
Macrophages in Glioblastoma Development and Therapy: A Double-Edged Sword

1
Integrative Cancer Center & Cancer Clinical Research Center, Sichuan Cancer Hospital & Institute, Sichuan Cancer Center, School of Medicine, University of Electronic Science and Technology of China, Chengdu 610041, China
2
Department of Radiation Oncology, Sichuan Cancer Hospital, Chengdu 610041, China
3
Key Laboratory of Cellular Physiology (Shanxi Medical University), Ministry of Education, Taiyuan 030001, China
4
Department of Physiology, Shanxi Medical University, Taiyuan 030001, China
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Life 2022, 12(8), 1225; https://doi.org/10.3390/life12081225
Submission received: 7 July 2022
/
Revised: 4 August 2022
/
Accepted: 5 August 2022
/
Published: 12 August 2022
(This article belongs to the Special Issue Tumor–Host Interaction during Cancer Progression)
Abstract
:Glioblastoma (GBM) is one of the leading lethal tumors, featuring aggressive malignancy and poor outcome to current standard temozolomide (TMZ) or radio-based therapy. Developing immunotherapies, especially immune checkpoint inhibitors, have improved patient outcomes in other solid tumors but remain fatigued in GBM patients. Emerging evidence has shown that GBM-associated macrophages (GAMs), comprising brain-resident microglia and bone marrow-derived macrophages, act critically in boosting tumor progression, altering drug resistance, and establishing an immunosuppressive environment. Based on its crucial role, evaluations of the safety and efficacy of GAM-targeted therapy are ongoing, with promising (pre)clinical evidence updated. In this review, we summarized updated literature related to GAM nature, the interplay between GAMs and GBM cells, and GAM-targeted therapeutic strategies.
1. Introduction
Glioblastoma (GBM) represents the most common primary intracranial tumor, accounting for approximately 48.6% of brain malignancies, according to the CBTRUS statistical report in 2020 [1]. GBM patients have a poor prognosis and a short survival time. Standard treatment for GBM consists of maximal safe surgical resection, followed by fractionated radiotherapy with concurrent or subsequent adjuvant temozolomide (TMZ)-based chemotherapy [2]. However, the response to current treatment remains limited in GBM patients.
In recent decades, immune-therapeutic approaches have achieved great clinical benefits in various solid tumors by restoring silenced or broken antitumor immune responses. However, favorable outcomes toward immunotherapy failed to be obtained in GBM patients, which is attributed to the unique intracranial environment of GBM. Compared to peripheral organs, the lack of a lymphatic network in the brain parenchyma and the existence of extensive vascular structures, including the blood–meningeal barrier (BMB), blood–cerebrospinal fluid barrier (BCSFB), and blood–brain barrier (BBB), endow GBM with “immune privilege” [3,4]. In this situation, GBM-associated macrophages (GAMs) are responsible for immunological surveillance and are comprised of ontogenetically distinct macrophage populations, including resident microglia and bone marrow-derived macrophages. During tumor evolution, GBM cells establish crosstalk with GAMs, stressing them into distinct phenotypes to affect tumor malignancy, vascular information, treatment response, and so on. In this review, updated research on the functional mechanism of GAMs is summarized, as well as related GAM-targeted therapy in GBM.
2. Biocharacters of GAMs
GAMs represent a mixed-cell collection exhibiting distinct ontology and phenotype, which can be provided by both intracranial microglia and macrophages from bone marrow (Figure 1). Microglia belong to the brain’s primary innate immune cells, originating from the primitive macrophage pool in the yolk sac. Microglia play a vital role in maintaining brain homeostasis by sensing environmental changes, removing cell debris, and providing neurotrophic factors [5].
Both the quantity and molecular characteristics of GAMs are highly plastic [6,7]. Evidence from single-cell sequencing demonstrated that GAMs constituted 59.05% and 27.87% of immunocytes in primary and recurrent GBMs, respectively [8]. The phenotype and activation state of GAMs are affected by multiple signaling molecules, growth factors, transcription factors, and epigenetic and posttranscriptional modifications [9,10,11]. Under tumoral or infective stimulation, unpolarized macrophages (M0 state) can be activated and polarized into two major subtypes, proinflammatory M1 and anti-inflammatory M2. M1 macrophages are characterized by increased secretion of proinflammatory cytokines, such as interleukin-1β (IL-1β), TNF, IL-12, and IL-18, which show strong antibacterial performance in mediating resistance to pathogens but can also lead to tissue destruction. Factors involved in proinflammation and immune stimulation are abundant in M1 GAMs, such as major histocompatibility complex class II (MHC-II), CD68 markers, CD80, and CD86 costimulatory molecules. In contrast, the M2 phenotype is generally supposed to participate in immunosuppression and tumor promotion, which is formed after being exposed to macrophage colony-stimulating factor 1 (CSF-1), interleukin 4 (IL-4), IL-10, and IL-13 [6,7]. For example, the expression of AEG-1 was positively associated with M2 markers in GBM tissues. Silencing AGE-1 in GBM decreased the M2 polarization of microglia and secretion of the tumor-supportive cytokines IL-6 and TGF-β1 [12]. However, accelerating findings have revealed that there are other versatile states of GAMs except for M1 or M2 phenotypes; a batch of GAMs performs as a mixture of M1 and M2 phenotypes [13]. M2 macrophages are divided into four subtypes: the M2a, M2b, M2c, and M2d subsets. The M2a subtype is activated by IL-4 and IL-13. M2b is elicited by IL-1R ligands or exposure to immune complexes plus LPS. M2c is induced by IL-10 and TGF-β [14], while M2d releases IL-10 and VEGF upon induction by TLR antagonists [15]. Among them, M2c GAMs are most closely related to GBM immune regulation, matrix deposition, and tissue remodeling [16]. Moreover, a nonpolarized M0 phenotype has also been observed in GAMs from GBM patients [14].
The abundance of intracranial GAMs predominantly determines the nature of the tumor immune environment. The M2-like GAM subtype plays a supportive role in constructing an immunosuppressive microenvironment by releasing inhibitory cytokines and chemokines to the antitumor immune response. When exposed to GBM-initiating cell-secreted factors, mTOR-STAT3-NF-κB signaling is activated and drives an immunosuppressive phenotype formation in microglia. Correspondingly, the infiltration and proliferation of effector T-cells are inhibited to block immune reactivity [17]. Broken secretion of CXCL9 and CXCL10 by GAMs suppresses T-cell infiltration into GBM tumors [18]. In addition, recruitment of M2 GAMs is accompanied by upregulated immune checkpoints, e.g., PD-L1, PD-L2, CD80, and CD86. An immune-exhausted state is formed, leading to an unsatisfactory response to anti-PD-1 therapy [18].
3. GAMs in Regulating Malignancy of GBM
Under normal conditions, microglia guarantee the intracranial steady state by sensing environmental changes, immune surveillance, and homeostatic maintenance [19,20]. These functions can be impaired by GBM cells to permit tumor initiation or growth, as verified by comparing the transcriptome of microglia from GBM-bearing mice and normal mice. A collection of genes encoding receptors that recognize various antigens, chemokines, and cytokines are downregulated in GAMs, corresponding to less sensitive microglia and impaired immune surveillance [21]. The homeostatic status of microglia can be disrupted by blocked SMAD3 signaling, leading to malfunction of self-renewal and grid-like distribution [22,23]. Increased PD-L1/PD-L2 expression has also been detected in microglia from GBM, suggesting enhanced immunologic tolerance in microglia [21].
During tumor development, GAMs continuously exert their significant protumorigenic functions through various cytokines, including transforming growth factor-beta (TGF-β), epidermal growth factor (EGF), platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF), hepatocyte growth factor (HGF), IL-1β, IL-6, IL-10, and matrix metallopeptidase-2 (MMP-2) (Figure 2). Among these, the effect of GAM-derived TGF-β on GBM has been extensively studied. Liu et al. found that TGF-β secreted by M2 phenotype GAMs upregulated phosphorylation of SMAD2/3, promoting epithelial-mesenchymal transition (EMT) and invasion of GBM cells [24]. In addition, GAMs participate in reciprocal molecular crosstalk with GBM stem cells (GSCs), displaying a more direct protumorigenic function by secreting TGF-β [25]. For example, integrin αvβ5 on GSCs can bind with TGF-β derived from GAMs in a paracrine way. Once combined, Src-STAT3 signaling is activated for protumorigenic effects.
GAM-derived TGF-β can also affect SMAD independently. For example, TGF-β signaling prevents proteasomal degradation of Sry-related high mobility group box (Sox) 9. Stabilization of Sox9 enhanced the migration and invasion of GBM cells, while downregulation of Sox9 inhibited the proliferation and development of xenograft GBM. After being activated by GM-CSF, GAMs can release the chemokine C-C ligand 5 (CCL5) to upregulate MMP2 secretion in GBM cells, consequently promoting tumor migration and invasion [26]. These findings may explain why the proliferation and migration of GBM cells were increased in the presence of microglia [27].
Infiltrating GAMs secrete IFNγ and elicit epigenetic immunoediting with stable expression of the myeloid-affiliated transcriptional program in GBM cells, which in turn leads to increased recruitment of GAMs. Moreover, similar epigenetic and transcriptional signatures have been identified in human mesenchymal subtype GSCs, which could indicate that epigenetic immunoediting may drive an acquired immune evasion program in the most aggressive mesenchymal GBM subtype by reshaping the tumor immune microenvironment [28].
4. GAMs in Angiogenesis of GBM
Microvascular hyperplasia is another hallmark of GBM, characterized by distorted vessels consisting of abnormal endothelial walls and mural cell (smooth muscle cells and pericytes) coverage. Normally, angiogenesis contains a series of steps, including breakdown of the basement membrane, remodeling of the extracellular matrix (ECM), and activation, proliferation, migration, and stabilization of endothelial cells. The best-characterized angiogenic factors include VEGFs, placental growth factor (PlGF), hepatocyte growth factor (HGF), basic fibroblast growth factor (bFGF), platelet-derived growth factor (PDGF), angiopoietins (Angs), TGF-β, and MMPs [29,30,31,32]. These neovessels formed even in hypoxic and necrotic areas of GBM and greatly support tumor cell growth and migration. However, highly proliferative GBM cells apart from vessels undergo extreme hypoxia and induce abnormal angiogenesis. These large and cross-linked pathological vessels are abnormal and functionally immature, resulting in exacerbated hypoxia with increased interstitial pressure (Figure 2).
There is a mutual effect between GAMs and angiogenesis. GAMs in hypoxic areas potently promote angiogenesis by secreting multiple angiogenic factors. These proangiogenic factors not only promote angiogenesis directly but also induce M2 polarization of GAMs, which contribute to further tumor angiogenesis. Hypoxia can induce the expression of hypoxia-inducible factor-1α (HIF-1α) in GAMs, a major proangiogenic factor that also significantly upregulates VEGF and VEGFR [33]. Interacting with VEGFR on endothelial cells stimulates MMP secretion to dissolve basement membrane and ECM components, which destabilizes endothelial-pericyte contact and facilitates the proliferation and migration of endothelial cells [34]. Beyond this, VEGF-α can foster the development of GAMs, interfere with the maturation of dendritic cells, limit T-cell recruitment into tumors, or promote T-cell exhaustion [35,36]. In particular, VEGF/VEGFR signaling induces TGF-β production in GAMs and promotes M2-like polarization [37]. In addition to VEGF, other proangiogenic factors are also implicated in tumor angiogenesis and immune suppression within the TME. Angiopoietin 2 (ANGPT2) negatively influences tumor immunity by stimulating Tie-2-expressing monocytes/macrophages to secrete IL-10, leading to the expansion of Treg cells and inhibiting effector T-cell activation [38,39]. Placental growth factor (PlGF), another member of the VEGF family, also promotes tumor-associated macrophage (TAM) repolarization to the M2 subtype. In a mouse glioma model, dual blockade of ANGPT2 and VEGF with the bispecific antibody A2 V has been shown to reprogram GAMs toward the antitumor M1 polarized subtype [40,41]. PlGF blockade induces vessel normalization and macrophage polarization from an M2-like to an M1-like phenotype [42,43]. Therefore, antiangiogenic therapy potentiates the normalization of the tumor immune microenvironment and may improve the effectiveness of immunotherapy during combination treatment [36]. Therapeutically, in the TAVAREC trial (NCT01164189), the monoclonal antibody bevacizumab targeting VEGF with TMZ chemotherapy has been shown to improve PFS in GBM compared to TMZ alone [44].
