The Maintenance of Mitochondrial DNA Integrity and Dynamics by Mitochondrial Membranes


1
Wellcome Centre for Mitochondrial Research, Faculty of Medical Sciences, Newcastle University, Newcastle upon Tyne NE2 4HH, UK
2
Biosciences Institute, Faculty of Medical Sciences, Newcastle University, Newcastle upon Tyne NE2 4HH, UK
3
Translational and Clinical Research Institute, Faculty of Medical Sciences, Newcastle University, Newcastle upon Tyne NE2 4HH, UK
*
Authors to whom correspondence should be addressed.
Life 2020, 10(9), 164; https://doi.org/10.3390/life10090164
Submission received: 27 July 2020
/
Revised: 20 August 2020
/
Accepted: 23 August 2020
/
Published: 26 August 2020
(This article belongs to the Special Issue Mitochondria: From Physiology to Pathology)
Abstract
:Mitochondria are complex organelles that harbour their own genome. Mitochondrial DNA (mtDNA) exists in the form of a circular double-stranded DNA molecule that must be replicated, segregated and distributed around the mitochondrial network. Human cells typically possess between a few hundred and several thousand copies of the mitochondrial genome, located within the mitochondrial matrix in close association with the cristae ultrastructure. The organisation of mtDNA around the mitochondrial network requires mitochondria to be dynamic and undergo both fission and fusion events in coordination with the modulation of cristae architecture. The dysregulation of these processes has profound effects upon mtDNA replication, manifesting as a loss of mtDNA integrity and copy number, and upon the subsequent distribution of mtDNA around the mitochondrial network. Mutations within genes involved in mitochondrial dynamics or cristae modulation cause a wide range of neurological disorders frequently associated with defects in mtDNA maintenance. This review aims to provide an understanding of the biological mechanisms that link mitochondrial dynamics and mtDNA integrity, as well as examine the interplay that occurs between mtDNA, mitochondrial dynamics and cristae structure.
1. Introduction
Mitochondria act as metabolic hubs within the cell to facilitate a myriad of essential cellular processes such as energy production, the regulation of apoptosis and cellular signalling pathways, amongst others. They are unique organelles in the fact that they harbour their own genome that is distinct from the nuclear genome. In human cells, this consists of a circular double-stranded DNA molecule, referred to as mitochondrial DNA (mtDNA). A typical cell possesses between a few hundred and several thousand copies of mtDNA that are replicated independently of the cell cycle within the mitochondrial matrix and segregated between mitochondria. These genomes are closely interlinked with the cristae ultrastructure of the mitochondrion. Once they have been replicated, mtDNA molecules are subsequently distributed around the mitochondrial network by processes that rely on the plastic nature of mitochondria to undertake fission and fusion events, as well as the modulation of cristae structure. Defects in the fission and fusion machinery, or in proteins associated with modulating cristae structure, disrupt the even allocation of mtDNA throughout the network and subsequently to daughter organelles and cells. Defects in mtDNA metabolism typically manifest as the accumulation of mtDNA molecules harbouring deletions and/or as a depletion in the number of copies of mtDNA per cell. The close association between mtDNA and cellular energy production means that the loss of mtDNA number and integrity limits the capacity for the mitochondria to meet the energy demands of an organism. From a clinical perspective, patients carrying mutations within dynamics or cristae-associated genes display heterogeneous neurological phenotypes. There is a clear requirement to understand the basic biological processes linking mitochondrial structure with mtDNA maintenance and its links to mitochondrial disease.
This review aims to disentangle the relationships that exist between mitochondrial dynamics, cristae structure and the organisation of mtDNA. In addition, the biological mechanisms that may prompt the disruption of mtDNA integrity following the impairment of mitochondrial dynamics are assessed. Finally, these mechanisms are discussed in context with observations from the clinic.
2. Mitochondrial DNA
2.1. The Origins of mtDNA
mtDNA is present in the mitochondria of almost all eukaryotic organisms, and the advent of genome sequencing has allowed its evolutionary origins to be dissected. It is now understood that mtDNA derives from an event whereby a host cell engulfed an alphaproteobacterium [1]. This endosymbiotic occurrence fostered a relationship in which the bacterium was utilised for its energy-producing capacity by the host cell. This gave rise to the first complex eukaryotic cells, and since then the course of evolution and the transfer of mitochondrial genes to the nucleus has led to mitochondrial genomes that vary significantly in both structure and size between modern eukaryotic organisms. For example, higher plants harbour genomes that are typically 200–300 kb in size made up of linear and small circular regions of DNA, whereas, algae and fungi have much smaller linear genomes in the region of 30–90 kb [2,3].
