
Temporal Dominance of B.1.1.7 over B.1.354 SARS-CoV-2 Variant: A Hypothesis Based on Areas of Variant Co-Circulation

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
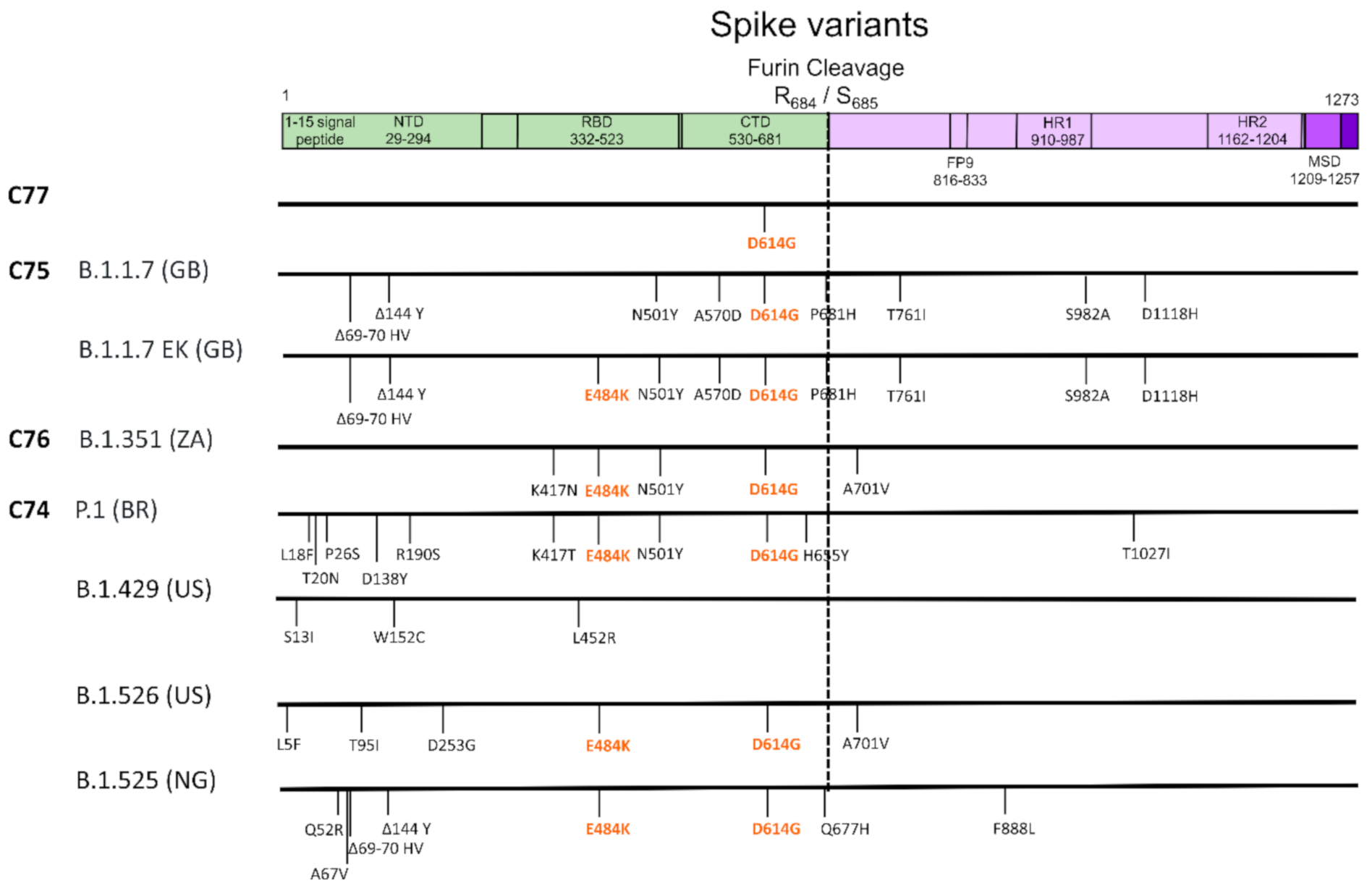
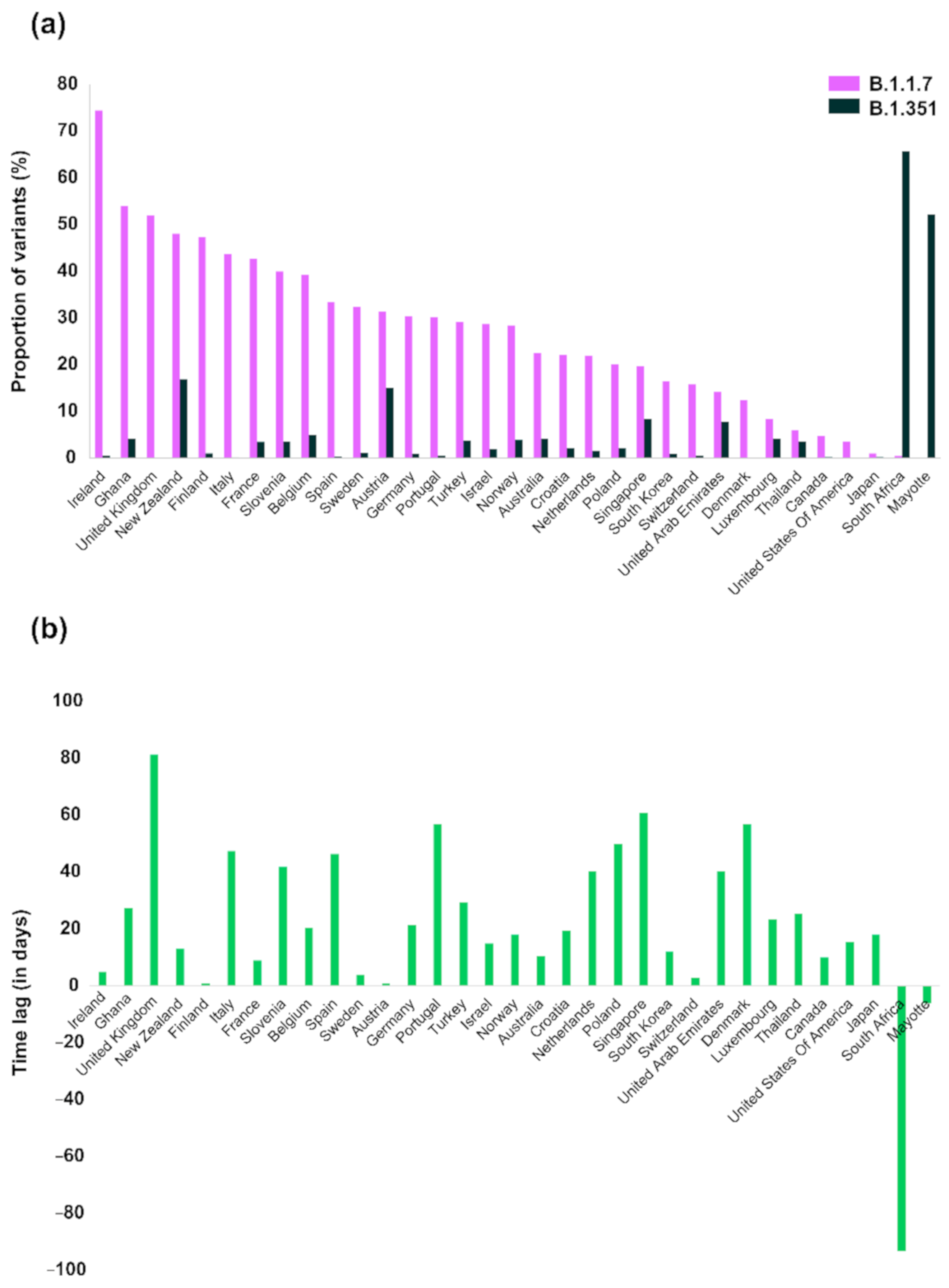
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Weekly Epidemiological Update—23 February 2021 Special Edition: Proposed Working Definitions of SARS-CoV-2 Variants of Interestand Variants of Concern; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Loman, N.; Pybus, O.; Barclay, W.; Barrett, J.; Carabelli, A.; Connor, T.; Peacock, T.; Robertson, D.L.; Volz, E. Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 2 April 2021).
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv 2020. [Google Scholar] [CrossRef]
- Faria, N.R.; Claro, I.M.; Candido, D.; Franco, L.A.M.; Andrade, P.S.; Coletti, T.M.; Silva, C.A.M.; Sales, F.C.; Manuli, E.R.; Aguiar, R.S.; et al. Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in Manaus: Preliminary Findings. Available online: https://virological.org/t/genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-manaus-preliminary-findings/586 (accessed on 2 April 2021).