Conditionally, M2-like GAMs can mediate angiogenesis in a VEGF-independent manner. Cat Eye Syndrome Critical Region Protein 1 (CECR1) is highly expressed by M2-like macrophages in GBM and is positively correlated with tumoral microvascular density. The proangiogenic properties of CECR1 in macrophages were partially mediated via paracrine activation of pericytes by PDGFB-PDGFRβ signaling. CECR1-PDGFB-PDGFRβ cross-activation between macrophages and pericytes promoted pericyte recruitment and tumor angiogenesis [45]. In addition, GBM cell-derived IL-8 and CCL2 stimulated GAMs to secrete TNF-α and activated endothelial cells (ECs), characterized by the expression of VCAM-1, ICAM-1, CXCL5, and CXCL10. EC activation was associated with a higher WHO grade of GBM, worse overall survival (OS), and resistance to antiangiogenic therapy. Inhibition of TNFα prevented EC activation and prolonged survival of GBM-bearing mice [46]. Additionally, M2-polarized microglia released insulin-like growth factor-binding protein 1 (IGFBP1), which was induced by upregulation of macrophage colony-stimulating factor (MCSF) in a spleen tyrosine kinase (SYK)-PI3K-NFκB-dependent manner in GBM and promoted angiogenesis. Silencing IGFBP1 in microglial cells reduced the ability to induce angiogenesis, which might be a promising target for macrophage-based antiangiogenic therapy [47].
In addition to angiogenesis, at least four other modalities involved in neovascularization in GBM have been proposed: vascular co-option, vasculogenesis, vascular mimicry, and glioblastoma-endothelial cell transdifferentiation. Among these modalities, the role of GAMs in vasculogenesis has been investigated. Vasculogenesis involves the differentiation of circulating endothelial progenitor cells (EPCs). Apart from EPCs, GAMs express CXCR4 and migrate in response to the chemokine stromal cell-derived factor 1α (SDF-1α) gradient into tumor sites to contribute to vasculogenesis [48].
5. GAMs in Drug Resistance of GBM
Resistance to TMZ remains a main clinical challenge in most GBM patients [49]. Apart from the genetic nature of GBM cells, accumulating evidence has demonstrated that GAMs are closely related to the clinical response to TMZ by releasing various soluble factors [50,51]. Both microglia and macrophages are responsible for secreting IL-11, which in turn activates STAT3-MYC signaling in GBM, conferring TMZ resistance [50]. Inhibition of GAM recruitment and IL-11 secretion by ablation or genetic inactivation of myeloid-specific phosphoinositide-3-kinase gamma isoform (PI3Kγ) reversed TMZ sensitivity in a murine glioblastoma model [50]. Other researchers highlighted the potentially distinct effects of different GAM subpopulations in altering treatment responses. For example, M2-like GAMs contribute to resistance against TMZ by secreting exosomal miR-21-5p. Downregulating miR-21-enriched exosomes from M2 GAMs successfully overcame TMZ resistance in patient-derived xenograft (PDX) models [52]. Meanwhile, induction of M1-like polarization of GAMs by GBM-derived extracellular HMGB1 restored the sensitivity of GBM to TMZ [53]. Apart from chemoresistance, studies have suggested that GAMs are also involved in resistance to radiotherapy and antiangiogenic therapy. The dynamics and plasticity of the GAM transcriptome during radiotherapy correspond with an altered quantity of monocyte-derived macrophages or microglia [54]. Adding GAM inhibitors to standard treatment is supposed to recover the response. Crosstalk between GAMs and GSCs has been shown to be closely associated with GBM malignant behavior and therapeutic resistance. Pleiotrophin (PTN) has also been observed to be secreted by GAMs to stimulate GSCs through its receptor PTPRZ1, supporting GSC maintenance and tumorigenic potential to promote malignancy of GBM [55]. The influence of GAMs on GSCs is mediated via paracrine signaling by exosomes derived from GAMs. Small extracellular vesicles (sEVs) derived from GAMs transferred miR-27a-3p, miR-22-3p, and miR-221-3p to GSCs, triggering the pro-neural-to-mesenchymal transition in GSCs and increasing radiotherapy resistance [56] (Figure 2).
Since the famous Checkmate 143 trial (NCT02017717) demonstrated a restricted efficacy of nivolumab in improving the OS of GBM patients, great efforts have been made to elucidate the mechanism of immune checkpoint blockade (ICB) resistance [57]. The first explanation is that once PD-1 and CTLA-4 are blocked, GAMs are induced to express PD-L1 to interact with CD80, an alternative binding partner of PD-L1 on T-cells, thereby leading to CD4+ T-cell suppression, Treg expansion, and thus ICB resistance [58]. Therefore, the triple ICB regimen (supplementing anti-PD-L1) resulted in decreased tumor growth and an enhanced response compared with the double ICB regimen (11/13 vs. 6/13) [58]. Studies conducted by Goswami S et al. demonstrated that a unique population of CD73 high-expressing macrophages persisted after ICB treatment. Knocking out CD73 prolonged survival in a murine model of GBM treated with anti-PD-1 and anti-CTLA-4, which provided potential rationales for combining macrophage-targeted therapy with ICB [59]. The PI3K/Akt pathway is critical in modifying the polarization of macrophages, which are predominantly activated in the M2 subpopulation [60]. IPI-549, a selective PI3K-γ inhibitor, shifted GAMs from the M2 to the M1 phenotype by blocking PI3K-γ [61]. Using a TMZ-resistant glioma xenograft model, the combination of IPI-549 with PD-1 antibody strongly inhibited tumor growth, suggesting that macrophage repolarization could be a potential approach to overcome TMZ resistance [62].
Bevacizumab, a humanized monoclonal antibody to VEGF, is the only FDA-approved anti-angiogenic drug for GBM, which still meets resistance issues. Higher numbers of tumor-infiltrated GAMs are correlated with poor survival in antiangiogenic agent-treated patients, implying that GAMs participate in escape from antiangiogenic therapy [63]. Studies analyzed differential transcriptional expression between bevacizumab-resistant GBM patients and bevacizumab-naïve patients and suggested that macrophage migration inhibitory factor (MIF) was significantly downregulated and correlated with increased M2-like macrophages localizing to the tumor edge and tumor growth. Overexpression of MIF in bevacizumab-resistant GBM xenograft models resulted in decreased tumor weight, decreased GAMs, increased M1/M2 ratio, and decreased angiogenesis [64]. Above all, detailed mechanisms and underlying therapeutic strategies related to vascular–immune crosstalk need to be investigated further to realize synergetic effects of antiangiogenic therapy and immunotherapy in GBM.
Furthermore, several efforts have been made to elucidate the complex and interconnected GBM hallmarks. GAMs, as the most abundant immune cells in the TME, are largely responsible for the nature of the TME and play a critical role in the complicated and large network. The effects are achieved mainly through cytokines released by GAMs, such as VEGF, PDGF, bFGF, HGF, ILs, and MMPs. These multifunctional cytokines interact with each other and construct a protumoral TME. However, the underlying mechanisms for the role of GAMs in interconnected hallmarks of GBM still need further research [65].
6. GAM-Targeted Therapy in GBM
6.1. Targeting Phagocytosis Checkpoints
Insufficient T-cell infiltration leads to unsatisfactory efficacy of immunotherapy checkpoint inhibitors (ICIs). Therefore, great attention has been given to macrophages, the most abundant immune cells in GBM, to reverse the immune “cold” environment (Figure 3).
A wide array of preclinical and clinical evidence has highlighted that phagocytosis checkpoints could be a potential target to induce an effective anticancer immune response of macrophages (Table 1). A series of phagocytosis checkpoint pairs have been identified, such as CD47/SIRPα, CD24/Siglec-10, PD-1/PD-L1, and MHC I-LILRB. Among them, CD47-SIRPα is the most well-studied phagocytosis checkpoint on macrophages to mediate “do not eat me” signaling [66,67]. During interactions between glioma and GAMs, CD47 has been found to be highly expressed in GBM cells [68,69]. The increased expression promotes proliferation and invasion of GBM cells and was positively correlated with glioma grade and negatively associated with clinical outcomes. CD47 is a transmembrane protein widely expressed on the surface of normal cells and solid tumors [70]. Structurally, it consists of an N-terminal extracellular variable region, five hydrophobic transmembrane helices, and a very short intracellular signal sequence. SIRPα is mainly expressed on the surface of myeloid cells (e.g., monocytes, macrophages, granulocytes), and its intracellular domain contains an immunoreceptor tyrosine inhibitory motif (ITIM). Binding of CD47 in normal cells and SIRPα on macrophages can cause phosphorylation of two SIRPα cytosolic ITIMs, which in turn recruit and activate Src homology-2 (SH2)-containing protein tyrosine phosphatases (SHP)-1/2 protein, leading to dephosphorylation of a series of intracellular proteins, inhibiting cytoskeleton rearrangement and cell motility, ultimately inhibiting macrophage phagocytosis and further impairing the antigen presentation and activation of adaptive immune responses [71,72]. Therefore, upregulated CD47-SIRPα signaling weakens phagocytosis of macrophages as an immune evasion mechanism. The blockade of CD47-SIRPa signaling significantly enhances phagocytosis by macrophages.
Therapeutic antibodies were invented, including Hu5F9-G4 [73]. Treating GBM with Hu5F9-G4 resulted in increased macrophage-mediated phagocytosis of GBM cells and GSCs and promoted the M1-phenotypic transition of macrophages [74]. In addition, Hu5F9-G4 significantly suppressed tumors and prolonged survival time in an immunocompetent allograft glioma mouse model [75,76]. In addition, the safety, pharmacokinetics, and pharmacodynamics of Hu5F9-G4 have been investigated in phase I clinical trials in adult patients with solid tumors (NCT02216409). The results showed that a treatment regimen for Hu5F9-G4 is well tolerated in patients with solid tumors and lymphoma. Another ongoing phase I clinical trial testing the safety of Hu5F9-G4 in patients with recurrent or progressive malignant brain tumors is currently recruiting participants (NCT05169944). Hu5F9-G4-mediated phagocytosis of GBM cells can even be significantly enhanced by irradiation and TMZ chemotherapy [77].
In addition to anti-CD47 antibodies, small molecule inhibitors targeting the CD47-SIRPα interaction or inhibiting CD47 expression are also being investigated. Compared with therapeutic antibodies, small molecule inhibitors have a low molecular weight [78] and enhanced permeability and retention effects [79,80,81], which might make them better candidates for brain cancer therapy. At present, the application of CD47 small molecule inhibitors in GBM remains under preclinical evaluation. RRx-001, a multipotent small molecule with vascular normalization effects, downregulated CD47 expression by inhibiting its transcription factor MYC directly [82,83]. A recent study demonstrated that administration of RRx-001 prior to TMZ or irinotecan results in significantly increased uptake of irinotecan and temozolomide in orthotopic glioma tumors of mice [84]. Other small molecule inhibitors, such as Pep-20, D4-2, and NCGC00138783, can directly block the binding of CD47 and SIRPα; [85,86,87] metformin, JQ1, and 4Mu can not only inhibit the CD47-SIRPα bond but also reduce CD47 expression at the transcriptional level [82,88,89,90]; PQ912 and SNE177 decrease CD47 expression by regulating posttranslational modification [91]; the widely used EGFR-TKI gefitinib, as a first-line treatment for patients with advanced EGFR mutation-positive non-small cell lung cancer, has also been shown to induce CD47 downregulation in vitro [92].