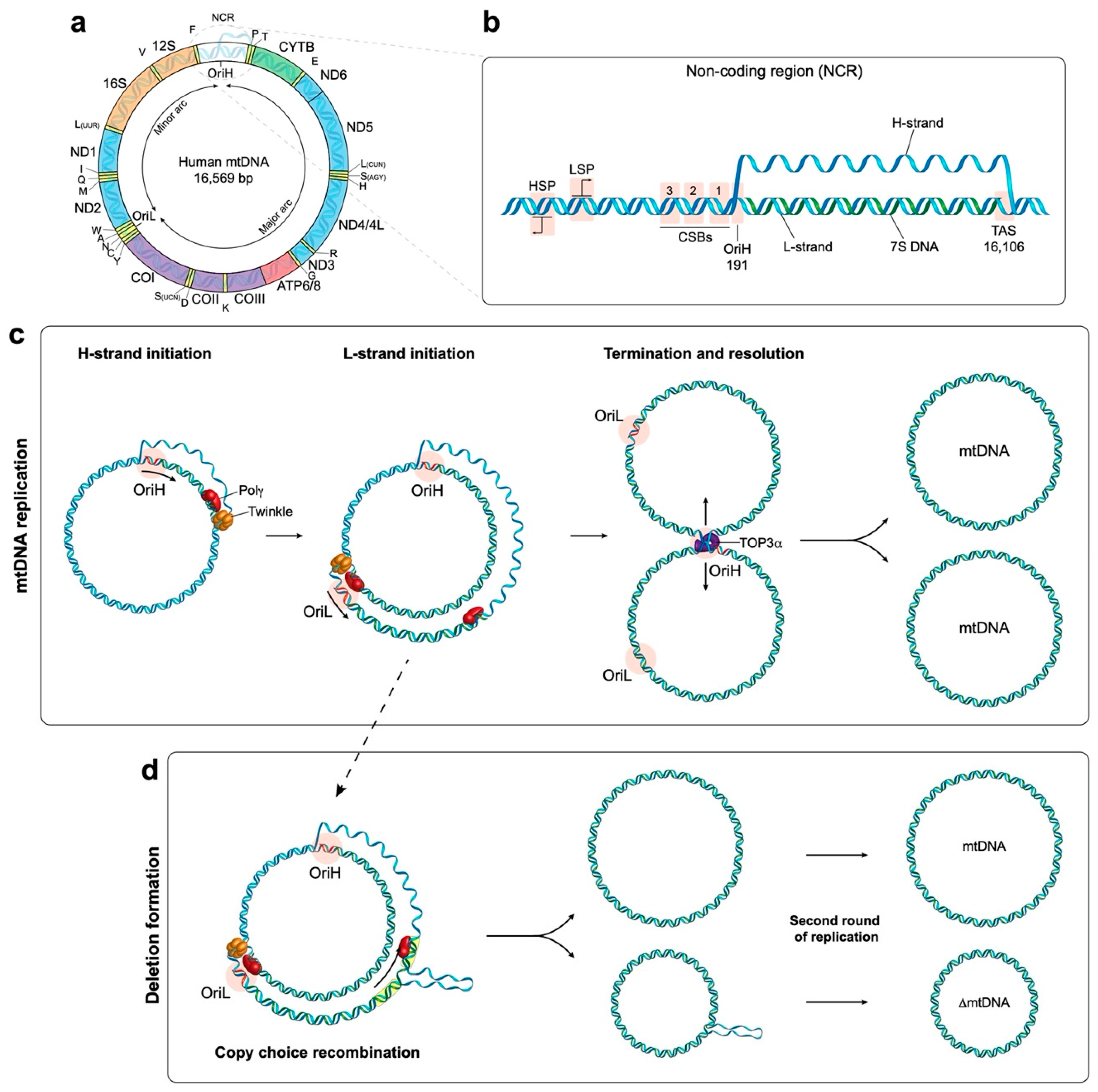
Human mtDNA is 16,569 base pairs in length and is organised into a double-stranded circular structure containing 37 genes which encodes for 13 mitochondrial proteins (Figure 1a) [4]. These proteins are all essential components of the oxidative phosphorylation (OXPHOS) system. The OXPHOS machinery is made up of four respiratory chain complexes and the ATP (adenosine triphosphate) synthase which are responsible for energy production in the currency of ATP [5]. In addition, mtDNA encodes 22 transfer RNA (tRNA) and 2 ribosomal RNA (rRNA) molecules which are components of the mitochondrial translation system [4]. The rest of the mitochondrial proteome, currently estimated at 1158 proteins [6], is encoded by the nucleus as the result of the lateral transfer of mitochondrial genes [1]. This evolutionary pressure towards mtDNA reduction means that human mtDNA possesses very few noncoding regions and contains areas of overlapping genes [4]. The OXPHOS system is made up of approximately 90 proteins, and as such is comprised of the products of both mitochondrial and nuclear genes [7]. This dual-genetic origin requires that nuclear-encoded subunits be translated in the cytosol prior to being imported into the mitochondria. Conversely, subunits encoded by mtDNA are synthesised and assembled within the mitochondria by a dedicated mitochondrial translation machinery. Once the nuclear subunits are imported, they are assembled alongside the mtDNA-encoded subunits to form the respiratory complexes that make up the electron transport chain.
2.2. mtDNA Replication and Segregation
Unlike nuclear DNA, the replication of mtDNA occurs throughout the cell cycle. The process of mtDNA replication utilises a different set of proteins to those which carry out replication of the nuclear genome [8]. These proteins are all nuclear-encoded and are imported into the mitochondrial matrix from the cytosol. The mitochondrial transcription machinery generates the primers for mtDNA synthesis and can therefore be considered essential components of the mtDNA replication machinery. The mechanisms of replication will be outlined in brief here; for further details there are a number of excellent reviews on the topic [9,10].
