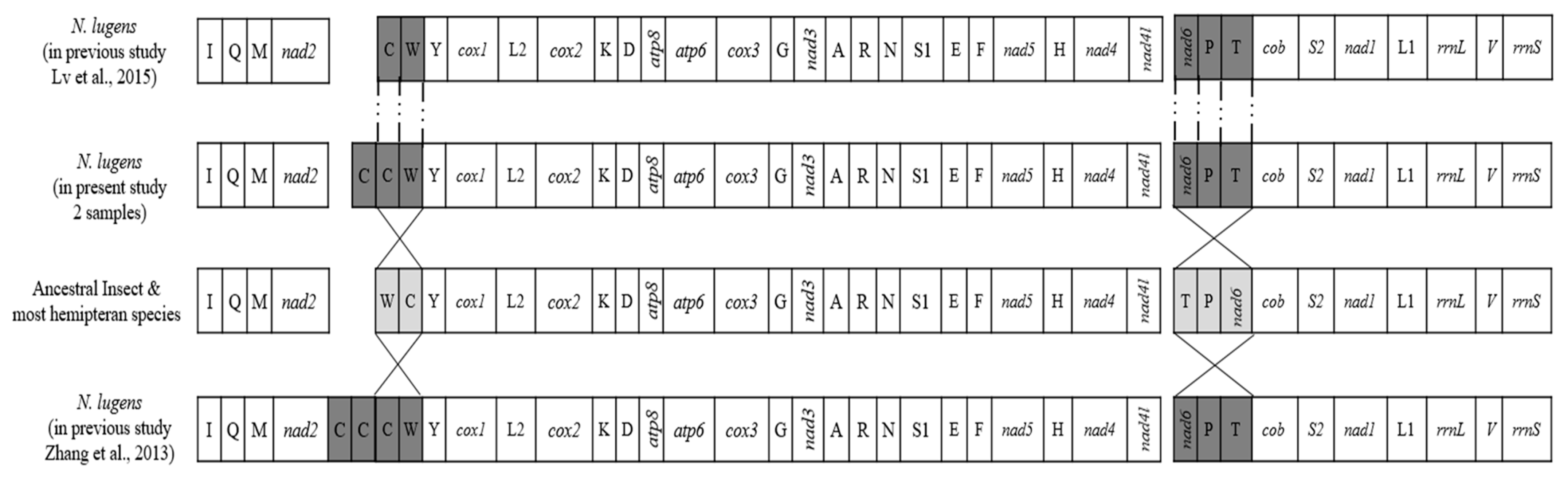
3.2. N. lugens Genome Organization and Rearrangement
3.3. N. lugens Nucleotide Composition and Skewness
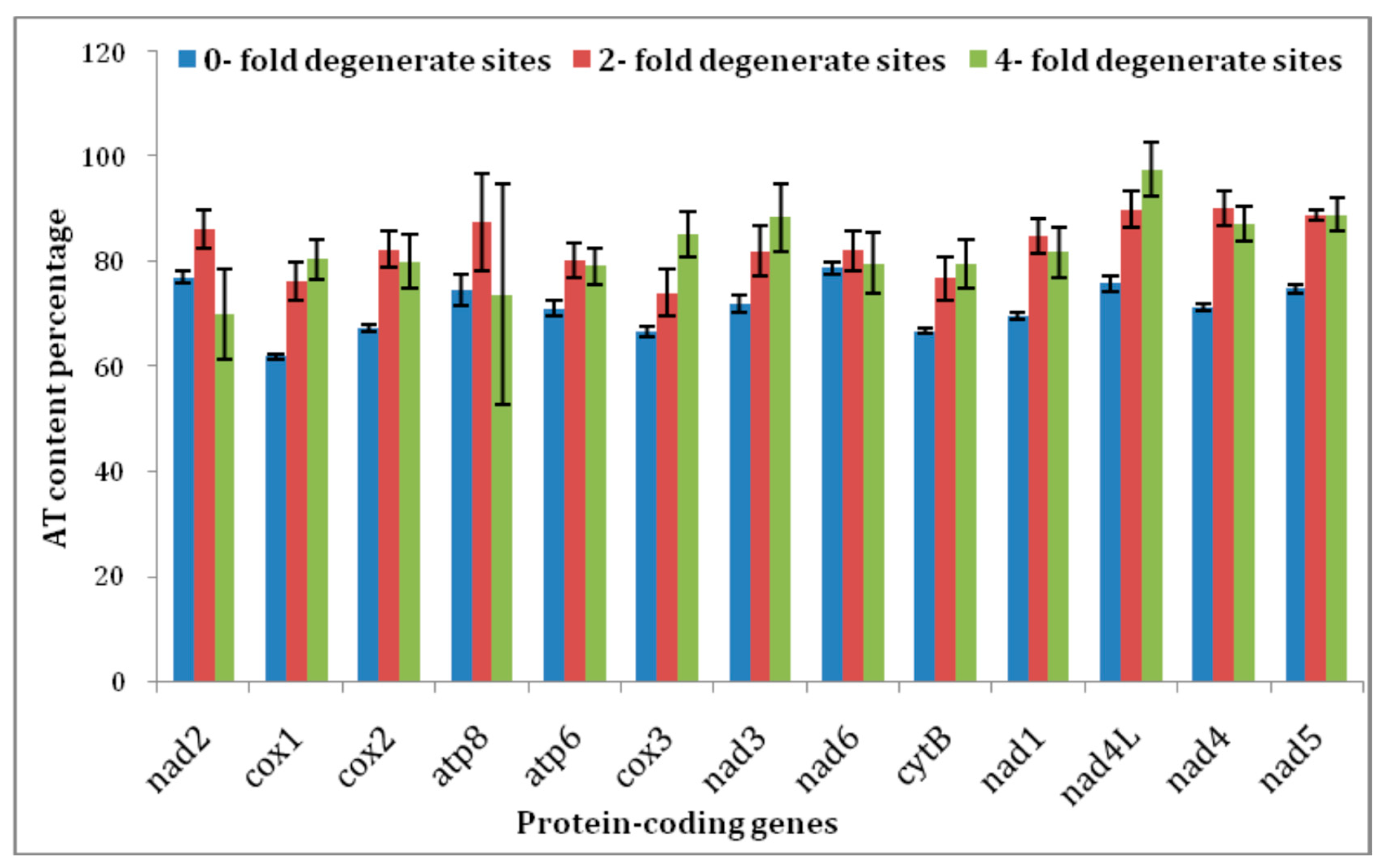
3.4. Protein-Coding Genes
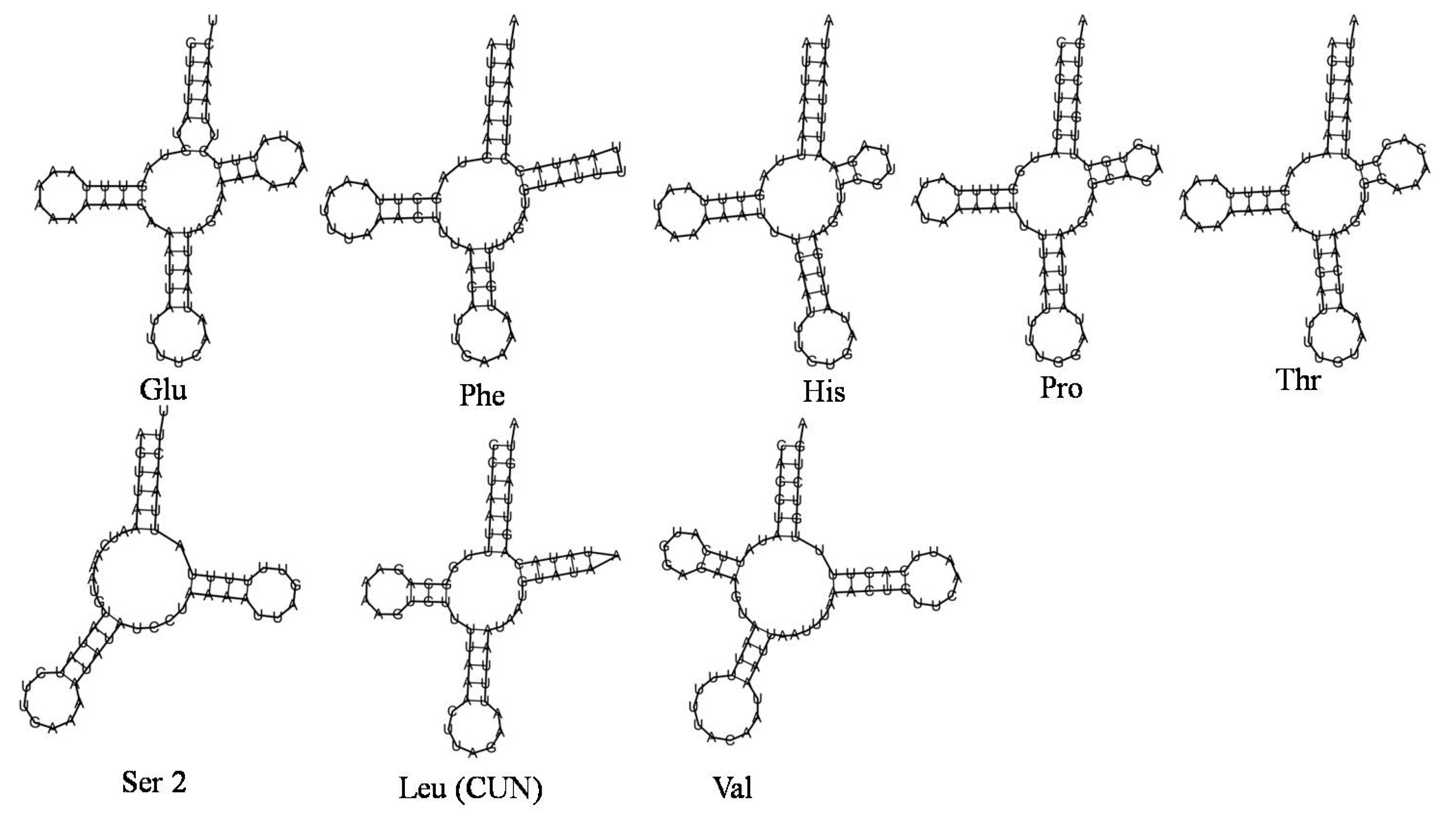
3.5. N. lugens Ribosomal RNA Genes, Transfer RNA Genes and Associated tRNA Structure
3.6. N. lugens Intergenic Spacers and Control Region
3.7. Nucleotide Diversity and Gene Evolutionary Rate
3.8. Base Composition and AT/GC-Skew of Mitogenome of Delphacidae
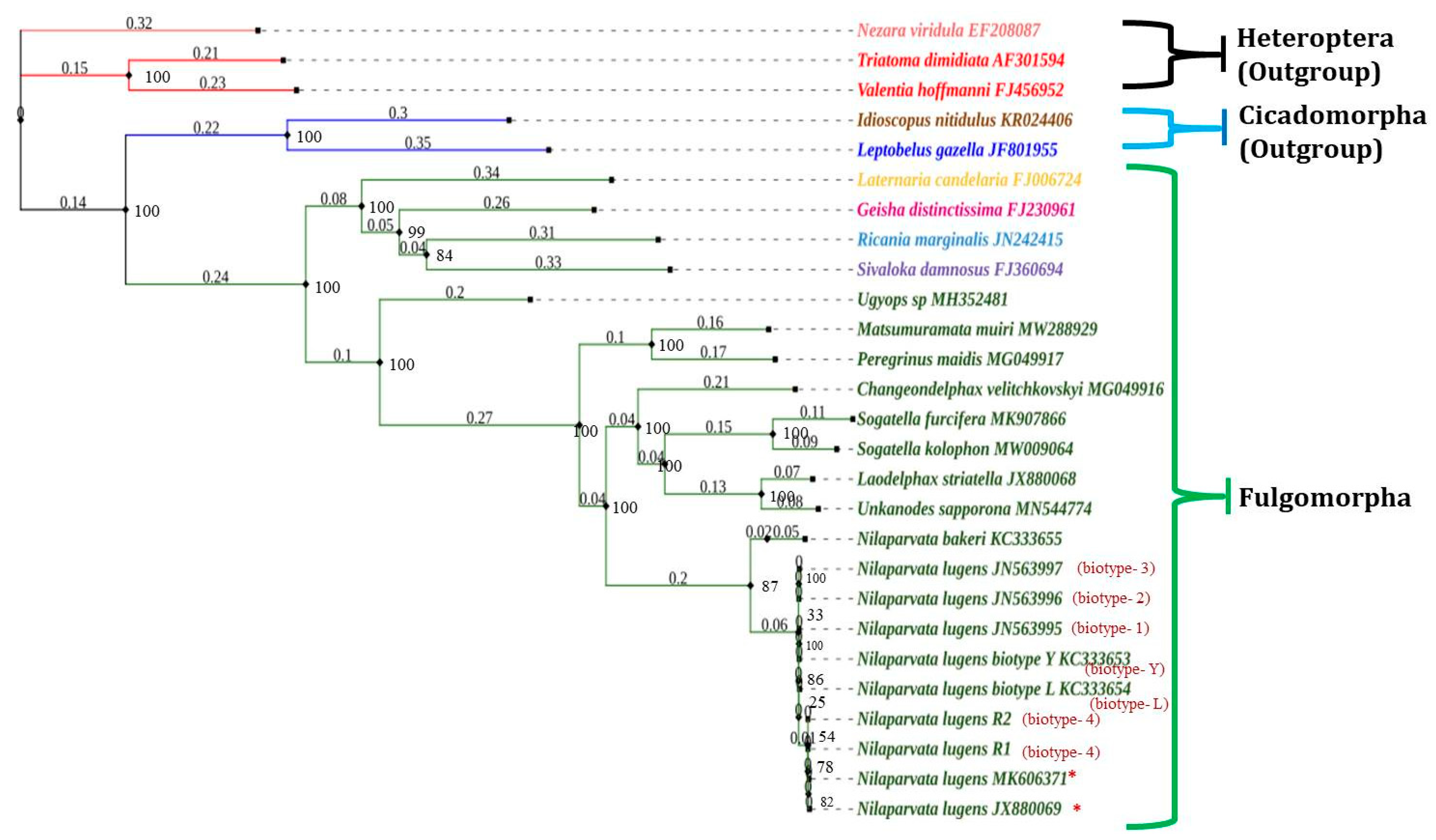
3.9. Phylogenetic Relationship
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pandi, G.; Chander, S.; Singh, M.P. Impact of elevated CO2 on rice brown planthopper Nilaparvata lugens (Stal.). Indian J. Entomol. 2017, 79, 82–85. [Google Scholar] [CrossRef]
- Jena, M.; Govindharaj, G.-P.-P.; Adak, T.; Rath, P.; Gowda G, B.; Patil, N.; Prasanthi, G.; Mohapatra, S. Paradigm shift of insect pests in rice ecosystem and their management strategy. ORYZA-An Int. J. Rice 2018, 55, 82–89. [Google Scholar] [CrossRef]
- Horgan, F.G.; Garcia, C.P.F.; Haverkort, F.; de Jong, P.W.; Ferrater, J.B. Changes in insecticide resistance and host range performance of planthoppers artificially selected to feed on resistant rice. Crop Prot. 2020, 127, 104963. [Google Scholar] [CrossRef]
- Guru-Pirasanna-Pandi, G.; Chander, S.; Pal, M.; Soumia, P.S. Impact of elevated CO2 on Oryza sativa phenology and brown planthopper, Nilaparvata lugens (Hemiptera: Delphacidae) population. Curr. Sci. 2018, 114, 1767–1777. [Google Scholar] [CrossRef]
- Anant, A.K.; Guru-Pirasanna-Pandi, G.; Jena, M.; Chandrakar, G.; Chidambaranathan, P.; Raghu, S.; Gowda, G.B.; Annamalai, M.; Patil, N.; Adak, T.; et al. Genetic dissection and identification of candidate genes for brown planthopper, Nilaparvata lugens (Delphacidae: Hemiptera) resistance in farmers’ varieties of rice in Odisha. Crop Prot. 2021, 144, 105600. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.-X.; Szymura, J.M.; Hewitt, G.M. Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 1995, 40, 382–391. [Google Scholar] [CrossRef]