- Zhang, W.; Davis, B.D.; Chen, S.S.; Martinez, J.M.S.; Plummer, J.T.; Vail, E. Emergence of a Novel SARS-CoV-2 Variant in Southern California. JAMA 2021, 325, 1324–1326. [Google Scholar] [CrossRef] [PubMed]
- Lasek-Nesselquist, E.; Lapierre, P.; Schneider, E.; George, K.S.; Pata, J. The localized rise of a B.1.526 SARS-CoV-2 variant containing an E484K mutation in New York State. medRxiv 2021. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.; Buzko, O.; Spilman, P.; Niazi, K.; Rabizadeh, S.; Soon-Shiong, P. Molecular dynamic simulation reveals E484K mutation enhances spike RBD-ACE2 affinity and the combination of E484K, K417N and N501Y mutations (501Y.V2 variant) induces conformational change greater than N501Y mutant alone, potentially resulting in an escape mutant. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kissler, S.; Fauver, J.R.; Mack, C.; Tai, C.G.; Breban, M.I.; Watkins, A.E.; Samant, R.M.; Anderson, D.J.; Ho, D.D.; Grubaugh, N.D.; et al. Densely sampled viral trajectories suggest longer duration of acute infection with B.1.1.7 variant relative to non-B.1.1.7 SARS-CoV-2. Harvard University’s DASH repository 2021. medRxiv 2021. [Google Scholar] [CrossRef]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7. Nature 2021, 2021, 1–6. [Google Scholar] [CrossRef]
- Cele, S.; Gazy, I.; Jackson, L.; Hwa, S.-H.; Tegally, H.; Lustig, G.; Giandhari, J.; Pillay, S.; Wilkinson, E.; Naidoo, Y.; et al. Escape of SARS-CoV-2 501Y.V2 from neutralization by convalescent plasma. Nature 2021, 2021, 1–9. [Google Scholar] [CrossRef]
- Wibmer, C.K.; Ayres, F.; Hermanus, T.; Madzivhandila, M.; Kgagudi, P.; Oosthuysen, B.; Lambson, B.E.; de Oliveira, T.; Vermeulen, M.; van der Berg, K.; et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat. Med. 2021, 27, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Wadman, M.; Cohen, J. Novavax vaccine delivers 89% efficacy against COVID-19 in U.K.—But is less potent in South Africa. Science 2021. [Google Scholar] [CrossRef]
- Callaway, E.; Mallapaty, S. Novavax offers first evidence that COVID vaccines protect people against variants. Nature 2021, 590, 17. [Google Scholar]
- Madhi, S.A.; Baillie, V.; Cutland, C.L.; Voysey, M.; Koen, A.L.; Fairlie, L.; Padayachee, S.D.; Dheda, K.; Barnabas, S.L.; Bhorat, Q.E.; et al. Efficacy of the ChAdOx1 nCoV-19 Covid-19 Vaccine against the B.1.351 Variant. N. Engl. J. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Baele, G.; Suchard, M.A.; Rambaut, A.; Lemey, P. Emerging Concepts of Data Integration in Pathogen Phylodynamics. Syst. Biol. 2017, 66, e47–e65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobey, S. Pathogen evolution and the immunological niche. Ann. N. Y. Acad. Sci. 2014, 1320, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| VOC | B.1.1.7 | B.1.351 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Countries | Proportion (%) | Proportion of SGTF 1—Week 8 of 2021 (%) | Earliest Variant Sequence | Number of Variant Sequences | Number of Sequences 2 | Proportion (%) | Earliest Variant Sequence | Number of Variant Sequences | Number of Sequences 2 |
| Ireland | 74.50 | 88.6 | 17/12/2020 | 1966 | 2639 | 0.62 | 22/12/2020 | 16 | 2594 |
| United Kingdom | 51.99 | NA 3 | 20/09/2020 | 111,140 | 213,761 | 0.16 | 10/12/2020 | 212 | 135,667 |
| Ghana | 54.03 | NA | 10/12/2020 | 67 | 124 | 4.21 | 06/01/2021 | 4 | 95 |