2.2.1. mtDNA Transcription
Human mtDNA consists of a ‘heavy’ (guanine rich) and a ‘light’ (cytosine rich) strand, with each strand having a single promoter region located close together within the noncoding region (NCR) of the genome (Figure 1b) [11]. These are referred to as the light strand promoter (LSP) and heavy strand promoter (HSP) respectively and facilitate polycistronic transcription of both strands of mtDNA almost in their entirety. Transcription initiation minimally requires the mitochondrial RNA polymerase (POLRMT) with mitochondrial transcription factor A (TFAM) and mitochondrial transcription factor B2 (TFB2M) [12,13]. Following initiation, the mitochondrial transcription elongation factor TEFM promotes POLRMT processivity [14,15]. RNA primers responsible for the initiation of DNA synthesis are generated when transcription from the LSP is terminated prematurely around a series of conserved sequence blocks (CSBs) downstream of LSP, forming an R-loop [16]. This allows the DNA synthesis machinery to assemble and leads to the initiation of DNA synthesis from OriH [17,18].
2.2.2. mtDNA Replication
The protein complex responsible for DNA synthesis is termed the replisome. At the core of the replisome is DNA polymerase-γ (POLγ) which is responsible for synthesising the DNA [19]. POLγ is a heterotrimer made up of a catalytic subunit (POLγA) which functions as a highly accurate proofreader of the newly synthesised DNA [20]. The processivity of POLγA is increased by two copies of the accessory subunit (POLγB) which interact with the DNA substrate [21,22]. In order for POLγ to access and replicate the DNA, it is necessary for the dsDNA to first be unwound. This is performed by the DNA helicase TWINKLE which travels in front of the replication fork and unwinds DNA in the 5′ to 3′ direction [23]. The activity of both TWINKLE and POLγ is enhanced by the mitochondrial single stranded DNA-binding protein (mtSSB) [24]. mtSSB has also been demonstrated to bind to and stabilise the unwound single stranded DNA behind the replication fork [23,24].
During the initial phase of DNA synthesis only the H-strand is replicated. Once the replisome has travelled approximately 12,000 bp it reaches the origin of L-strand replication (OriL), which becomes single stranded and folds into a stem-loop structure [25]. This structure prevents mtSSB from binding and provides a stretch of poly(T) ssDNA that is accessible to POLRMT [26,27]. POLRMT transcribes a short 25–75 bp primer on the single-stranded template, from which synthesis of the L-strand is initiated [27]. See Figure 1c for a schematic overview of mtDNA replication. The strand displacement model proposes that the long tract of exposed H-strand ssDNA is coated and protected by mtSSB until DNA synthesis is initiated from OriL. This model has been reviewed in depth elsewhere [9]. In addition to this model, the bootlace model proposes that the displaced single-stranded lagging-strand template DNA is instead coated by RNA transcripts [28]. Fully double-stranded replication intermediates reminiscent of coupled leading and lagging-strand DNA replication have also been observed and characterised using two-dimensional agarose gel electrophoresis [29,30,31].
2.2.3. Termination of mtDNA Replication
Once DNA synthesis is complete, the primers at OriL and OriH must be removed to allow for ligation of the DNA ends. Studies have revealed that RNase H1 is involved in the process of primer removal as the loss of RNase H1 in mouse embryonic fibroblasts (MEF) leads to the retention of the primers at sites including both origins of replication; OriL and OriH [32]. The role of RNase H1 in removing primers has been reconstituted in vitro at OriL [33]. Furthermore, the loss of RNase H1 in vivo in mice is embryonically lethal as a consequence of significant mtDNA loss [34]. Following primer removal, the DNA ends need to be prepared to allow ligation by DNA ligase III to occur [35,36]. In a reconstituted system, the mitochondrial genome maintenance exonuclease 1 (MGME1) protein facilitates efficient ligation by modifying single stranded DNA overhangs that occur on the 5′ end of the newly synthesised DNA following primer removal [36]. The loss of MGME1 in vivo has been shown to result in diminished ligation at OriH [37].
Once replication is complete, the two genomes remain connected at the OriH region by a single stranded linkage (termed a hemicatenane) and must be separated (see Figure 1c for a schematic overview). Recent work has revealed that topoisomerase 3α, a type 1A topoisomerase, cleaves the single strand linkage to allow passage of one of the strands to occur, resulting in the separation of the two mtDNA molecules [38].
2.2.4. Formation of mtDNA Deletions
Common manifestations of defects in mtDNA maintenance are deletions and rearrangements of mtDNA. Single, large-scale deletions can cause mitochondrial DNA diseases if they undergo clonal expansion to accumulate beyond a biochemical threshold, typically 60–90% [39]. Such deletions are sporadic and believed to be formed during embryonic development. The most well-studied single deletion is the 4977 bp “common deletion”, underlying Pearson’s syndrome and Kearns–Sayre syndrome in early life, and chronic progressive external ophthalmoplegia (CPEO) in later life [40]. Alternatively, multiple mtDNA deletions can occur secondarily to disease-causing mutations in nuclear genes that encode factors involved in mtDNA replication, nucleotide metabolism and mitochondrial dynamics [41]. Multiple deletions are also observed in post-mitotic tissues during normal ageing [42,43]. Both the mechanism of deletion formation and the mechanism of clonal expansion of deletions have been the subject of debate. The clonal expansion of mtDNA deletions has been recently reviewed extensively elsewhere [44].