- Shao, R.; Campbell, N.J.H.; Barker, S.C. Numerous Gene Rearrangements in the Mitochondrial Genome of the Wallaby Louse, Heterodoxus macropus (Phthiraptera). Mol. Biol. Evol. 2001, 18, 858–865. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, J.S.; Naaz, N.; Prabhakar, C.S.; Rao, M.S.; Das, B. The mitochondrial genome of the peach fruit fly, Bactrocera zonata (Saunders) (Diptera: Tephritidae): Complete DNA sequence, genome organization, and phylogenetic analysis with other tephritids using next generation DNA sequencing. Gene 2015, 569, 191–202. [Google Scholar] [CrossRef]
- Taanman, J.-W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys Acta-Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef] [Green Version]
- Cameron, S.L.; Lambkin, C.L.; Barker, S.C.; Whiting, M.F. Utility of Mitochondrial Genomes as Phylogenetic Markers for Insect intraordinal relationships A case study from flies (Diptera). Syst. Entomol. 2007, 32, 40–59. [Google Scholar] [CrossRef]
- Zrzavy, J. Evolution of antennae and historical ecology of the hemipteran insects(Paraneoptera). Acta Entomol. Bohemoslov. 1992, 89, 77–86. [Google Scholar]
- Hennig, W. Insect Phylogeny; John Wiley & Sons: Hoboken, NJ, USA, 1981. [Google Scholar]
- Bourgoin, T.; Campbell, B.C. Inferring a Phylogeny for Hemiptera: Falling into the ‘Autapomorphic Trap’. Denisia 2002, 4, 67–82. [Google Scholar]
- Urban, J.M.; Cryan, J.R. Evolution of the planthoppers (Insecta: Hemiptera: Fulgoroidea). Mol. Phylogenet. Evol. 2007, 42, 556–572. [Google Scholar] [CrossRef]
- Xie, Q.; Tian, Y.; Zheng, L.; Bu, W. 18S rRNA hyper-elongation and the phylogeny of Euhemiptera (Insecta: Hemiptera). Mol. Phylogenet. Evol. 2008, 47, 463–471. [Google Scholar] [CrossRef]
- Urban, J.M.; Cryan, J.R. Entomologically famous, evolutionarily unexplored: The first phylogeny of the lanternfly family Fulgoridae (Insecta: Hemiptera: Fulgoroidea). Mol. Phylogenet. Evol. 2009, 50, 471–484. [Google Scholar] [CrossRef]