Similar to CD47, CD24 is overexpressed in a variety of solid tumors, including triple-negative breast cancer (TNBC), ovarian cancer, and GBM [93,94,95,96]. In glioma, CD24 expression levels are positively correlated with pathological grade and negatively correlated with outcomes [95]. Its partner Siglec-10 is highly expressed on tumor-associated macrophages with an ITIM in its cytoplasmic domain [97]. Some studies have confirmed that its expression is also increased in glioma and associated with poor prognosis of patients [98]. Mechanistically, the CD24-Siglec10 interaction induces the inhibition of phagocytosis in a similar way to CD47-SIRPa, which is mediated by SHP-1/SHP-2 once the ITIM region is phosphorylated [99].
Therapeutic strategies targeting other phagocytosis checkpoints are being explored. Amira A. Barkal et al. observed that both genetic ablation of CD24 or Siglec-10 and monoclonal antibody blockade of the CD24-Siglec-10 interaction robustly augmented the phagocytosis of all CD24-expressing human tumors tested. Dual treatment with CD24 and CD47 blocking antibodies revealed an increased induction of phagocytosis to nearly 30-fold that of baseline in some cancers [97]. Some anti-CD24 drugs targeting tumors have been put into clinical trials, such as SWA11 (mAb), which is related to ovarian cancer and pancreatic cancer, and rG7S-MICA (mAb), which is related to liver cancer. However, anticancer therapy using anti-CD24 antibodies in GBM to boost the innate immune system has not been tested in clinical trials. In summary, clinical and preclinical trials are mainly focused on targeting CD47/SIRPα and CD24/Siglec-10 to boost GAM phagocytic ability. More phagocytosis checkpoint pairs and related therapies are under development.
6.2. Targeting GAM Depletion
Colony-stimulating factor-1 receptor (CSF-1R) is a receptor tyrosine kinase expressed on macrophages that plays an important role in regulating the survival, proliferation, differentiation, and polarization of GAMs [100]. Binding of CSF-1R with its ligands, such as CSF-1 and IL-34, activates the CSF-1R pathway [101]. CSF-1R-positive macrophages correlate with poor prognosis in various solid cancers [102]. Targeting CSF-1R can effectively reduce the number of GAMs in the TME and promote GAM repolarization, thus promoting the activation of cytotoxic T-cells, inhibiting tumor growth, and preventing glioblastoma recurrence [103,104,105,106,107,108]. The small molecule inhibitor PLX-3397 was assessed in recurrent glioblastoma in a phase II clinical trial. However, no significant survival benefit was observed with PLX3397 monotherapy [109]. Therefore, considering the complexity and heterogeneity of the GBM TME, a macrophage-targeted strategy alone would not be enough to induce potent antitumor effects, and thus, more research focused on combination treatment is ongoing. Some preclinical evidence has demonstrated the feasibility and potential of the combination of anti-CSF-1R with other therapies. Administration of anti-CSF-1R and anti-PD1 to treat glioma in a mouse model indicated prolonged survival [110]. In addition, radiotherapy is also expected to function synergistically with immunotherapy in GBM, as ionizing radiation-induced DNA damage and cell death may be able to enhance the immunogenicity of tumor tissues and activate an immune response. Studies conducted by Akkari L and colleagues identified GAM gene expression signatures of different stages after radiotherapy in murine gliomas and found that targeting GAM populations using a colony-stimulating factor-1 receptor (CSF-1R) inhibitor combined with radiotherapy substantially enhanced survival in preclinical models [54]. In addition, a phase 1b/2 clinical trial (NCT02880371) evaluated ARRY-382, another CSF-1R inhibitor, plus pembrolizumab in patients with advanced solid tumors. Unfortunately, recently published results still failed to show a significant clinical benefit [111]. In summary, as mentioned above, the number of GAMs in the GBM TME is negatively correlated with prognosis in GBM patients, and therapeutics aimed at GAM depletion have been explored in GBM. The most well-studied target is CSF-1R. Although some favorable clinical and preclinical results have been obtained, studies regarding the efficacy, side effects, and combination therapy of CSF-1R blockade are still needed in the future.
6.3. Targeting GAM Reprogramming
In addition to CSF-1R, there are many other key molecules that regulate the survival and functions of GAMs in the TME, which have been demonstrated to be potential targets in macrophage-based therapies. For example, CD40, a costimulatory molecule, is expressed in most immune cells, including monocytes, macrophages, DCs, B cells, and nonimmune cells, such as endothelial cells, epithelial cells, and tumor cells [112]. The ligand CD40 L is expressed on the surfaces of activated T-cells and macrophages. Activated CD40/CD40 L pathways have broad immunostimulatory effects on APCs, B cells [112], and T-cells [113]. For macrophages, repolarization is induced to a tumor-suppressive type characterized by the production and release of proinflammatory cytokines, including IL-1β [114], TNFa [115], and IL-6 [116]. Therapeutically, agonistic CD40 antibodies (αCD40) are used in various solid tumors. In glioma, studies based on diverse experimental models and treatment regimens showed inconsistent results of their effectiveness [117,118]. More exploration of prime combinatorial regimens and the target population will be needed in the future. In addition, neuropilin-1 (NRP-1) is a coreceptor for class III semaphorins (SEMA3s) and members of the vascular endothelial growth factor (VEGF) family, and its b1 domain can interact and augment the VEGF-A and TGFβ pathways, thus promoting the protumorigenic M2 polarization of GAMs in the TME [119,120]. Miyauchi J. T. et al. found that the small molecule inhibitor EG00229, which inhibits its b1 domain, blocks the polarization of macrophages and increases the number of proinflammatory TAMs in the TME, resulting in an inhibitory effect on tumor growth [121]. Moreover, it has also been found that inhibition of β-amyloid precursor protein cleaving enzyme 1 (BACE1) with MK-8931 can effectively reprogram pTAM to sTAM and promote macrophage phagocytosis of glioma cells [122]. Moreover, some existing widely used drugs for nonmalignant diseases were also found to have the ability to regulate macrophage-mediated antitumor immune responses in glioma. The antibiotic minocycline inhibits microglial MMP expression and attenuates glioma invasion. The phase 1 clinical trial (NCT01580969) indicated that the combination of minocycline with radiation and bevacizumab was well-tolerated in patients with recurrent GBM [123,124]. Cyclosporine A, an immunosuppressant drug, has shown efficacy in attenuating glioma tumor growth and angiogenesis by inhibiting microglial infiltration in an experimental murine model [125]. Propentofylline, a drug with purported neuroprotective effects, has also been shown to reduce tumor growth in GBM by directly targeting microglia [126]. As mentioned above, reprogramming GAMs into antitumoral types is a wise approach to manipulate. Therapies targeting CD40, NRP-1, BACE1, and some existing widely used drugs for nonmalignant diseases are found to repolarize GAMs and show some promising clinical significance. However, elucidating the complicated regulatory mechanisms is a prerequisite for developing novel therapies.
6.4. Chimeric Antigen Receptor-Macrophage (CAR-M) Therapy
As great success for chimeric antigen T-cell therapy has been achieved in hematological malignancies, an increasing number of studies have attempted to copy this success in solid tumors. However, the results are far behind the expectations. Especially for GBM, some factors that compromise the efficacy of CAR-T therapy are nonnegligible. The existence of the BBB and lack of lymphatic networks restrict CAR-T cells from penetrating the GBM TME, and the immunosuppressive glioma TME decreases the viability of CAR-T cells and neutralizes their effects. Target-specific CAR-T cells have limited cytotoxicity in the heterogeneous GBM TME. To overcome the challenges of T-cell-based CAR therapy, researchers have focused their attention on macrophages. Therefore, strategies for the transduction of CARs into macrophages are under research, which is an approach using genetically engineered approaches to modify macrophages. Compared to CAR-T therapy, CAR-Ms have two major advantages. Unlike the poor infiltration of T-cells, macrophages exist abundantly in the GBM TME. Compared to the rapid development into exhaustion phenotypes of CAR-T cells after infiltrating the TME, the phenotypic plasticity of macrophages makes them changeable when faced with environmental stimuli.
Similar to CAR-T cells, CAR-Ms consist of an extracellular antigen-binding domain, hinge region, transmembrane domain, and intracellular domain. Structurally, the intracellular domain includes CD3ζ as used in CAR-T cells, the γ subunit of Fc receptor (FcRγ), and multiple epidermal growth factor-like domains protein 10 (Megf10), which contain immunoreceptor tyrosine-based activation motifs (ITAMs) with the ability to transduce phagocytic signals in macrophages. Similar to second- and third-generation CAR-T cells, an additional signaling domain is designed to enhance phagocytosis by macrophages. Studies have shown that the addition of a PI3K-recruiting domain significantly enhances phagocytosis by macrophages [127].
Studies conducted by Morrisey et al. in 2018 and Klichinsky in 2020 et al. show that CAR-Ms are able to phagocytize target antigen-expressing tumor cells, repolarize macrophages toward the antitumor M1 phenotype, promote T-cell recruitment, and stimulate the cells of the adaptive immune system. CAR-Ms against HER2 suggested a significant decrease in the metastatic tumor burden and longer overall survival in the mouse model. To our knowledge, there are no studies related to the use of CAR-Ms in GBM. However, there are still sufficient reasons to develop CAR-Ms strategies to treat GBM. Induced by the immunosuppressive cytokines secreted by GBM cells, most GAMs polarize into M2-like subtypes. CAR-Ms have been shown to convert the protumoral M2 phenotype into the proinflammatory M1 phenotype, which subsequently makes the GBM immune microenvironment “cold” into “hot”. GBM is an ideal candidate for this novel approach. Based on the normalized immune microenvironment, harnessing the immune system to defeat GBM will be easier to realize.
6.5. Other GAM-Based Therapies
Oncolytic virus (OV) is a novel way to elicit lytic tumor cell death to activate the immune response [128,129,130]. Beyond direct GBM cell lysis, the effect of OVs on macrophage modulation has been investigated. Van den Bossche, Wouter BL, and colleagues found that the oncolytic adenovirus Delta24-RGD, also known as DNX-2401, shifted the murine GBM macrophage phenotype from the pro-tumoral M2 toward the antitumoral and pro-inflammatory M1 phenotype, thereby disabling a major tumor-maintaining mechanism [131]. Recently, a phase I clinical trial of DNX-2401 treating patients with recurrent GBM suggested increased numbers of macrophages and proinflammatory factors, including IL-6 and TNF-α, in posttreatment tumor specimens [132]. Virus vector-mediated cancer gene therapy aimed at macrophage reprogramming is also under investigation. An adeno-associated virus (AAV) vector was used to selectively deliver antitumor transgenes encoding secreted antitumor proteins to tumor stromal cells, including macrophages, which could repolarize the target macrophage and promote a proinflammatory phenotype within the TME [133].
The SRC proto-oncogene nonreceptor tyrosine kinase (SRC) signaling pathway is constitutively activated in hypoxia in GBM. The increase in SRC activity causes VEGF, MMPs, and TGF-β upregulation. SRC inhibition could improve the GAM-orchestrated immunosuppressive TME. A series of SRC tyrosine kinase inhibitors (STKIs) have been developed, such as dasatinib, PP2, SI221, and bosutinib. However, despite encouraging preclinical results, most clinical trials in GBM have failed thus far [134,135].