The formation of mtDNA deletions has been proposed to occur either during mtDNA replication or as the result of double-strand breaks [45]. An early model proposed a slip-replication mechanism for the formation of the common deletion [46]. This model involves the annealing of the displaced H-strand to a downstream repeat sequence in the leading-strand template, leading to the removal of the sequence between the two repeats [46]. A more recent model that is supported by in vitro reconstitution experiments suggests that deletions are the result of copy-choice recombination [45,47]. Deletions in mtDNA predominantly form in the major arc between OriH and OriL in the direction of replication. During the synthesis of the L-strand, replication slippage can occur. Specifically, the 3′ end of the nascent L-strand becomes dissociated from the H-strand template at one repeat sequence, and subsequently reanneals to another repeat sequence further along the template. This produces a heteroduplex molecule consisting of a complete H-strand and a shortened L-strand harbouring the deletion. Subsequent rounds of replication will produce shortened mtDNA molecules containing the deletion, see Figure 1d [47].
It has also been observed that inducing high levels of double-strand breaks in mtDNA, which are normally rapidly degraded [48], can result in the formation of deletions [49,50]. Therefore, it has been suggested that limited nucleolytic processing of double-strand breaks could lead to the annealing of repeat sequences and the generation of deletion-containing mtDNA molecules [45,51]. As mitochondria have not been found to possess repair pathways for double-strand breaks comparable to those in the nucleus, this mechanism would presumably operate by a distinct mechanism.
2.3. mtDNA Packaging
A typical mammalian cell harbours between 1000 and 10,000 copies of the mtDNA genome that exist in a DNA-protein complex, termed a nucleoid [52]. Studies using super-resolution microscopy have revealed that nucleoids generally contain a single copy of mtDNA and have a diameter of approximately 100 nm [53,54]. This small size is attributed to the compaction of mtDNA by TFAM. Specifically, TFAM binds along the length of mtDNA at a ratio of 1 subunit per 16–17 bp, instigating bending and loop formation along the DNA backbone that results in compaction [55,56,57,58]. The stable protein filaments that are formed prevent POLRMT and TWINKLE from accessing the DNA [52,55]. Reconstituted nucleoids in vitro were demonstrated to become progressively more compact with increasing levels of TFAM, and a wide range of packaging densities were observed at typical physiological concentrations of TFAM [52]. It has therefore been speculated that TFAM may regulate the overall transcription and replication rate of mtDNA by controlling its accessibility to relevant proteins. TFAM is the most abundant protein found associated with nucleoids, however, a number of proteins related to replication, transcription and translation are also commonly found in association with nucleoids [59,60,61]. It has been proposed that these proteins are located within the “inner core” of the nucleoid, whereas other proteins such as mitochondrial chaperones and membrane binding proteins constitute the peripheral layer [62].
3. Membrane Dynamics and the Organisation of mtDNA
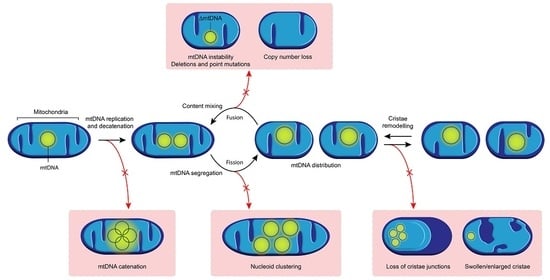
Once the mitochondrial genomes have been separated, they need to be segregated and dispersed around the mitochondrial network. Because the OXPHOS complexes are composed of both mtDNA-encoded and nuclear-encoded subunits, the complexes are assembled in situ proximal to the nucleoid [63,64]. The capacity for mtDNA to spatially diffuse by itself is limited and so nucleoids require mitochondrial membrane structure and dynamics to aid in their distribution around the mitochondrial network [65]. The disruption of nucleoid distribution can lead to a mosaic pattern of respiratory activity, in which only regions of the mitochondrial network that contain nucleoids are capable of assembling OXPHOS complexes and are therefore capable of oxidative ATP production [66]. Structurally, mitochondria are composed of two phospholipid membranes arranged as an outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM). The space between these two membranes is referred to as the intermembrane space (IMS). The IMM is intricately folded to form the cristae structures that harbour the respiratory chain complexes. The interior space enclosed by the IMM is referred to as the matrix and contains the mitochondrial genome. mtDNA is closely associated with the IMM and is suggested to be physically attached, a relationship that likely aids in its distribution [67,68,69]. Early studies identified a protein complex bound to the OriH region of mtDNA and to the IMM, although the factors responsible were not identified [67,70]. It has since been found visually using electron microscopy and immuno-gold labelling that mtDNA is found in close apposition to the IMM [69]. A number of plausible candidates for mtDNA tethering are discussed later in this review. This section aims to evaluate how the dynamic nature of mitochondrial membranes facilitate the distribution of the replicated genomes around the network.
3.1. Mitochondrial Fission and Its Role in mtDNA Distribution
Mitochondria cannot be synthesised de novo; they must have the capacity to grow and divide in order to distribute the replicated genomes to daughter mitochondria. The appearance of mitochondria within a cell can vary significantly; they can exist as isolated entities or be fused together in vast sprawling networks [71]. This versatile nature allows them to sustain energy production as well as act as signaling platforms in complex cellular processes such as apoptosis, autophagy and senescence [72,73,74]. There are two main mechanisms that underlie mitochondrial dynamics: fission and fusion. First, the relationship between fission and mtDNA distribution and maintenance will be discussed.