- Wilson, M.R. Handbook for the Identification of Leafhoppers and Planthoppers of Rice; CAB International: Wallingford, UK, 1991. [Google Scholar]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, J.S.; Naaz, N.; Lemtur, M.; Das, B.; Singh, A.K.; Bhatt, B.P.; Prabhakar, C.S. Genetic analysis of Bactrocera zonata (Diptera: Tephritidae) populations from India based on cox1 and nad1 gene sequences. Mitochondrial DNA Part A 2018, 29, 727–736. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.-J.; Zhu, W.-C.; Rong, X.; Zhang, Y.-K.; Ding, X.-L.; Liu, J.; Chen, D.-S.; Du, Y.; Hong, X.-Y. The complete mitochondrial genomes of two rice planthoppers, Nilaparvata lugens and Laodelphax striatellus: Conserved genome rearrangement in Delphacidae and discovery of new characteristics of atp8 and tRNA genes. BMC Genom. 2013, 14, 417. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Gascuel, O. A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Chilana, P.; Sharma, A.; Rai, A. Insect genomic resources: Status, availability and future. Curr. Sci. 2012, 102, 571–580. [Google Scholar]
- Ribeiro, F.J.; Przybylski, D.; Yin, S.; Sharpe, T.; Gnerre, S.; Abouelleil, A.; Berlin, A.M.; Montmayeur, A.; Shea, T.P.; Walker, B.J.; et al. Finished bacterial genomes from shotgun sequence data. Genome Res. 2012, 22, 2270–2277. [Google Scholar] [CrossRef] [Green Version]
- Knaus, B.J.; Cronn, R.; Liston, A.; Pilgrim, K.; Schwartz, M.K. Mitochondrial genome sequences illuminate maternal lineages of conservation concern in a rare carnivore. BMC Ecol. 2011, 11, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Prasad, M.N.V.; Rajkumar, M.; Freitas, H. Plant growth promoting rhizobacteria and endophytes accelerate phytoremediation of metalliferous soils. Biotechnol. Adv. 2011, 29, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Coates, B.S. Assembly and annotation of full mitochondrial genomes for the corn rootworm species, Diabrotica virgifera virgifera and Diabrotica barberi (Insecta: Coleoptera: Chrysomelidae), using Next Generation Sequence data. Gene 2014, 542, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Peng, X.; Jing, S.; Liu, B.; Zhu, L.; He, G. Intraspecific and Interspecific Variations in the Mitochondrial Genomes ofNilaparvata(Hemiptera: Delphacidae). J. Econ. Èntomol. 2015, 108, 2021–2029. [Google Scholar] [CrossRef]