The existence of the BBB prevents 98% of drugs from reaching the brain, which contributes to the limited effectiveness of chemotherapy. Novel biomaterials have been explored to address this issue. In a recent study, a pH-sensitive nanocomposite micelle composed of TfR-T12-PEG-PLGA and TATH7-PEG-PLGA was automatically assembled as a novel drug delivery system for sending chemotherapeutic paclitaxel (PTX) and immunomodulator Toll-7 receptor agonist R837, which successfully delivered PTX and R837 through the BBB and was rapidly ingested by tumor cells and tumor-associated immune cells in an acidic tumor microenvironment with the assistance of transferrin receptor (TfR) [136]. Macrophages infiltrated in tumors decreased significantly, and the immunosuppressive phenotype was relieved, characterized by increased TNF-α and decreased TGF-β. More effective and precise vectors should be developed for treatment, and the efficiency also needs to be demonstrated in clinical research.
7. Conclusions
Harnessing the host immune system to achieve potent antitumor responses with mild adverse reactions is the ultimate goal to pursue. Considering the immunosuppressive nature of the glioma microenvironment and the failure of T-cell-based therapies in glioma treatment, macrophages, as the most abundant immune cell in the glioma TME, might be an appropriate candidate to target. Overall, it is clear that crosstalk between GAMs and glioma plays a crucial role in glioma progression, angiogenesis, and treatment resistance. Understanding the underlying molecular mechanisms is necessary to develop macrophage-targeted therapies for glioma. More attention should be focused on the novel mechanisms and GAM-based clinical translational research in the future.
Author Contributions
M.W. and Y.S. contributed equally to this work. Conceptualization, C.X. and Y.S.; Data Curation, M.W., L.Z., L.C. and X.Z.; Writing—Original Draft Preparation, M.W., L.Z., L.C. and X.Z.; Writing—Review and Editing, Y.S.; Supervision, C.X.; Funding Acquisition, C.X. and Y.S. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81873048), Sichuan Provincial Science Fund for Distinguished Young Scholars of China (No. 2020JDJQ0065), and Medico-Engineering Cooperation Funds from University of Electronic Science and Technology of China (No. ZYGX2021YGCX004) to CX; The Fundamental Research Funds for the Central Universities (No. ZYGX2020KYQD002) and Open Fund from Key Laboratory of Cellular Physiology (Shanxi Medical University), Ministry of Education, China (No. CELLPHYSIOL/SXMU-2021-CPOF202102) to YS.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable.
Acknowledgments
Cartoons in the figures were created with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-Oncology 2020, 22, iv1–iv96. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Alves de Lima, K.; Rustenhoven, J.; Kipnis, J. Meningeal Immunity and Its Function in Maintenance of the Central Nervous System in Health and Disease. Annu. Rev. Immunol. 2020, 38, 597–620. [Google Scholar] [CrossRef] [PubMed]
- Carson, M.J.; Doose, J.M.; Melchior, B.; Schmid, C.D.; Ploix, C.C. CNS immune privilege: Hiding in plain sight. Immunol. Rev. 2006, 213, 48–65. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune microenvironment of gliomas. Lab Investig. 2017, 97, 498–518. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Allavena, P.; Locati, M.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Fu, W.; Wang, W.; Li, H.; Jiao, Y.; Huo, R.; Yan, Z.; Wang, J.; Wang, S.; Wang, J.; Chen, D.; et al. Single-Cell Atlas Reveals Complexity of the Immunosuppressive Microenvironment of Initial and Recurrent Glioblastoma. Front. Immunol. 2020, 11, 835. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, X. Pivotal regulators of tissue homeostasis and cancer: Macrophages. Exp. Hematol. Oncol. 2017, 6, 23. [Google Scholar] [CrossRef]
- T’Jonck, W.; Guilliams, M.; Bonnardel, J. Niche signals and transcription factors involved in tissue-resident macrophage development. Cell Immunol. 2018, 330, 43–53. [Google Scholar] [CrossRef]
- Collins, E.J.; Cervantes-Silva, M.P.; Timmons, G.A.; O’Siorain, J.R.; Curtis, A.M.; Hurley, J.M. Post-transcriptional circadian regulation in macrophages organizes temporally distinct immunometabolic states. Genome Res. 2021, 31, 1–15. [Google Scholar] [CrossRef]
- Li, J.; Sun, Y.; Sun, X.; Zhao, X.; Ma, Y.; Wang, Y.; Zhang, X. AEG-1 silencing attenuates M2-polarization of glioma-associated microglia/macrophages and sensitizes glioma cells to temozolomide. Sci. Rep. 2021, 11, 17348. [Google Scholar] [CrossRef]
- Szulzewsky, F.; Pelz, A.; Feng, X.; Synowitz, M.; Markovic, D.; Langmann, T.; Holtman, I.R.; Wang, X.; Eggen, B.J.L.; Boddeke, H.W.G.M.; et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS ONE 2015, 10, e0116644. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1, e85841. [Google Scholar] [CrossRef]
- Ferrante, C.J.; Pinhal-Enfield, G.; Elson, G.; Cronstein, B.N.; Hasko, G.; Outram, S.; Leibovich, S.J. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Rα) signaling. Inflammation 2013, 36, 921–931. [Google Scholar] [CrossRef]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef]
- Dumas, A.A.; Pomella, N.; Rosser, G.; Guglielmi, L.; Vinel, C.; Millner, T.O.; Rees, J.; Aley, N.; Sheer, D.; Wei, J.; et al. Microglia promote glioblastoma via mTOR-mediated immunosuppression of the tumour microenvironment. EMBO J. 2020, 39, e103790. [Google Scholar] [CrossRef]
- Chen, Q.; Jin, J.; Huang, X.; Wu, F.; Huang, H.; Zhan, R. EMP3 mediates glioblastoma-associated macrophage infiltration to drive T cell exclusion. J. Exp. Clin. Cancer Res. 2021, 40, 160. [Google Scholar] [CrossRef]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Maas, S.L.N.; Abels, E.R.; van de Haar, L.L.; Zhang, X.; Morsett, L.; Sil, S.; Guedes, J.; Sen, P.; Prabhakar, S.; Hickman, S.E.; et al. Glioblastoma hijacks microglial gene expression to support tumor growth. J. Neuroinflamm. 2020, 17, 120. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef]
- Bohlen, C.J.; Friedman, B.A.; Dejanovic, B.; Sheng, M. Microglia in Brain Development, Homeostasis, and Neurodegeneration. Annu. Rev. Genet. 2019, 53, 263–288. [Google Scholar] [CrossRef]
- Liu, Z.; Kuang, W.; Zhou, Q.; Zhang, Y. TGF-beta1 secreted by M2 phenotype macrophages enhances the stemness and migration of glioma cells via the SMAD2/3 signalling pathway. Int. J. Mol. Med. 2018, 42, 3395–3403. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Yu-Ju Wu, C.; Chen, C.H.; Lin, C.Y.; Feng, L.Y.; Lin, Y.C.; Wei, K.C.; Huang, C.Y.; Fang, J.Y.; Chen, P.Y. CCL5 of glioma-associated microglia/macrophages regulates glioma migration and invasion via calcium-dependent matrix metalloproteinase 2. Neuro Oncol. 2020, 22, 253–266. [Google Scholar] [CrossRef]
- Yeini, E.; Ofek, P.; Pozzi, S.; Albeck, N.; Ben-Shushan, D.; Tiram, G.; Golan, S.; Kleiner, R.; Sheinin, R.; Israeli Dangoor, S.; et al. P-selectin axis plays a key role in microglia immunophenotype and glioblastoma progression. Nat. Commun. 2021, 12, 1912. [Google Scholar] [CrossRef]
- Gangoso, E.; Southgate, B.; Bradley, L.; Rus, S.; Galvez-Cancino, F.; McGivern, N.; Güç, E.; Kapourani, C.-A.; Byron, A.; Ferguson, K.M.; et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell 2021, 184, 2454–2470. [Google Scholar] [CrossRef]
- Norden, A.D.; Drappatz, J.; Muzikansky, A.; David, K.; Gerard, M.; McNamara, M.B.; Phan, P.; Ross, A.; Kesari, S.; Wen, P.Y. An exploratory survival analysis of anti-angiogenic therapy for recurrent malignant glioma. J. Neuro-Oncol. 2009, 92, 149–155. [Google Scholar] [CrossRef]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef]
- Ferrari, G.; Cook, B.D.; Terushkin, V.; Pintucci, G.; Mignatti, P. Transforming growth factor-beta 1 (TGF-beta1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J. Cell. Physiol. 2009, 219, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Mazzone, M.; Jonckx, B.; Carmeliet, P. FLT1 and its ligands VEGFB and PlGF: Drug targets for anti-angiogenic therapy? Nat. Rev. Cancer 2008, 8, 942–956. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef]
- Lapeyre-Prost, A.; Terme, M.; Pernot, S.; Pointet, A.L.; Voron, T.; Tartour, E.; Taieb, J. Immunomodulatory Activity of VEGF in Cancer. Int. Rev. Cell Mol. Biol. 2017, 330, 295–342. [Google Scholar] [CrossRef]
- Lee, W.S.; Yang, H.; Chon, H.J.; Kim, C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp. Mol. Med. 2020, 52, 1475–1485. [Google Scholar] [CrossRef]
- Min, A.K.T.; Mimura, K.; Nakajima, S.; Okayama, H.; Saito, K.; Sakamoto, W.; Fujita, S.; Endo, H.; Saito, M.; Saze, Z.; et al. Therapeutic potential of anti-VEGF receptor 2 therapy targeting for M2-tumor-associated macrophages in colorectal cancer. Cancer Immunol. Immunother. 2021, 70, 289–298. [Google Scholar] [CrossRef]