3.1.1. Mitochondrial Fission
Mitochondrial fission is the division of a mitochondrion into multiple distinct mitochondria and has been implicated in the distribution of nucleoids around the mitochondrial network [75,76,77]. Fission requires the progressive constriction, and eventual scission, of the IMM and OMM. The initial constriction occurs at contact sites between the OMM and the endoplasmic reticulum (ER), where actin polymerisation provides the force required to contract the mitochondrial membrane [78,79,80]. Further constriction is primarily fulfilled by dynamin-related protein 1 (DRP1), a cytosolic protein that translocates to the OMM and interacts with several adaptor proteins [81]. DRP1 binds with mitochondrial fission factor 1 (MFF), mitochondrial dynamics protein of 49kDa (MID49), MID51 and mitochondrial fission 1 protein (FIS1) [82,83,84,85]. Utilising GTP (guanine triphosphate), polymerisation of DRP1 with MID49 and MID51 occurs leading to the formation of linear filaments. GTP hydrolysis induces the oligomerisation of DRP1 and filament shortening to create rings which constrict around the mitochondria [79,86]. It has now been established that DRP1-mediated constriction is sufficient for the final scission step to separate mitochondria [87,88]. Recent studies have also highlighted the importance of additional interorganelle contacts for mitochondrial fission, with roles for lysosomes in fission regulation and with Golgi-derived vesicles during final scission [89,90].
It has been observed that ER-OMM contact sites that mark sites of mitochondrial division are often also spatially located adjacent to replicated nucleoids [75,76,77], suggesting a role for fission in mtDNA segregation. Furthermore, the visualisation of DRP1 and MFF using confocal microscopy has demonstrated colocalisation at sites adjacent to the nucleoid [69,77,91]. Where division occurs between replicated nucleoids, the daughter mtDNA molecules are subsequently observed to be located at the tips of the separated mitochondria [75,76]. This mechanism ensures that following division each mitochondrion receives a copy of the genome, and secondly functions to disperse nucleoids throughout the mitochondrial network. At this stage, it is unclear what signalling takes place to ensure that division occurs between the two replicated nucleoids.
3.1.2. Defects in Mitochondrial Fission and Human Disease
At this point in time, the prevalence of human diseases secondary to defects in mitochondrial fission is not known. However, they would appear to be much less common than diseases that are related to mitochondrial fusion. The most common clinical manifestations that occur in relation to disruption of mitochondrial fission genes are subtypes of Charcot–Marie–Tooth (CMT) neuropathy and optic neuropathy, as identified following mutations that occur in DRP1, GDAP1, INF2, MFF and SLC25A46 [92,93,94,95,96]. Severe neurological presentations, such as neurodevelopmental delay and epilepsy, are observed following mutations of the DRP1 gene [97]. Leigh-like syndrome has been observed in patients harbouring mutations in MFF and SLC25A46 [98,99]. Mutations in DNM2 have been linked to CPEO and central core myopathy [100,101], a form of myotubular myopathy. Extra-neurological involvements are relatively uncommon, except cardiac arrhythmia and neutropenia, which have been associated with mutations in DNM2 [101,102], and glomerular disease, described in INF2-related disease (Appendix A Table A1).
Mitochondrial respiratory chain deficiencies and multiple mtDNA deletions have been demonstrated in muscle biopsies taken from patients harbouring DNM2 mutations [103]. Normal qualitative and quantitative assessments of mtDNA were reported in the skeletal muscle or fibroblasts that contain mutant forms of other mitochondrial fission-associated genes such as GDAP1, INF2 and MFF (Table A1) [93,94,98,104,105,106,107,108,109]. These observations would suggest that the biological consequences of these genetic defects are tissue specific, given that only DNM2 mutations primarily manifest with a myopathic phenotype [100,101]. Whilst other fission-associated genes predominantly cause peripheral neuropathy including optic neuropathy or CNS involvement, it is important to note that these affected tissues are far less accessible for further characterisation compared to muscle biopsy.
3.1.3. Mitochondrial Fission and mtDNA Integrity
The disruption of mitochondrial fission by genetic knockdown of the fission factors DRP1 or MFF results in an elongated and fused mitochondrial network that prevents the even distribution of nucleoids around the network, resulting in the clustering of nucleoids [66,77]. Reports in the literature differ on its effects on mtDNA copy number and respiratory activity. In human cells, one study reported that the loss of DRP1 induced mtDNA loss and a reduction in respiratory activity as measured by ATP production [110], while a separate study found that the knockdown of DRP1 or MFF using siRNA did not affect the overall mtDNA copy number [77]. In mice, mtDNA copy number and respiratory function were shown to be unaffected in Drp1 knockout MEFs [111], but a whole-body knockout of Drp1 is embryonically lethal [111,112]. In a tissue-specific knockout of Drp1 in the heart, mice survived for 11 days and displayed exacerbated mitochondrial fusion, nucleoid clustering, reduced mtDNA and respiratory defects [66]. Interestingly, in this model immunofluorescent staining of the mtDNA-encoded cytochrome c oxidase 1 subunit (COX1) revealed that there was an increased staining intensity in regions where nucleoids were clustered together, and a decreased intensity in areas with a sparse presence of nucleoids [66]. These data suggest that nucleoid clustering leads to a mosaic pattern of respiratory subunit distribution in the mitochondrial network. Similarly, the knockout of MFF is associated with premature death and defective mitochondrial respiratory activity [113]. In some cases, it was reported that DRP1 knockout was associated with enlarged nucleoid size [77]. However, this may be a limitation associated with the microscope resolution used, as it would not be expected that fission would affect the decatenation of the DNA molecules. As such, these apparently enlarged nucleoids may represent decatenated mtDNA molecules located in close proximity, which are beyond the limits of detection, although this remains to be confirmed.