- Hua, J.; Li, M.; Dong, P.; Cui, Y.; Xie, Q.; Bu, W. Phylogenetic analysis of the true water bugs (Insecta: Hemiptera: Heteroptera: Nepomorpha): Evidence from mitochondrial genomes. BMC Evol. Biol. 2009, 9, 134. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, J.; Jiang, L.-Y.; Qiao, G.-X. Hemipteran Mitochondrial Genomes: Features, Structures and Implications for Phylogeny. Int. J. Mol. Sci. 2015, 16, 12382–12404. [Google Scholar] [CrossRef] [Green Version]
- Jeyaprakash, A.; Hoy, M.A. The mitochondrial genome of the predatory mite Metaseiulus occidentalis (Arthropoda: Chelicerata: Acari: Phytoseiidae) is unexpectedly large and contains several novel features. Gene 2007, 391, 264–274. [Google Scholar] [CrossRef]
- Shao, R.; Mitani, H.; Barker, S.C.; Takahashi, M.; Fukunaga, M. Novel mitochondrial gene content and gene arrangement indicate illegitimate inter-mtDNA recombination in the chigger mite, Leptotrombidium pallidum. J. Mol. Evol. 2005, 60, 764–773. [Google Scholar] [CrossRef]
- Cha, S.Y.; Yoon, H.J.; Lee, E.M.; Yoon, M.H.; Hwang, J.S.; Jin, B.R.; Han, Y.S.; Kim, I. The complete nucleotide sequence and gene organization of the mitochondrial genome of the bumblebee, Bombus ignitus (Hymenoptera: Apidae). Gene 2007, 392, 206–220. [Google Scholar] [CrossRef]
- Hong, G.; Jiang, S.; Yu, M.; Yang, Y.; Li, F.; Xue, F.; Wei, Z. The complete nucleotide sequence of the mitochondrial genome of the cabbage butterfly, Artogeia melete (Lepidoptera: Pieridae). Acta Biochim. Et Biophys. Sin. 2009, 41, 446–455. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wei, Z.-J.; Hong, G.-Y.; Jiang, S.-T.; Wen, L.-P. The complete nucleotide sequence of the mitochondrial genome of Phthonandria atrilineata (Lepidoptera: Geometridae). Mol. Biol. Rep. 2009, 36, 1441–1449. [Google Scholar] [CrossRef]
- Song, N.; Liang, A.-P. Complete Mitochondrial Genome of the Small Brown Planthopper, Laodelphax striatellus(Delphacidae: Hemiptera), with a Novel Gene Order. Zool. Sci. 2009, 26, 851–860. [Google Scholar] [CrossRef]