- Khan, K.A.; Kerbel, R.S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nature Rev. Clin. Oncol. 2018, 15, 310–324. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Chen, Y.-Y.; Muthana, M.; Welford, A.F.; Tal, A.O.; Scholz, A.; Plate, K.H.; Reiss, Y.; Murdoch, C.; De Palma, M.; et al. Angiopoietin 2 stimulates TIE2-expressing monocytes to suppress T cell activation and to promote regulatory T cell expansion. J. Immunol. 2011, 186, 4183–4190. [Google Scholar] [CrossRef]
- Kloepper, J.; Riedemann, L.; Amoozgar, Z.; Seano, G.; Susek, K.; Yu, V.; Dalvie, N.; Amelung, R.L.; Datta, M.; Song, J.W.; et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc. Natl. Acad. Sci. USA 2016, 113, 4476–4481. [Google Scholar] [CrossRef]
- Peterson, T.E.; Kirkpatrick, N.D.; Huang, Y.; Farrar, C.T.; Marijt, K.A.; Kloepper, J.; Datta, M.; Amoozgar, Z.; Seano, G.; Jung, K.; et al. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 4470–4475. [Google Scholar] [CrossRef]
- De Falco, S. The discovery of placenta growth factor and its biological activity. Exp. Mol. Med. 2012, 44, 1–9. [Google Scholar] [CrossRef]
- Odorisio, T.; Schietroma, C.; Zaccaria, M.L.; Cianfarani, F.; Tiveron, C.; Tatangelo, L.; Failla, C.M.; Zambruno, G. Mice overexpressing placenta growth factor exhibit increased vascularization and vessel permeability. J. Cell Sci. 2002, 115, 2559–2567. [Google Scholar] [CrossRef]
- van den Bent, M.J.; Klein, M.; Smits, M.; Reijneveld, J.C.; French, P.J.; Clement, P.; de Vos, F.Y.F.; Wick, A.; Mulholland, P.J.; Taphoorn, M.J.B.; et al. Bevacizumab and temozolomide in patients with first recurrence of WHO grade II and III glioma, without 1p/19q co-deletion (TAVAREC): A randomised controlled phase 2 EORTC trial. Lancet Oncol. 2018, 19, 1170–1179. [Google Scholar] [CrossRef]
- Zhu, C.; Chrifi, I.; Mustafa, D.; van der Weiden, M.; Leenen, P.J.M.; Duncker, D.J.; Kros, J.M.; Cheng, C. CECR1-mediated cross talk between macrophages and vascular mural cells promotes neovascularization in malignant glioma. Oncogene 2017, 36, 5356–5368. [Google Scholar] [CrossRef]
- Wei, Q.; Singh, O.; Ekinci, C.; Gill, J.; Li, M.; Mamatjan, Y.; Karimi, S.; Bunda, S.; Mansouri, S.; Aldape, K.; et al. TNFα secreted by glioma associated macrophages promotes endothelial activation and resistance against anti-angiogenic therapy. Acta Neuropathol. Commun. 2021, 9, 67. [Google Scholar] [CrossRef]
- Nijaguna, M.B.; Patil, V.; Urbach, S.; Shwetha, S.D.; Sravani, K.; Hegde, A.S.; Chandramouli, B.A.; Arivazhagan, A.; Marin, P.; Santosh, V.; et al. Glioblastoma-derived Macrophage Colony-stimulating Factor (MCSF) Induces Microglial Release of Insulin-like Growth Factor-binding Protein 1 (IGFBP1) to Promote Angiogenesis. J. Biol. Chem. 2015, 290, 23401–23415. [Google Scholar] [CrossRef]
- Hardee, M.E.; Zagzag, D. Mechanisms of glioma-associated neovascularization. Am. J. Pathol. 2012, 181, 1126–1141. [Google Scholar] [CrossRef]
- Urbantat, R.M.; Jelgersma, C.; Brandenburg, S.; Nieminen-Kelha, M.; Kremenetskaia, I.; Zollfrank, J.; Mueller, S.; Rubarth, K.; Koch, A.; Vajkoczy, P.; et al. Tumor-Associated Microglia/Macrophages as a Predictor for Survival in Glioblastoma and Temozolomide-Induced Changes in CXCR2 Signaling with New Resistance Overcoming Strategy by Combination Therapy. Int. J. Mol. Sci. 2021, 22, 11180. [Google Scholar] [CrossRef]
- Li, J.; Kaneda, M.M.; Ma, J.; Li, M.; Shepard, R.M.; Patel, K.; Koga, T.; Sarver, A.; Furnari, F.; Xu, B.; et al. PI3Kgamma inhibition suppresses microglia/TAM accumulation in glioblastoma microenvironment to promote exceptional temozolomide response. Proc. Natl. Acad Sci. USA 2021, 118, e2009290118. [Google Scholar] [CrossRef]
- Hudson, A.L.; Parker, N.R.; Khong, P.; Parkinson, J.F.; Dwight, T.; Ikin, R.J.; Zhu, Y.; Chen, J.; Wheeler, H.R.; Howell, V.M. Glioblastoma Recurrence Correlates With Increased APE1 and Polarization Toward an Immuno-Suppressive Microenvironment. Front. Oncol. 2018, 8, 314. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.Y.; Su, Y.K.; Liu, H.W.; Chen, C.H.; Chiu, S.C.; Cho, D.Y.; Lin, S.Z.; Chen, Y.S.; Lin, C.M. Preclinical Evidence of STAT3 Inhibitor Pacritinib Overcoming Temozolomide Resistance via Downregulating miR-21-Enriched Exosomes from M2 Glioblastoma-Associated Macrophages. J. Clin. Med. 2019, 8, 959. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Fu, W.J.; Chen, X.Q.; Wang, S.; Deng, R.S.; Tang, X.P.; Yang, K.D.; Niu, Q.; Zhou, H.; Li, Q.R.; et al. Autophagy-based unconventional secretion of HMGB1 in glioblastoma promotes chemosensitivity to temozolomide through macrophage M1-like polarization. J. Exp. Clin. Cancer Res. 2022, 41, 74. [Google Scholar] [CrossRef] [PubMed]
- Akkari, L.; Bowman, R.L.; Tessier, J.; Klemm, F.; Handgraaf, S.M.; de Groot, M.; Quail, D.F.; Tillard, L.; Gadiot, J.; Huse, J.T.; et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 2020, 12, eaaw7843. [Google Scholar] [CrossRef]
- Shi, Y.; Ping, Y.-F.; Zhou, W.; He, Z.-C.; Chen, C.; Bian, B.-S.-J.; Zhang, L.; Chen, L.; Lan, X.; Zhang, X.-C.; et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat. Commun. 2017, 8, 15080. [Google Scholar] [CrossRef]
- Zhang, Z.; Xu, J.; Chen, Z.; Wang, H.; Xue, H.; Yang, C.; Guo, Q.; Qi, Y.; Guo, X.; Qian, M.; et al. Transfer of MicroRNA via Macrophage-Derived Extracellular Vesicles Promotes Proneural-to-Mesenchymal Transition in Glioma Stem Cells. Cancer Immunol. Res. 2020, 8, 966–981. [Google Scholar] [CrossRef]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro-Oncology 2017, 20, 674–686. [Google Scholar] [CrossRef]
- Aslan, K.; Turco, V.; Blobner, J.; Sonner, J.K.; Liuzzi, A.R.; Núñez, N.G.; de Feo, D.; Kickingereder, P.; Fischer, M.; Green, E.; et al. Heterogeneity of response to immune checkpoint blockade in hypermutated experimental gliomas. Nat. Commun. 2020, 11, 931. [Google Scholar] [CrossRef]
- Goswami, S.; Walle, T.; Cornish, A.E.; Basu, S.; Anandhan, S.; Fernandez, I.; Vence, L.; Blando, J.; Zhao, H.; Yadav, S.S.; et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nature Med. 2020, 26, 39–46. [Google Scholar] [CrossRef]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef]
- Amano, M.T.; Castoldi, A.; Andrade-Oliveira, V.; Latancia, M.T.; Terra, F.F.; Correa-Costa, M.; Breda, C.N.S.; Felizardo, R.J.F.; Pereira, W.O.; da Silva, M.B.; et al. The lack of PI3Kγ favors M1 macrophage polarization and does not prevent kidney diseases progression. Int. Immunopharmacol. 2018, 64, 151–161. [Google Scholar] [CrossRef]
- Miyazaki, T.; Ishikawa, E.; Matsuda, M.; Sugii, N.; Kohzuki, H.; Akutsu, H.; Sakamoto, N.; Takano, S.; Matsumura, A. Infiltration of CD163-positive macrophages in glioma tissues after treatment with anti-PD-L1 antibody and role of PI3Kγ inhibitor as a combination therapy with anti-PD-L1 antibody in in vivo model using temozolomide-resistant murine glioma-initiating cells. Brain Tumor Pathol. 2020, 37, 41–49. [Google Scholar] [CrossRef]
- Lu-Emerson, C.; Snuderl, M.; Kirkpatrick, N.D.; Goveia, J.; Davidson, C.; Huang, Y.; Riedemann, L.; Taylor, J.; Ivy, P.; Duda, D.G.; et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro-Oncology 2013, 15, 1079–1087. [Google Scholar] [CrossRef]
- Castro, B.A.; Flanigan, P.; Jahangiri, A.; Hoffman, D.; Chen, W.; Kuang, R.; de Lay, M.; Yagnik, G.; Wagner, J.R.; Mascharak, S.; et al. Macrophage migration inhibitory factor downregulation: A novel mechanism of resistance to anti-angiogenic therapy. Oncogene 2017, 36, 3749–3759. [Google Scholar] [CrossRef]
- Torrisi, F.; Alberghina, C.; D’Aprile, S.; Pavone, A.M.; Longhitano, L.; Giallongo, S.; Tibullo, D.; Di Rosa, M.; Zappalà, A.; Cammarata, F.P.; et al. The Hallmarks of Glioblastoma: Heterogeneity, Intercellular Crosstalk and Molecular Signature of Invasiveness and Progression. Biomedicines 2022, 10, 806. [Google Scholar] [CrossRef]
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.-X.; Weissman, I.L. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 568–586. [Google Scholar] [CrossRef]
- Logtenberg, M.E.W.; Scheeren, F.A.; Schumacher, T.N. The CD47-SIRPα Immune Checkpoint. Immunity 2020, 52, 742–752. [Google Scholar] [CrossRef]
- Li, F.; Lv, B.; Liu, Y.; Hua, T.; Han, J.; Sun, C.; Xu, L.; Zhang, Z.; Feng, Z.; Cai, Y.; et al. Blocking the CD47-SIRPα axis by delivery of anti-CD47 antibody induces antitumor effects in glioma and glioma stem cells. Oncoimmunology 2018, 7, e1391973. [Google Scholar] [CrossRef]
- Yang, J.; Yao, Y.; Tong, L.; Zhu, Z.; Wang, L.; Yang, J. CD47 is highly expressed in gliomas and targeting CD47 is a promising therapeutic strategy. Eur. J. Inflamm. 2021, 19, 1–19. [Google Scholar] [CrossRef]
- Fenalti, G.; Villanueva, N.; Griffith, M.; Pagarigan, B.; Lakkaraju, S.K.; Huang, R.Y.C.; Ladygina, N.; Sharma, A.; Mikolon, D.; Abbasian, M.; et al. Structure of the human marker of self 5-transmembrane receptor CD47. Nat. Commun. 2021, 12, 5218. [Google Scholar] [CrossRef]
- Veillette, A.; Chen, J. SIRPα–CD47 Immune Checkpoint Blockade in Anticancer Therapy. Trends Immunol. 2018, 39, 173–184. [Google Scholar] [CrossRef]