Cardiolipin is an integral structural component of mitochondrial membranes that is synthesised from phosphatidic acid (PA). The IMM contains approximately 20% cardiolipin, the presence of which is considered a signature of the IMM. Both cardiolipin and PA have been implicated in facilitating the fission and fusion processes (this topic has been reviewed in depth elsewhere [114]). Briefly, DRP1 binds to both cardiolipin and PA, cardiolipin at the OMM can stimulate oligomerisation of DRP1 and subsequent GTP hydrolysis to induce fission [115,116,117]. It has also been shown that DRP1 binding to cardiolipin induces reorganisation of the membrane to an inverted hexagonal, nonbilayer configuration that promotes membrane constriction [118]. Conversely, the activity of DRP1 can be restrained at the OMM by the reversion of cardiolipin to PA by MitoPLD (mitochondria-localised phospholipase D), as the enhanced level of PA inhibits oligomerisation-stimulated GTP hydrolysis that is responsible for membrane constriction [119]. As such, the loss of cardiolipin is associated with a reduced capacity to correctly segregate and guide nucleoids to the daughter mitochondria; this leads to a lack of mtDNA inheritance between replicating cells, resulting in a dysfunctional respiratory phenotype in daughter cells [120,121,122]. Modulating the level of cardiolipin has also been associated with loss of mtDNA and subsequent mitochondrial dysfunction [121,123]. Clearly, fission plays an important role in facilitating the dissemination of mtDNA around the mitochondrial network and to subsequent daughter organelles (see Figure 2a for a schematic overview). Furthermore, these observations also highlight how mitochondrial fission maintains respiratory function independently of mtDNA copy number or integrity. It is clear that if the fission process is not tightly regulated then cellular respiration will be affected and can contribute to the mitochondrial disease phenotypes discussed earlier.
3.2. Mitochondrial Fusion and Its Role in mtDNA Maintenance and Distribution
Together with mitochondrial fission, mitochondrial fusion has been implicated in the dissemination of nucleoids around the network. Furthermore, fusion is recognized as an important mediator of mtDNA maintenance. Defects in mtDNA maintenance may manifest as either a quantitative reduction in mtDNA copy number (depletion) or as an accumulation of rearranged mtDNA molecules (deletions and/or duplications), all of which may be indicative of impaired mtDNA replication. Defects in the proteins that regulate mitochondrial fusion dynamics have been implicated in a range of genetic diseases that will be discussed here and are summarised in Table A1. Their contribution to the development of disease will be described in relation to their role in mtDNA maintenance.
3.2.1. Mitochondrial Fusion
Mitochondrial fusion is the joining of two separate mitochondria and is important to enable the sharing of contents between neighbouring mitochondria [124]. The fusion of the OMM and IMM occur sequentially, therefore the outer membranes are joined first. This is regulated by two outer membrane proteins, the dynamin related GTPases mitofusin 1 & 2 (MFN1 and MFN2). MFN1 is responsible for tethering the adjoining mitochondria together in a GTP-dependent dimerization process, prompting a conformational change which in turn mediates GTP hydrolysis by MFN1 to pull the membranes together, resulting in mitochondrial fusion [125,126,127].The role of MFN2 is less clear, although it has been implicated in interactions between mitochondria, as well as between mitochondria and the ER [128,129,130]. Following OMM fusion the IMM can be fused. IMM fusion is controlled by optic atrophy 1 (OPA1). OPA1 exists in multiple forms of different sizes which are regulated by proteolytic processing; long OPA1 is IMM-anchored while the short OPA1 is soluble. The presence of OPA1 in its long isoform has been associated with promoting IMM fusion [131]. Processing of OPA1 to its short isoform either by YME1L1 or OMA1 is associated with mitochondrial fission [132,133].
There is evidence that OMM fusion proteins also play a role in the dissemination of nucleoids. It has been reported that the knockout of OMM fusion proteins Mfn1 & 2 in MEFs leads to the clustering of nucleoids [134]. Using super-resolution microscopy these clusters were confirmed to be separate individual nucleoids in close proximity rather than a cluster of interlinked DNA molecules. The role of IMM fusion in nucleoid distribution is less clear. It has been reported by some groups that the loss of OPA1 is associated with a reduction in the number of nucleoids per cell, and in some cases a clustered phenotype [77,135,136]. In contrast, a recent study found that in MEFs the removal of Opa1 was not associated with any alterations in mtDNA distribution, although there was a reduction in copy number [134]. This normal nucleoid distribution following Opa1 knockout may be explained by the role of MFN2 in forming a tether between the OMM and ER [137,138]. As discussed earlier, it is recognized that the ER localises at positions of nucleoid replication and is implicated in coordinating mitochondrial constriction at these sites [75]. Taken together, this highlights the importance of the OMM acting as a signalling platform for coordinating the circulation of nucleoids around the mitochondrial network.