- Lessinger, A.C.; Junqueira, A.C.M.; Lemos, T.A.; Kemper, E.L.; da Silva, F.R.; Vettore, A.L.; Arruda, P.; Azeredo-Espin, A.M.L. The mitochondrial genome of the primary screwworm fly Cochliomyia hominivorax (Diptera: Calliphoridae). Insect Mol. Biol. 2000, 9, 521–529. [Google Scholar] [CrossRef]
- Hou, W.-R.; Chen, Y.; Wu, X.; Hu, J.-C.; Peng, Z.-S.; Yang, J.; Tang, Z.-X.; Zhou, C.-Q.; Li, Y.-M.; Yang, S.-K.; et al. A complete mitochondrial genome sequence of Asian black bear Sichuan subspecies (Ursus thibetanus mupinensis). Int. J. Biol. Sci. 2007, 3, 85–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.-T.; Hong, G.-Y.; Yu, M.; Li, N.; Yang, Y.; Liu, Y.-Q.; Wei, Z.-J. Characterization of the complete mitochondrial genome of the giant silkworm moth, Eriogyna pyretorum (Lepidoptera: Saturniidae). Int. J. Biol. Sci. 2009, 5, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-M.; Li, Y.; Chen, M.; Wang, H.; Li, Q.; Xia, R.-X.; Zeng, C.-Y.; Li, Y.-P.; Liu, Y.-Q.; Qin, L. Complete mitochondrial genome of the atlas moth, Attacus atlas (Lepidoptera: Saturniidae) and the phylogenetic relationship of Saturniidae species. Gene 2014, 545, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-X.; Qin, D.-Z. First mitogenome for the tribe Saccharosydnini (Hemiptera: Delphacidae: Delphacinae) and the phylogeny of three predominant rice planthoppers. Eur. J. Èntomol. 2018, 115, 242–248. [Google Scholar] [CrossRef]
- Salvato, P.; Simonato, M.; Battisti, A.; Negrisolo, E. The complete mitochondrial genome of the bag-shelter moth Ochrogaster lunifer (Lepidoptera, Notodontidae). BMC Genom. 2008, 9, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thao, M.L.; Baumann, L.; Baumann, P. Organization of the mitochondrial genomes of whiteflies, aphids, and psyllids (Hemiptera, Sternorrhyncha). BMC Evol. Biol. 2004, 4, 25. [Google Scholar] [CrossRef] [Green Version]
- Djoumad, A.; Nisole, A.; Zahiri, R.; Freschi, L.; Picq, S.; Gundersen-Rindal, D.E.; Sparks, M.E.; Dewar, K.; Stewart, D.; Maaroufi, H.; et al. Comparative analysis of mitochondrial genomes of geographic variants of the gypsy moth, Lymantria dispar, reveals a previously undescribed genotypic entity. Sci. Rep. 2017, 7, 14245. [Google Scholar] [CrossRef] [Green Version]