- Matozaki, T.; Murata, Y.; Okazawa, H.; Ohnishi, H. Functions and molecular mechanisms of the CD47–SIRPα signalling pathway. Trends Cell Biol. 2009, 19, 72–80. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Zhao, F.; Tseng, S.; Narayanan, C.; Shura, L.; Willingham, S.; Howard, M.; Prohaska, S.; Volkmer, J.; et al. Pre-Clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PLoS ONE 2015, 10, e0137345. [Google Scholar] [CrossRef]
- Zhang, M.; Hutter, G.; Kahn, S.A.; Azad, T.D.; Gholamin, S.; Xu, C.Y.; Liu, J.; Achrol, A.S.; Richard, C.; Sommerkamp, P.; et al. Anti-CD47 Treatment Stimulates Phagocytosis of Glioblastoma by M1 and M2 Polarized Macrophages and Promotes M1 Polarized Macrophages in vivo. PLoS ONE 2016, 11, e0153550. [Google Scholar] [CrossRef]
- Hu, J.; Xiao, Q.; Dong, M.; Guo, D.; Wu, X.; Wang, B. Glioblastoma Immunotherapy targeting the innate immune checkpoint CD47-SIRPα axis. Front. Immunol. 2020, 11, 593219. [Google Scholar] [CrossRef]
- Hutter, G.; Theruvath, J.; Graef, C.M.; Zhang, M.; Schoen, M.K.; Manz, E.M.; Bennett, M.L.; Olson, A.; Azad, T.D.; Sinha, R. Microglia are effector cells of CD47-SIRPα antiphagocytic axis disruption against glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 997–1006. [Google Scholar] [CrossRef]
- Gholamin, S.; Youssef, O.A.; Rafat, M.; Esparza, R.; Kahn, S.; Shahin, M.; Giaccia, A.J.; Graves, E.E.; Weissman, I.; Mitra, S. Irradiation or temozolomide chemotherapy enhances anti-CD47 treatment of glioblastoma. Innate Immun. 2020, 26, 130–137. [Google Scholar] [CrossRef]
- Sun, G.; Rong, D.; Li, Z.; Sun, G.; Wu, F.; Li, X.; Cao, H.; Cheng, Y.; Tang, W.; Sun, Y. Role of Small Molecule Targeted Compounds in Cancer: Progress, Opportunities, and Challenges. Front. Cell Dev. Biol. 2021, 9, 694363. [Google Scholar] [CrossRef]
- Imai, K.; Takaoka, A. Comparing antibody and small-molecule therapies for cancer. Nat. Rev. Cancer 2006, 6, 714–727. [Google Scholar] [CrossRef]
- Hoelder, S.; Clarke, P.A.; Workman, P. Discovery of small molecule cancer drugs: Successes, challenges and opportunities. Mol. Oncol. 2012, 6, 155–176. [Google Scholar] [CrossRef]
- Baker, M. Upping the ante on antibodies. Nat. Biotechnol. 2005, 23, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Oronsky, B.; Scicinski, J.; Cabrales, P.; Minchinton, A. RRx-001, an epigenetic-based radio-and chemosensitizer, has vascular normalizing effects on SCCVII and U87 tumors. Clin. Epigenetics 2016, 8, 53. [Google Scholar] [CrossRef] [PubMed]
- Oronsky, B.; Reid, T.; Cabrales, P. Vascular priming with RRx-001 to increase the uptake and accumulation of temozolomide and irinotecan in orthotopically implanted gliomas. J. Drug Target. 2021, 29, 998–1003. [Google Scholar] [CrossRef]
- Burgess, T.L.; Amason, J.D.; Rubin, J.S.; Duveau, D.Y.; Lamy, L.; Roberts, D.D.; Farrell, C.L.; Inglese, J.; Thomas, C.J.; Miller, T.W. A homogeneous SIRPα-CD47 cell-based, ligand-binding assay: Utility for small molecule drug development in immuno-oncology. PLoS ONE 2020, 15, e0226661. [Google Scholar] [CrossRef]
- Wang, H.; Sun, Y.; Zhou, X.; Chen, C.; Jiao, L.; Li, W.; Gou, S.; Li, Y.; Du, J.; Chen, G.; et al. CD47/SIRPα blocking peptide identification and synergistic effect with irradiation for cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000905. [Google Scholar] [CrossRef]
- Hazama, D.; Yin, Y.; Murata, Y.; Matsuda, M.; Okamoto, T.; Tanaka, D.; Terasaka, N.; Zhao, J.; Sakamoto, M.; Kakuchi, Y.; et al. Macrocyclic Peptide-Mediated Blockade of the CD47-SIRPα Interaction as a Potential Cancer Immunotherapy. Cell Chem. Biol. 2020, 27, 1181–1191.e7. [Google Scholar] [CrossRef]
- Tan, W.; Tang, H.; Jiang, X.; Ye, F.; Huang, L.; Shi, D.; Li, L.; Huang, X.; Li, L.; Xie, X.; et al. Metformin mediates induction of miR-708 to inhibit self-renewal and chemoresistance of breast cancer stem cells through targeting CD47. J. Cell. Mol. Med. 2019, 23, 5994–6004. [Google Scholar] [CrossRef]
- Rodríguez, M.M.; Fiore, E.; Bayo, J.; Atorrasagasti, C.; García, M.; Onorato, A.; Domínguez, L.; Malvicini, M.; Mazzolini, G. 4Mu Decreases CD47 Expression on Hepatic Cancer Stem Cells and Primes a Potent Antitumor T Cell Response Induced by Interleukin-12. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 2738–2750. [Google Scholar] [CrossRef]
- Kim, H.S.; Won, Y.J.; Shim, J.H.; Kim, H.J.; Kim, B.S.; Hong, H.N. Role of EphA2-PI3K signaling in vasculogenic mimicry induced by cancer-associated fibroblasts in gastric cancer cells. Oncol. Lett. 2019, 18, 3031–3038. [Google Scholar] [CrossRef]
- Logtenberg, M.E.W.; Jansen, J.H.M.; Raaben, M.; Toebes, M.; Franke, K.; Brandsma, A.M.; Matlung, H.L.; Fauster, A.; Gomez-Eerland, R.; Bakker, N.A.M.; et al. Glutaminyl cyclase is an enzymatic modifier of the CD47- SIRPα axis and a target for cancer immunotherapy. Nat. Med. 2019, 25, 612–619. [Google Scholar] [CrossRef]
- Nigro, A.; Ricciardi, L.; Salvato, I.; Sabbatino, F.; Vitale, M.; Crescenzi, M.A.; Montico, B.; Triggiani, M.; Pepe, S.; Stellato, C.; et al. Enhanced Expression of CD47 Is Associated With Off-Target Resistance to Tyrosine Kinase Inhibitor Gefitinib in NSCLC. Front. Immunol. 2019, 10, 3135. [Google Scholar] [CrossRef]
- Tarhriz, V.; Bandehpour, M.; Dastmalchi, S.; Ouladsah Eb Madarek, E.; Zarredar, H.; Eyvazi, S. Overview of CD24 as a new molecular marker in ovarian cancer. J. Cell. Physiol. 2019, 234, 2134–2142. [Google Scholar] [CrossRef]
- Kristiansen, G.; Winzer, K.-J.; Mayordomo, E.; Bellach, J.; Schlüns, K.; Denkert, C.; Dahl, E.; Pilarsky, C.; Altevogt, P.; Guski, H. CD24 expression is a new prognostic marker in breast cancer. Clin. Cancer Res. 2003, 9, 4906–4913. [Google Scholar]
- Deng, J.; Gao, G.; Wang, L.; Wang, T.; Yu, J.; Zhao, Z. CD24 Expression as a Marker for Predicting Clinical Outcome in Human Gliomas. J. Biomed. Biotechnol. 2012, 2012, 517172. [Google Scholar] [CrossRef]
- Tiburcio, P.D.B.; Locke, M.C.; Bhaskara, S.; Chandrasekharan, M.B.; Huang, L.E. The neural stem-cell marker CD24 is specifically upregulated in IDH-mutant glioma. Transl. Oncol. 2020, 13, 100819. [Google Scholar] [CrossRef]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef]
- Mao, R.; Zhou, L.; Yang, Y.; Wang, P.; Lin, H.; Zheng, J.; Lv, G.; Zhou, D. Regulation of prognosis-related Siglecs in the glioma microenvironment. J. Cancer Res. Clin. Oncol. 2021, 147, 3343–3357. [Google Scholar] [CrossRef]
- Yin, S.-S.; Gao, F.-H. Molecular Mechanism of Tumor Cell Immune Escape Mediated by CD24/Siglec-10. Front. Immunol. 2020, 11, 1324. [Google Scholar] [CrossRef]
- Stanley, E.R.; Chitu, V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb. Perspect. Biol. 2014, 6, a021857. [Google Scholar] [CrossRef]
- Achkova, D.; Maher, J. Role of the colony-stimulating factor (CSF)/CSF-1 receptor axis in cancer. Biochem. Soc. Trans. 2016, 44, 333–341. [Google Scholar] [CrossRef]
- Zhang, Q.-W.; Liu, L.; Gong, C.-Y.; Shi, H.-S.; Zeng, Y.-H.; Wang, X.-Z.; Zhao, Y.-W.; Wei, Y.-Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef]
- Laoui, D.; Van Overmeire, E.; De Baetselier, P.; Van Ginderachter, J.A.; Raes, G. Functional relationship between tumor-associated macrophages and macrophage colony-stimulating factor as contributors to cancer progression. Front. Immunol. 2014, 5, 489. [Google Scholar] [CrossRef]
- Tan, I.L.; Arifa, R.D.N.; Rallapalli, H.; Kana, V.; Lao, Z.; Sanghrajka, R.M.; Sumru Bayin, N.; Tanne, A.; Wojcinski, A.; Korshunov, A.; et al. CSF1R inhibition depletes tumor-associated macrophages and attenuates tumor progression in a mouse sonic Hedgehog-Medulloblastoma model. Oncogene 2021, 40, 396–407. [Google Scholar] [CrossRef]
- Barca, C.; Foray, C.; Hermann, S.; Herrlinger, U.; Remory, I.; Laoui, D.; Schäfers, M.; Grauer, O.M.; Zinnhardt, B.; Jacobs, A.H. The Colony Stimulating Factor-1 Receptor (CSF-1R)-Mediated Regulation of Microglia/Macrophages as a Target for Neurological Disorders (Glioma, Stroke). Front. Immunol. 2021, 12, 787307. [Google Scholar] [CrossRef]
- Yan, D.; Kowal, J.; Akkari, L.; Schuhmacher, A.J.; Huse, J.T.; West, B.L.; Joyce, J.A. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene 2017, 36, 6049–6058. [Google Scholar] [CrossRef]
- Butowski, N.; Colman, H.; de Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-Oncology 2016, 18, 557–564. [Google Scholar] [CrossRef]
- Przystal, J.M.; Becker, H.; Canjuga, D.; Tsiami, F.; Anderle, N.; Keller, A.-L.; Pohl, A.; Ries, C.H.; Schmittnaegel, M.; Korinetska, N.; et al. Targeting CSF1R Alone or in Combination with PD1 in Experimental Glioma. Cancers 2021, 13, 2400. [Google Scholar] [CrossRef]
- Johnson, M.; Dudek, A.Z.; Sukari, A.; Call, J.; Kunk, P.R.; Lewis, K.; Gainor, J.F.; Sarantopoulos, J.; Lee, P.; Golden, A.; et al. ARRY-382 in Combination with Pembrolizumab in Patients With Advanced Solid Tumors: Results From a Phase 1b/2 Study. Clin. Cancer Res. 2022, 28, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Bazan, F.; Blanchard, D.; Brière, F.; Galizzi, J.P.; van Kooten, C.; Liu, Y.J.; Rousset, F.; Saeland, S. The CD40 antigen and its ligand. Annu. Rev. Immunol. 1994, 12, 881–922. [Google Scholar] [CrossRef] [PubMed]