| Finland | 47.43 | NA | 18/12/2020 | 268 | 565 | 1.07 | 19/12/2020 | 6 | 563 |
| Italy | 43.67 | NA | 14/12/2020 | 2244 | 5139 | 0.21 | 30/01/2021 | 8 | 3788 |
| France | 42.75 | 65.8 | 13/12/2020 | 1847 | 4320 | 3.48 | 22/12/2020 | 144 | 4137 |
| Slovenia | 40.00 | NA | 29/12/2020 | 46 | 115 | 3.64 | 09/02/2021 | 2 | 55 |
| Belgium | 39.33 | 46.3 | 30/11/2020 | 2064 | 5248 | 5.02 | 20/12/2020 | 244 | 4857 |
| Spain | 33.53 | 25–30 | 08/11/2020 | 1323 | 3946 | 0.34 | 24/12/2020 | 10 | 2930 |
| Sweden | 32.39 | 41.5 4 | 20/12/2020 | 458 | 1414 | 1.22 | 24/12/2020 | 16 | 1310 |
| New Zealand | 48.09 | NA | 16/12/2020 | 63 | 131 | 16.96 | 29/12/2020 | 19 | 112 |
| Portugal | 30.17 | 50.5 | 09/11/2020 | 746 | 2473 | 0.51 | 04/01/2021 | 9 | 1782 |
| Germany | 30.44 | 54.5 | 30/11/2020 | 4427 | 14,543 | 1.03 | 21/12/2020 | 143 | 13,817 |
| Israel | 28.74 | ~90 5 | 16/12/2020 | 434 | 1510 | 1.92 | 31/12/2020 | 16 | 833 |
| Turkey | 29.15 | NA | 24/12/2020 | 479 | 1643 | 3.78 | 22/01/2021 | 54 | 1428 |
| Norway | 28.49 | 72.5 | 09/12/2020 | 337 | 1183 | 3.92 | 27/12/2020 | 40 | 1021 |
| Netherlands | 22.00 | 64.3 5 | 12/11/2020 | 1697 | 7715 | 1.63 | 22/12/2020 | 99 | 6091 |
| Croatia | 22.06 | NA | 20/01/2021 | 60 | 272 | 2.13 | 09/02/2021 | 2 | 94 |
| Australia | 22.52 | NA | 30/11/2020 | 134 | 595 | 4.14 | 10/12/2020 | 23 | 556 |
| Poland | 20.08 | 9 | 22/12/2020 | 151 | 752 | 2.11 | 10/02/2021 | 2 | 95 |
| Austria | 31.49 | 63.2 | 22/12/2020 | 336 | 1067 | 15.09 | 23/12/2020 | 159 | 1054 |
| South Korea | 16.45 | NA | 14/12/2020 | 90 | 547 | 0.97 | 26/12/2020 | 4 | 413 |
| Switzerland | 15.80 | 40.5 5 | 09/11/2020 | 1983 | 12,550 | 0.63 | 12/11/2020 | 77 | 12,313 |
| Denmark | 12.47 | 76.5 | 09/11/2020 | 4889 | 39,191 | 0.06 | 04/01/2021 | 12 | 19,655 |
| Singapore | 19.70 | NA | 08/12/2020 | 66 | 335 | 8.45 | 07/02/2021 | 6 | 71 |
| United Arab Emirates | 14.19 | NA | 16/11/2020 | 21 | 148 | 7.81 | 26/12/2020 | 5 | 64 |
| Canada | 4.83 | NA | 15/12/2020 | 54 | 1117 | 0.26 | 25/12/2020 | 2 | 761 |
| Luxembourg | 8.40 | 65.5 | 24/12/2020 | 32 | 381 | 4.17 | 16/01/2021 | 2 | 48 |
| United States of America | 3.58 | 26.2 | 17/12/2020 | 2652 | 74,129 | 0.06 | 01/01/2021 | 38 | 62,390 |
| Thailand | 6.06 | NA | 08/01/2021 | 8 | 132 | 3.61 | 03/02/2021 | 3 | 83 |
| Japan | 1.07 | NA | 01/12/2020 | 59 | 5527 | 0.26 | 19/12/2020 | 9 | 3441 |
| Mayotte | 0.19 | NA | 13/01/2021 | 1 | 518 | 52.24 | 07/01/2021 | 338 | 647 |
| South Africa | 0.47 | NA | 09/01/2021 | 1 | 213 | 65.74 | 08/10/2020 | 1086 | 1652 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostaki, E.G.; Tseti, I.; Tsiodras, S.; Pavlakis, G.N.; Sfikakis, P.P.; Paraskevis, D. Temporal Dominance of B.1.1.7 over B.1.354 SARS-CoV-2 Variant: A Hypothesis Based on Areas of Variant Co-Circulation. Life 2021, 11, 375. https://doi.org/10.3390/life11050375
Kostaki EG, Tseti I, Tsiodras S, Pavlakis GN, Sfikakis PP, Paraskevis D. Temporal Dominance of B.1.1.7 over B.1.354 SARS-CoV-2 Variant: A Hypothesis Based on Areas of Variant Co-Circulation. Life. 2021; 11(5):375. https://doi.org/10.3390/life11050375
Chicago/Turabian StyleKostaki, Evangelia Georgia, Ioulia Tseti, Sotirios Tsiodras, George N. Pavlakis, Petros P. Sfikakis, and Dimitrios Paraskevis. 2021. "Temporal Dominance of B.1.1.7 over B.1.354 SARS-CoV-2 Variant: A Hypothesis Based on Areas of Variant Co-Circulation" Life 11, no. 5: 375. https://doi.org/10.3390/life11050375