- Ohtsuki, T.; Kawai, G.; Watanabe, K. The minimal tRNA: Unique structure of Ascaris suum mitochondrial tRNASerUCU having a short T arm and lacking the entire D arm. FEBS Lett. 2002, 514, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Sheffield, N.C.; Song, H.; Cameron, S.L.; Whiting, M.F. Nonstationary Evolution and Compositional Heterogeneity in Beetle Mitochondrial Phylogenomics. Syst. Biol. 2009, 58, 381–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, J.; Li, M.; Dong, P.; Cui, Y.; Xie, Q.; Bu, W. Comparative and phylogenomic studies on the mitochondrial genomes of Pentatomomorpha (Insecta: Hemiptera: Heteroptera). BMC Genom. 2008, 9, 610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.; Kang, J.; Jung, C.; Hoelmer, K.; Lee, S.H.; Lee, S. Complete mitochondrial genome of brown marmorated stink bug Halyomorpha halys (Hemiptera: Pentatomidae), and phylogenetic relationships of hemipteran suborders. Mol. Cells 2009, 28, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, H.; Shi, A.; Štys, P.; Zhou, X.; Cai, W. The Complete Mitochondrial Genome and Novel Gene Arrangement of the Unique-Headed Bug Stenopirates sp. (Hemiptera: Enicocephalidae). PLoS ONE 2012, 7, e29419. [Google Scholar] [CrossRef] [Green Version]
- Castellana, S.; Vicario, S.; Saccone, C. Evolutionary Patterns of the Mitochondrial Genome in Metazoa: Exploring the Role of Mutation and Selection in Mitochondrial Protein–Coding Genes. Genome Biol. Evol. 2011, 3, 1067–1079. [Google Scholar] [CrossRef] [Green Version]
- Boore, J.L.; Macey, J.R.; Medina, M. Sequencing and comparing whole mitochondrial genomes of animals. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2005; Volume 395, pp. 311–348. [Google Scholar]
- Cameron, S.L.; Beckenbach, A.T.; Dowton, M.P.; Whiting, M.F. Evidence from mitochondrial genomics on interordinal relationships in insects. Arthropod Syst. Phylogeny 2006, 64, 27–34. [Google Scholar]