Interestingly, the knockdown of MFN1 & 2 in conjunction with DRP1 has been found to prevent the clustering of nucleoids in human cells [77]. Mechanistically, how the impairment of both fission and fusion would restore nucleoid distribution is not clear as it may be expected that this would render the mitochondria unable to be dynamic, and thus unable to evenly distribute nucleoids. Another study in vivo demonstrated that the dual knockout of Mff and Mfn1 in mice could completely rescue the heart dysfunction, shortened life span, and respiratory chain dysfunction associated with Mff knockout [113], although this rescue was tissue specific. It was suggested that this dual knockout reinstates a balance of fission and fusion, rather than hyperfusion or fragmentation. The relationship between mitochondrial dynamics, nucleoid segregation and maintenance of the respiratory chain is complex and therefore further study is still required to disentangle its intricacies. See Figure 2 for an overview of the interplay between fission and fusion in mtDNA distribution.
3.2.2. Defects in Mitochondrial Fusion and Human Disease
In recent years, genetic defects involved in mitochondrial fusion have emerged to be the common cause of several neurological and ophthalmological disorders. OPA1 mutations account for 60% of dominant optic atrophy (DOA) cases [139], and the prevalence of the disease has recently been revised to 1 in 34,000 [140]. More complex neurological phenotypes such as cerebellar ataxia, spasticity, CPEO [141,142] and more recently, Parkinsonism and dementia, have been identified in patients with OPA1 mutations [143]. MFN2 mutations are the fourth most common cause of CMT neuropathy [144,145], accounting for 20% of CMT2 [146], a form of dominant axonal neuropathy. Mutations in SPG7 were initially described in hereditary spastic paraplegia associated with mitochondrial OXPHOS defects [147]. Independent cohort studies have subsequently shown that cerebellar ataxia could be the most prominent clinical feature without evidence of upper motor signs in cases of SPG7 mutations, and are the most common or second most frequent cause of recessive genetic ataxia in European populations [148,149]. Interestingly, a recent Spanish study suggested that around one-fifth of SPG7 cases exhibited Parkinsonism [150].
AFG3L2 and SPG7 together form the subunits of the m-AAA metalloprotease complex, which is crucial for the maturation, maintenance and quality control of the mitochondrial proteome [151]. Heterozygous mutations in AFG3L2 cause spinocerebellar ataxia type 28 (SCA28) and mitochondrial respiratory chain deficiency [151]. Given the close interaction between AFG3L2 and paraplegin (SPG7), there is little surprise that genetic defects result in many overlapping clinical features of neurodegeneration and the classic phenotypes of mitochondrial dysfunction such as CPEO and multiple mtDNA deletions in the muscle [152,153].
Severe, childhood-onset encephalopathy has been observed in mutations in OPA1 (recessive), FBXL4 and YME1L1 (Table A1). On the other hand, mitochondrial DNA depletion is rare in defects of mitochondrial fusion and has only been identified in several cases of severe childhood disease secondary to mutations in MFN2 and FBXL4 [154,155]. Intriguingly, multiple lipomatosis, as previously observed in myoclonic epilepsy and ragged-red fibres (MERRF) syndrome but no other forms of primary mtDNA mutations, has been identified in several families of MFN2 mutations [156,157,158].
3.2.3. Mitochondrial Fusion and mtDNA Copy Number
A number of studies have highlighted that mitochondrial fusion is critical for the maintenance of mtDNA copy number. In yeast, the loss of fusion activity leads to a loss of mtDNA copy number [159,160]. Consistent with this observation, mtDNA content and respiratory activity is reduced in MEFs following the knockout of OMM fusion factors Mfn1 &2 either alone or together or following knockout of the IMM fusion factor Opa1 [134,135]. Mice carrying a mutation in Opa1 display mtDNA loss and reduced mitochondrial function in the heart [161], while whole-body knockouts of MFN1 & 2 are embryonically lethal [162]. The roles of both MFN1 & 2 have therefore been studied in a tissue-specific context. Work in mice has shown that the knockout of Mfn1 & 2 in heart and skeletal muscle leads to a drop in overall mtDNA copy number and OXPHOS deficiency [134,136]. Cardiolipin also has a role in regulating fusion as it is necessary for the biogenesis of OPA1 and for the formation of higher order OPA1 oligomers which are required for fusion [163,164,165]. The cardiolipin precursor PA has also been associated with the induction of fusion in an MFN dependent manner [166]. The disruption of cardiolipin levels is linked with a reduction in mtDNA copy number [121,123]. ATAD3 (ATPase family AAA-domain containing protein 3 A) has also been implicated in maintaining mitochondrial fusion as its manipulation either by knockdown or overexpression leads to mitochondrial fragmentation [167,168,169,170]. This fragmented phenotype is mediated by the recruitment of DRP1 to the OMM via oligomerisation of the ATAD3 coiled-coil domain [170]. Furthermore, it was demonstrated that in addition to activating DRP1, the dimerisation of ATAD3 provokes mtDNA instability by disrupting the binding of TFAM and mtDNA. Indeed, the knockdown of ATAD3 has been associated with a reduction in mtDNA content [170]. In human fibroblasts that are either deficient for ATAD3 or harbour a duplication of the ATAD3 gene cluster, nucleoids were found to be enlarged and clustered together suggesting a role of ATAD3 and fusion in mtDNA distribution [171,172]. The knockout of ATAD3 in skeletal muscle has been associated with the progressive formation of mtDNA deletions and copy number depletion [173].