- Khong, A.; Brown, M.D.; Vivian, J.B.; Robinson, B.W.; Currie, A.J. Agonistic anti-CD40 antibody therapy is effective against postoperative cancer recurrence and metastasis in a murine tumor model. J. Immunother. 2013, 36, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.H., Jr.; Stout, R.D.; Suttles, J. Role of the CD40-CD40 ligand interaction in CD4+ T cell contact-dependent activation of monocyte interleukin-1 synthesis. Eur. J. Immunol. 1994, 24, 3148–3154. [Google Scholar] [CrossRef]
- Stout, R.D.; Suttles, J.; Xu, J.; Grewal, I.S.; Flavell, R.A. Impaired T cell-mediated macrophage activation in CD40 ligand-deficient mice. J. Immunol. 1996, 156, 8–11. [Google Scholar]
- Mukundan, L.; Bishop, G.A.; Head, K.Z.; Zhang, L.; Wahl, L.M.; Suttles, J. TNF receptor-associated factor 6 is an essential mediator of CD40-activated proinflammatory pathways in monocytes and macrophages. J. Immunol. 2005, 174, 1081–1090. [Google Scholar] [CrossRef]
- Chonan, M.; Saito, R.; Shoji, T.; Shibahara, I.; Kanamori, M.; Sonoda, Y.; Watanabe, M.; Kikuchi, T.; Ishii, N.; Tominaga, T. CD40/CD40L expression correlates with the survival of patients with glioblastomas and an augmentation in CD40 signaling enhances the efficacy of vaccinations against glioma models. Neuro-Oncology 2015, 17, 1453–1462. [Google Scholar] [CrossRef]
- van Hooren, L.; Vaccaro, A.; Ramachandran, M.; Vazaios, K.; Libard, S.; van de Walle, T.; Georganaki, M.; Huang, H.; Pietilä, I.; Lau, J.; et al. Agonistic CD40 therapy induces tertiary lymphoid structures but impairs responses to checkpoint blockade in glioma. Nat. Commun. 2021, 12, 4127. [Google Scholar] [CrossRef]
- Glinka, Y.; Stoilova, S.; Mohammed, N.; Prud’homme, G.J. Neuropilin-1 exerts co-receptor function for TGF-beta-1 on the membrane of cancer cells and enhances responses to both latent and active TGF-beta. Carcinogenesis 2011, 32, 613–621. [Google Scholar] [CrossRef]
- Jarvis, A.; Allerston, C.K.; Jia, H.; Herzog, B.; Garza-Garcia, A.; Winfield, N.; Ellard, K.; Aqil, R.; Lynch, R.; Chapman, C.; et al. Small molecule inhibitors of the neuropilin-1 vascular endothelial growth factor A (VEGF-A) interaction. J. Med. Chem. 2010, 53, 2215–2226. [Google Scholar] [CrossRef]
- Miyauchi, J.T.; Chen, D.; Choi, M.; Nissen, J.C.; Shroyer, K.R.; Djordevic, S.; Zachary, I.C.; Selwood, D.; Tsirka, S.E. Ablation of Neuropilin 1 from glioma-associated microglia and macrophages slows tumor progression. Oncotarget 2016, 7, 9801–9814. [Google Scholar] [CrossRef]
- Zhai, K.; Huang, Z.; Huang, Q.; Tao, W.; Fang, X.; Zhang, A.; Li, X.; Stark, G.R.; Hamilton, T.A.; Bao, S. Pharmacological inhibition of BACE1 suppresses glioblastoma growth by stimulating macrophage phagocytosis of tumor cells. Nat. Cancer 2021, 2, 1136–1151. [Google Scholar] [CrossRef]
- Hu, F.; Ku, M.-C.; Markovic, D.; Dzaye, O.D.a.; Lehnardt, S.; Synowitz, M.; Wolf, S.A.; Kettenmann, H. Glioma-associated microglial MMP9 expression is upregulated by TLR2 signaling and sensitive to minocycline. Int. J. Cancer 2014, 135, 2569–2578. [Google Scholar] [CrossRef]
- Cohen, A.L.; Anker, C.J.; Salzman, K.; Jensen, R.L.; Shrieve, D.C.; Colman, H. A phase 1 study of repeat radiation, minocycline, and bevacizumab in patients with recurrent glioma (RAMBO). J. Clin. Oncol. 2014, 32, 2066. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Ellert-Miklaszewska, A.; Lipko, M.; Sielska, M.; Frankowska, M.; Kaminska, B. Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. PLoS ONE 2011, 6, e23902. [Google Scholar] [CrossRef]
- Jacobs, V.L.; Landry, R.P.; Liu, Y.; Romero-Sandoval, E.A.; de Leo, J.A. Propentofylline decreases tumor growth in a rodent model of glioblastoma multiforme by a direct mechanism on microglia. Neuro-Oncology 2012, 14, 119–131. [Google Scholar] [CrossRef]
- Morrissey, M.A.; Williamson, A.P.; Steinbach, A.M.; Roberts, E.W.; Kern, N.; Headley, M.B.; Vale, R.D. Chimeric antigen receptors that trigger phagocytosis. eLife 2018, 7, e36688. [Google Scholar] [CrossRef]
- Russell, S.J.; Peng, K.-W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental Therapy of Human Glioma by Means of a Genetically Engineered Virus Mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef]
- Hofman, L.; Lawler, S.E.; Lamfers, M.L.M. The Multifaceted Role of Macrophages in Oncolytic Virotherapy. Viruses 2021, 13, 1570. [Google Scholar] [CrossRef]
- van den Bossche, W.B.L.; Kleijn, A.; Teunissen, C.E.; Voerman, J.S.A.; Teodosio, C.; Noske, D.P.; van Dongen, J.J.M.; Dirven, C.M.F.; Lamfers, M.L.M. Oncolytic virotherapy in glioblastoma patients induces a tumor macrophage phenotypic shift leading to an altered glioblastoma microenvironment. Neuro-Oncology 2018, 20, 1494–1504. [Google Scholar] [CrossRef]
- van Putten, E.H.P.; Kleijn, A.; van Beusechem, V.W.; Noske, D.; Lamers, C.H.J.; de Goede, A.L.; Idema, S.; Hoefnagel, D.; Kloezeman, J.J.; Fueyo, J.; et al. Convection Enhanced Delivery of the Oncolytic Adenovirus Delta24-RGD in Patients with Recurrent GBM: A Phase I Clinical Trial Including Correlative Studies. Clin. Cancer Res. 2022, 28, 1572–1585. [Google Scholar] [CrossRef]
- Volak, A.; LeRoy, S.G.; Natasan, J.S.; Park, D.J.; Cheah, P.S.; Maus, A.; Fitzpatrick, Z.; Hudry, E.; Pinkham, K.; Gandhi, S.; et al. Virus vector-mediated genetic modification of brain tumor stromal cells after intravenous delivery. J. Neuro-Oncol. 2018, 139, 293–305. [Google Scholar] [CrossRef]
- Torrisi, F.; Vicario, N.; Spitale, F.M.; Cammarata, F.P.; Minafra, L.; Salvatorelli, L.; Russo, G.; Cuttone, G.; Valable, S.; Gulino, R.; et al. The Role of Hypoxia and SRC Tyrosine Kinase in Glioblastoma Invasiveness and Radioresistance. Cancers 2020, 12, 2860. [Google Scholar] [CrossRef]
- Cirotti, C.; Contadini, C.; Barilà, D. SRC Kinase in Glioblastoma News from an Old Acquaintance. Cancers 2020, 12, 1558. [Google Scholar] [CrossRef]
- Sun, P.; Wu, Z.; Xiao, Y.; Wu, H.; Di, Q.; Zhao, X.; Quan, J.; Tang, H.; Wang, Q.; Chen, W. TfR-T12 short peptide and pH sensitive cell transmembrane peptide modified nano-composite micelles for glioma treatment viaby remodeling tumor microenvironment. Nanomed. Nanotechnol. Biol. Med. 2022, 41, 102516. [Google Scholar] [CrossRef]
Figure 1.
Ontology and polarization of glioblastoma-associated macrophages. Microglia originate from erythromyeloid progenitors (EMP) in the yolk sac. EMP-derived cells are incorporated into the CNS and take on an amoeboid morphology followed by transitioning toward a ramified state. GBM-associated macrophages are derived from monocytes, which arise from hematopoietic stem cells (HSCs) in bone marrow. In the GBM microenvironment, macrophages and microglia are induced by GBM-derived cytokines (such as CSF-1, IL-4/10/13, and TGF-β) and polarized into GAMs.
Figure 1.
Ontology and polarization of glioblastoma-associated macrophages. Microglia originate from erythromyeloid progenitors (EMP) in the yolk sac. EMP-derived cells are incorporated into the CNS and take on an amoeboid morphology followed by transitioning toward a ramified state. GBM-associated macrophages are derived from monocytes, which arise from hematopoietic stem cells (HSCs) in bone marrow. In the GBM microenvironment, macrophages and microglia are induced by GBM-derived cytokines (such as CSF-1, IL-4/10/13, and TGF-β) and polarized into GAMs.

Figure 2.
Functions of glioblastoma-associated macrophages. GAMs release many cytokines that promote the malignant phenotype of GBM, including tumor malignancy, angiogenesis, and treatment resistance (TMZ chemotherapy, radiation, anti-angiogenesis, and immunotherapy).
Figure 2.
Functions of glioblastoma-associated macrophages. GAMs release many cytokines that promote the malignant phenotype of GBM, including tumor malignancy, angiogenesis, and treatment resistance (TMZ chemotherapy, radiation, anti-angiogenesis, and immunotherapy).

Figure 3.
GAM-targeted therapy in GBM. Targeting the phagocytosis checkpoint is designed to block phagocytosis pairs, such as CD47/SIRPα and CD24/Siglec-10, to enhance the phagocytosis of tumor cells by macrophages; GAM depletion is aimed at reducing the number of GAMs in the TME. Targeting GAM reprogramming is to repolarize protumoral M2-like GAMs into tumor-suppressive M1-like macrophages; chimeric antigen receptor-macrophage (CAR-M) therapy is an approach using genetically engineered approaches to modify macrophages, which present enhanced phagocytosis, enhanced antigen presentation, and repolarization to M1-like macrophages.
Figure 3.
GAM-targeted therapy in GBM. Targeting the phagocytosis checkpoint is designed to block phagocytosis pairs, such as CD47/SIRPα and CD24/Siglec-10, to enhance the phagocytosis of tumor cells by macrophages; GAM depletion is aimed at reducing the number of GAMs in the TME. Targeting GAM reprogramming is to repolarize protumoral M2-like GAMs into tumor-suppressive M1-like macrophages; chimeric antigen receptor-macrophage (CAR-M) therapy is an approach using genetically engineered approaches to modify macrophages, which present enhanced phagocytosis, enhanced antigen presentation, and repolarization to M1-like macrophages.