- Huang, Y.-X.; Qin, D.-Z. The complete mitochondrial genome sequence of the corn planthopper, Peregrinus maidis (Hemiptera: Fulgoroidea). Mitochondrial DNA Part B 2017, 2, 783–784. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| U | C | A | G | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU | ||||
| UUU | Phe | 25.5 | 1.70 | UCU | Ser | 8.5 | 2.27 | UAU | Tyr | 9.6 | 1.66 | UGU | Cys | 3.0 | 1.86 |
| UUC | Phe | 04.5 | 0.30 | UCC | Ser | 2.5 | 0.66 | UAC | Tyr | 2.0 | 0.34 | UGC | Cys | 0.2 | 0.14 |
| UUA | Leu | 25.7 | 3.88 | UCA | Ser | 7.3 | 1.96 | UAA | Stop | 0.0 | 0.00 | UGA | Trp | 5.2 | 1.72 |
| UUG | Leu | 03.5 | 0.52 | UCG | Ser | 0.5 | 0.12 | UAG | Stop | 0.0 | 0.00 | UGG | Trp | 0.8 | 0.28 |
| CUU | Leu | 04.8 | 0.73 | CCU | Pro | 3.4 | 1.36 | CAU | His | 3.5 | 1.41 | CGU | Arg | 1.1 | 1.30 |
| CUC | Leu | 00.8 | 0.12 | CCC | Pro | 2.9 | 1.18 | CAC | His | 1.5 | 0.59 | CGC | Arg | 0.1 | 0.09 |
| CUA | Leu | 04.5 | 0.67 | CCA | Pro | 3.4 | 1.36 | CAA | Gln | 3.1 | 1.67 | CGA | Arg | 1.8 | 2.14 |
| CUG | Leu | 00.5 | 0.07 | CCG | Pro | 0.2 | 0.09 | CAG | Gln | 0.6 | 0.33 | CGG | Arg | 0.4 | 0.47 |
| AUU | Ile | 26.1 | 1.76 | ACU | Thr | 4.5 | 1.62 | AAU | Asn | 11.2 | 1.66 | AGU | Ser | 3.9 | 1.05 |
| AUC | Ile | 03.5 | 0.24 | ACC | Thr | 2.2 | 0.81 | AAC | Asn | 2.3 | 0.34 | AGC | Ser | 0.5 | 0.14 |
| AUA | Met | 16.5 | 1.72 | ACA | Thr | 4.1 | 1.48 | AAA | Lys | 8.9 | 1.72 | AGA | Ser | 5.9 | 1.59 |
| AUG | Met | 02.7 | 0.28 | ACG | Thr | 0.2 | 0.08 | AAG | Lys | 1.5 | 0.28 | AGG | Ser | 0.8 | 0.21 |
| GUU | Val | 04.8 | 1.84 | GCU | Ala | 2.8 | 1.89 | GAU | Asp | 3.3 | 1.48 | GGU | Gly | 4.8 | 1.56 |
| GUC | Val | 00.6 | 0.23 | GCC | Ala | 1.0 | 0.68 | GAC | Asp | 1.2 | 0.52 | GGC | Gly | 0.5 | 0.15 |
| GUA | Val | 04.5 | 1.69 | GCA | Ala | 2.0 | 1.37 | GAA | Glu | 5.5 | 1.80 | GGA | Gly | 5.1 | 1.63 |
| GUG | Val | 00.6 | 0.23 | GCG | Ala | 0.1 | 0.05 | GAG | Glu | 0.6 | 0.20 | GGG | Gly | 2.1 | 0.67 |
| Protein-Coding Genes | Rates of Non-Synonymous Substitutions (Ka) | Rates of Synonymous Substitutions (Ks) | Ka/Ks Ratio | Rates of Non-Synonymous Substitutions Jukes-Cantor Adjusted J(Ka) | Rates of Synonymous Substitutions Jukes-Cantor Adjusted J(Ks) | JKa/JKs Ratio |
|---|---|---|---|---|---|---|
| nad2 | 0.15659 | 0.40221 | 0.38932 | 0.18751 | 0.72155 | 0.25987 |
| cox1 | 0.04354 | 0.46370 | 0.09390 | 0.04562 | 0.98025 | 0.04654 |
| cox2 | 0.09142 | 0.43730 | 0.20906 | 0.10113 | 0.86819 | 0.11648 |
| atp8 | 0.18724 | 0.39761 | 0.47091 | 0.23575 | 0.73457 | 0.32094 |
| atp6 | 0.18595 | 0.40582 | 0.45821 | 0.25526 | 0.71953 | 0.35476 |
| cox3 | 0.11981 | 0.44117 | 0.27157 | 0.13657 | 0.85826 | 0.15912 |
| nad3 | 0.32060 | 0.54762 | 0.58544 | 0.37831 | 0.96928 | 0.39029 |
| nad6 | 0.19594 | 0.36638 | 0.53480 | 0.24547 | 0.59248 | 0.41431 |
| cytB | 0.08143 | 0.47195 | 0.17254 | 0.08905 | 1.00887 | 0.08827 |
| nad1 | 0.10299 | 0.39094 | 0.26344 | 0.11561 | 0.65987 | 0.17520 |
| nad4L | 0.16987 | 0.29200 | 0.58175 | 0.20935 | 0.40849 | 0.51250 |
| nad4 | 0.14236 | 0.33225 | 0.42847 | 0.16642 | 0.49613 | 0.33544 |
| nad5 | 0.14155 | 0.32656 | 0.43346 | 0.16546 | 0.48086 | 0.34409 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Govindharaj, G.-P.-P.; Babu, S.B.; Choudhary, J.S.; Asad, M.; Chidambaranathan, P.; Gadratagi, B.-G.; Rath, P.C.; Naaz, N.; Jaremko, M.; Qureshi, K.A.; et al. Genome Organization and Comparative Evolutionary Mitochondriomics of Brown Planthopper, Nilaparvata lugens Biotype 4 Using Next Generation Sequencing (NGS). Life 2022, 12, 1289. https://doi.org/10.3390/life12091289
Govindharaj G-P-P, Babu SB, Choudhary JS, Asad M, Chidambaranathan P, Gadratagi B-G, Rath PC, Naaz N, Jaremko M, Qureshi KA, et al. Genome Organization and Comparative Evolutionary Mitochondriomics of Brown Planthopper, Nilaparvata lugens Biotype 4 Using Next Generation Sequencing (NGS). Life. 2022; 12(9):1289. https://doi.org/10.3390/life12091289
Chicago/Turabian StyleGovindharaj, Guru-Pirasanna-Pandi, Soumya Bharti Babu, Jaipal Singh Choudhary, Muhammad Asad, Parameswaran Chidambaranathan, Basana-Gowda Gadratagi, Prakash Chandra Rath, Naiyar Naaz, Mariusz Jaremko, Kamal Ahmad Qureshi, and et al. 2022. "Genome Organization and Comparative Evolutionary Mitochondriomics of Brown Planthopper, Nilaparvata lugens Biotype 4 Using Next Generation Sequencing (NGS)" Life 12, no. 9: 1289. https://doi.org/10.3390/life12091289