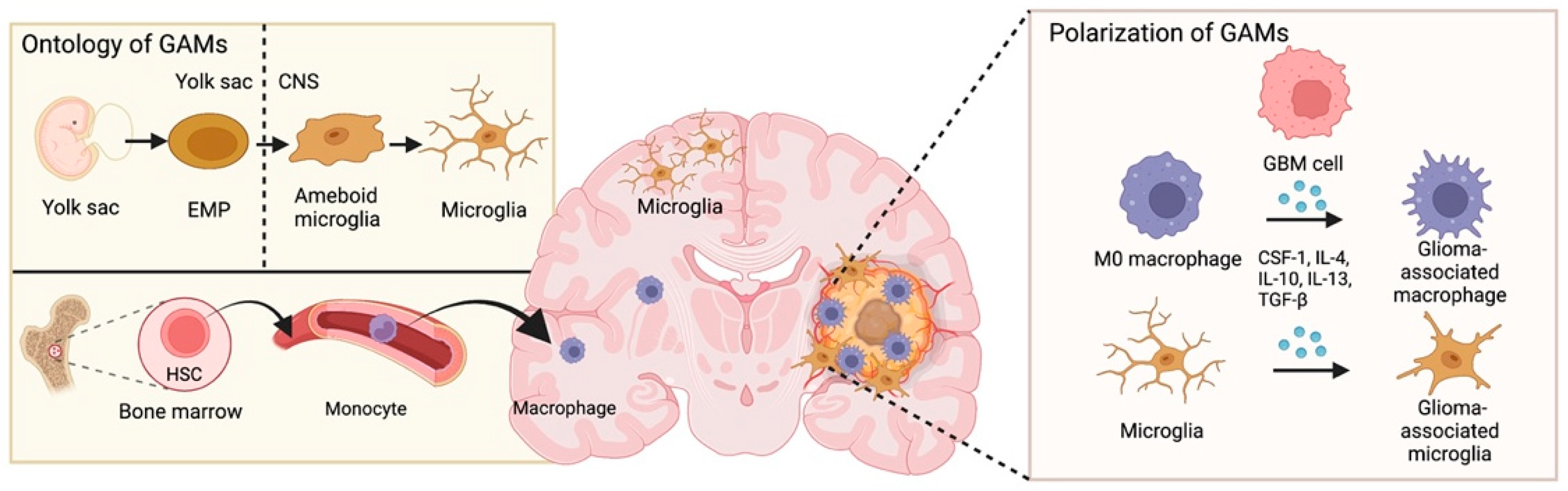{kind=link}
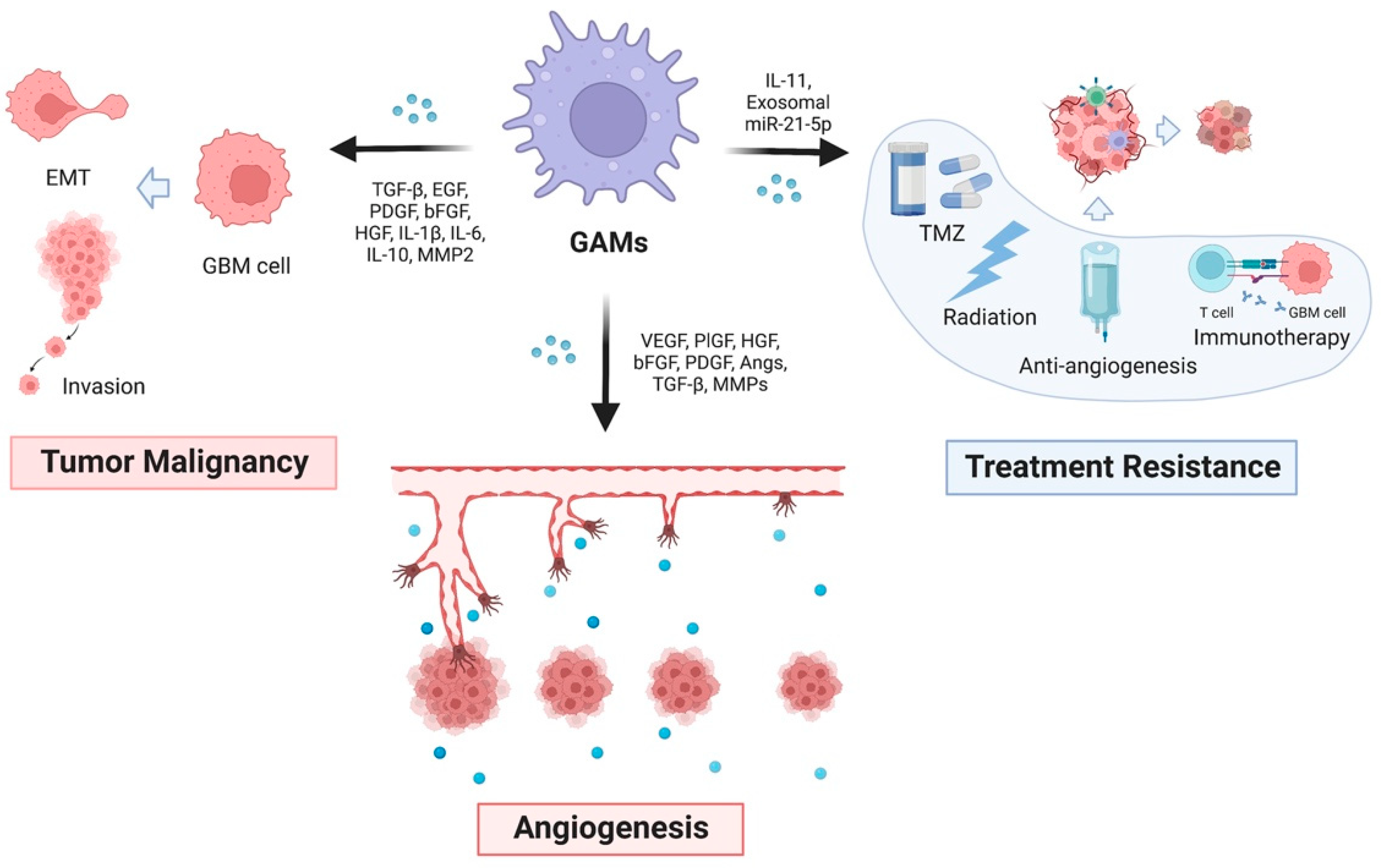{kind=link}
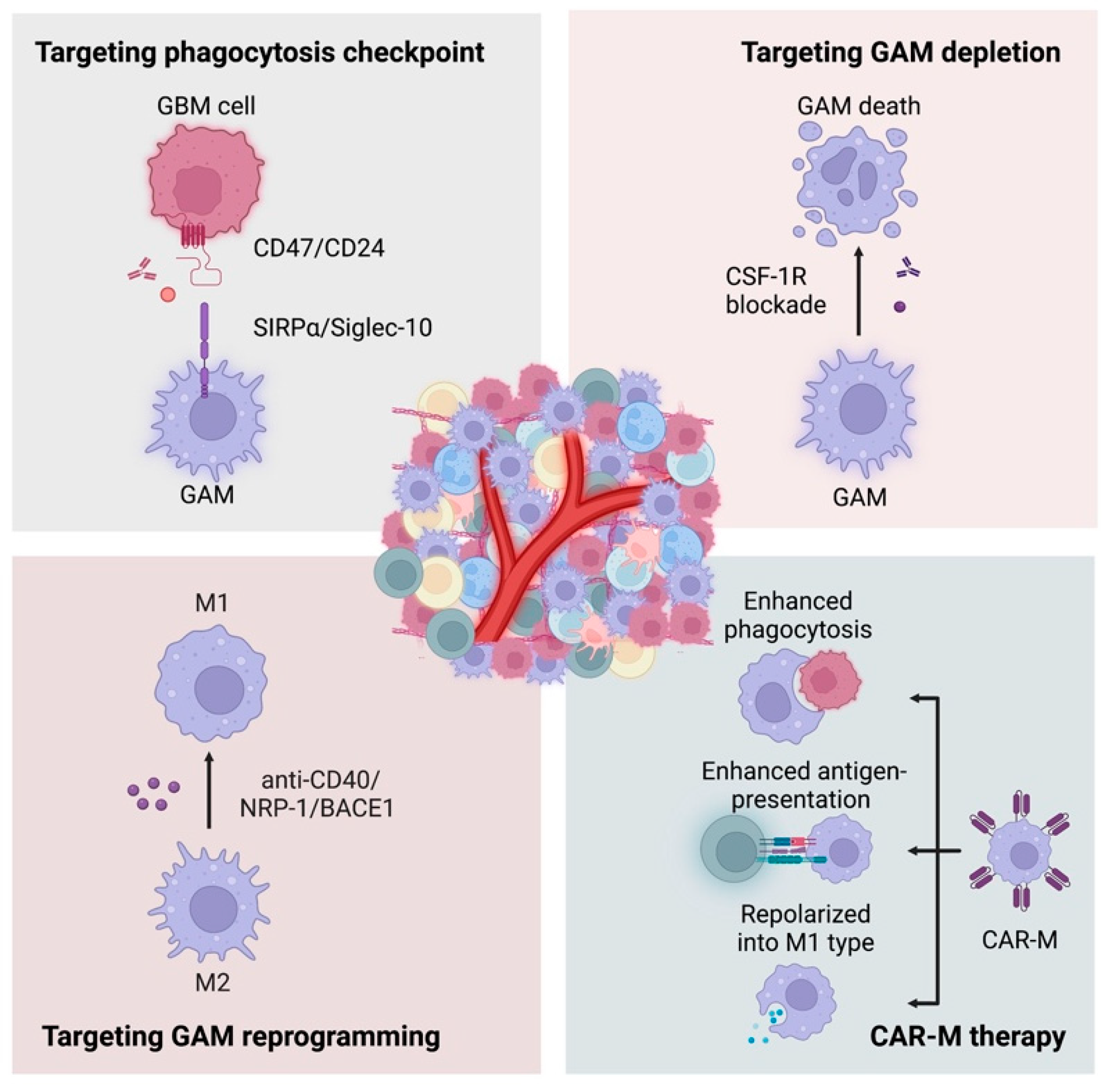{kind=link}
Table 1.
Clinical trials of macrophage-targeted therapies in glioblastoma.
| Category | Therapeutic Approach | Conditions | Phases | Enrollment | Study Number | Status | |
|---|---|---|---|---|---|---|---|
| Phagocytosis Checkpoints Blockade | CD47 | Magrolimab | Brain Cancer | Phase 1 | 24 | NCT05169944 | Not yet recruiting |
| RRx-001 | Brain Tumor | Phase 1 | 24 | NCT04525014 | Recruiting | ||
| RRx-001 | Newly Diagnosed GBM | Phase 1 | 19 | NCT02871843 | Completed | ||
| IBI188 | Advanced Malignancies | Phase 1 | 49 | NCT03717103 | Completed | ||
| AK117 | Neoplasms Malignant | Phase 1 | 162 | NCT04728334 | Recruiting | ||
| AK118 | Neoplasms Malignant | Phase 1 | 159 | NCT04349969 | Not yet recruiting | ||
| HX009 | Advanced Solid Tumor | Phase 1 | 21 | NCT04097769 | Active, not recruiting | ||
| IBC0966 | Advanced Malignant Tumors | Phase 1 | 228 | NCT04980690 | Not yet recruiting | ||
| IBI322 | Advanced Solid Tumor | Phase 1 | 36 | NCT04912466 | Not yet recruiting | ||
| IBI323 | Advanced Solid Tumor | Phase 1 | 218 | NCT04328831 | Recruiting | ||
| IBI324 | Advanced Malignancies | Phase 1 | 45 | NCT04338659 | Not yet recruiting | ||
| SRF231 | Advanced Solid Cancers Hematologic Cancers | Phase 1 | 148 | NCT03512340 | Completed | ||
| STI-6643 | Solid Tumor Relapsed Solid Neoplasm Refractory Tumor | Phase 1 | 24 | NCT04900519 | Recruiting | ||
| TQB2928 | Advanced Cancer | Phase 1 | 180 | NCT05192512 | Recruiting | ||
| SIRPα | BI 765063 | Solid Tumor, Adult | Phase 1 | 116 | NCT03990233 | Recruiting | |
| CC-95251 | Neoplasms | Phase 1 | 230 | NCT03783403 | Recruiting | ||
| GAM-depletion | CSF-1R | Cabiralizumab | Malignant Glioma, and other solid tumors | Phase 1 | 313 | NCT02526017 | Completed |
| PLX3397 | Recurrent GBM | Phase 2 | 38 | NCT01349036 | Terminated | ||
| PLX3397 | Newly Diagnosed GBM | Phase 1/2 | 65 | NCT01790503 | Completed | ||
| ARRY-382 | Advanced Solid Tumors | Phase 2 | 82 | NCT02880371 | Completed | ||
| CXCR4 | USL311 | Relapsed/Recurrent GBM | Phase 2 | 26 | NCT02765165 | Terminated | |
| Other GAM-based therapy | Oncolytic virus | DNX-2401 | Recurrent High-Grade Glioma | Phase 1 | 36 | NCT03896568 | Recruiting |
| Brainstem Glioma | Phase 1 | 24 | NCT03178032 | Active, not recruiting | |||
| GBM | Phase 1 | 37 | NCT02197169 | Completed | |||
| Recurrent Malignant Gliomas | Phase 1 | 37 | NCT00805376 | Completed | |||
| Recurrent GBM | Phase 1 | 31 | NCT01956734 | Completed | |||
| GBM, and other brain tumors | Phase 2 | 49 | NCT02798406 | Completed |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wu, M.; Shi, Y.; Zhu, L.; Chen, L.; Zhao, X.; Xu, C. Macrophages in Glioblastoma Development and Therapy: A Double-Edged Sword. Life 2022, 12, 1225. https://doi.org/10.3390/life12081225
AMA Style
Wu M, Shi Y, Zhu L, Chen L, Zhao X, Xu C. Macrophages in Glioblastoma Development and Therapy: A Double-Edged Sword. Life. 2022; 12(8):1225. https://doi.org/10.3390/life12081225
Chicago/Turabian StyleWu, Mengwan, Ying Shi, Luyi Zhu, Luoyi Chen, Xinchen Zhao, and Chuan Xu. 2022. "Macrophages in Glioblastoma Development and Therapy: A Double-Edged Sword" Life 12, no. 8: 1225. https://doi.org/10.3390/life12081225
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.