The mechanisms underlying the loss of mtDNA when fusion is impaired have long been unclear, although recent work has shed light on potential links between the two. In general, the loss of copy number indicates that there is a defect with the process of mtDNA replication. It has been demonstrated that mitochondrial fusion is necessary to facilitate high levels of replication through content mixing to ensure a proper stoichiometry of replisome components. The loss of OPA1 alone or the collective loss of MFN1 & 2 together prompts an imbalance of replisome factors and thus leads to a reduced rate of mtDNA replication, leading to mtDNA depletion [134].
3.2.4. Mitochondrial Fusion and mtDNA Integrity
It has been observed that aside from being required to maintain copy number, fusion appears to play an important role in maintaining the integrity of the mitochondrial genome. For example, mutations in the OPA1 gene have been associated with the accumulation of mtDNA deletions and OXPHOS defects in the skeletal muscle of patients [141,174,175]. Similarly, patients with mutations in the OMM fusion protein MFN2 display evidence of deletion-containing mtDNA genomes [154,176]. In animal models, the knockout of Mfn1 & 2 in the skeletal muscle of mice is associated with an increased occurrence of point mutations and deletions of mtDNA [136]. On the other hand, another group studying the cardiac tissue of mice with MFN1 & 2 genetically removed found no differences in levels of mtDNA mutations or deletions [134]. These studies highlight that effects may be tissue-specific, and that care must be taken when comparing effects in patients carrying missense mutations with effects seen in knockout animal models. However, from a clinical perspective, mutations in the fusion machinery have a clear association with the progressive onset of mtDNA mutations and deletions (see Table A1).
Multiple lines of evidence indicate that fusion plays a protective role against mutations. Work using fibroblasts derived from MFN2 patients found that these cells display a reduced capacity to repair mtDNA damage [176]. This finding potentially suggests that fusion may preserve mtDNA integrity by enabling the repair of DNA damage or by facilitating the clearance of mitochondria harbouring damaged DNA. In support of this notion, excessive mitochondrial fragmentation (which would occur with impaired fusion) has been associated with an increased production of reactive oxygen species [177]. Indeed, in an Opa1 mutant mouse it was observed that there was an increased level of ROS coupled with a reduced antioxidant capacity [161]. A separate study used the mutator mouse, which has a mutation in the proofreading domain of polymerase PolgA, and consequently rapidly accumulates mtDNA mutations, crossed with a knockout of MFN1 [178]. The mutator mouse or the MFN1 knockout mouse individually survive into adulthood, however crosses between the two resulted in embryonic lethality [136]. In this case it is plausible that fusion exerts a protective effect through its ability to “dilute” mutation-containing mtDNA via content mixing and therefore preserve mitochondrial function.
It remains unclear how defects in fusion contribute to the molecular mechanism of deletion formation. Deletions may conceivably accumulate at an increased rate either because of impaired mtDNA replication or because of increased mtDNA damage. It has been speculated that the primary purpose of mitochondrial fusion is to enable the sharing of contents between two mitochondria. Indeed, it has been published that matrix proteins and mtDNA are transferred between fused mitochondria [179]. Conversely, mitochondria from dual MFN knockout cells were found to have increased protein heterogeneity compared to their wild-type counterparts [136]. It may therefore be that the loss of content mixing associated with defective fusion impairs mtDNA replication through an imbalance of replisome components [134]. This could then promote replication stalling and copy-choice recombination, leading to increased deletion formation. Alternatively, it is possible that increased mtDNA damage in the absence of fusion promotes the formation of deletion-containing mtDNA molecules via double-strand break formation. However, a key limitation in the idea of mtDNA repair underlying the formation of deletions is that at this stage the proteins that are responsible for DNA repair have not been identified as being present in mitochondria.
It is also conceivable that deletions accumulate in the absence of fusion because their removal is impaired. However, the observation that defects in fusion are associated with an accumulation of mtDNA mutations and deletions appears counterintuitive, as it would be expected that a fragmented network would be optimal for selective mitophagy to clear dysfunctional mitochondria away. Indeed, in a Drosophila melanogaster model of mtDNA heteroplasmy, the knockdown of MFN promoted the mitophagy of fragmented mitochondria and a reduction in the mutant mtDNA load [180]. Similarly, a perpetually fused network due to the inhibition of fission by DRP1 or FIS1 in human cells was associated with a shift in heteroplasmy towards mutant mtDNA, possibly because of reduced mitophagy, although this was not addressed [181]. As such, it appears that the accumulation of mtDNA mutations and deletions in the absence of fusion is not related to a reduction in mitophagy.
Collectively, these studies demonstrate that IMM and OMM fusion have an important role in maintaining the integrity of mtDNA and coordinating its replication. However, the relationship is complex and further study is certainly required. It seems likely that the role of fusion in facilitating content mixing between mitochondria is important to maintain the balance of proteins required for mtDNA replication [134,136]. There is also evidence to suggest that fusion also has a protective role against the effects of ROS and DNA damage [161,177]. This, coupled with an imbalance of replisome proteins leading to replication stalling/slippage, could underlie the onset of mutations and deletions associated with fusion defects, as well as contribute to mtDNA copy number loss.
4. The Relationship between mtDNA and Mitochondrial Cristae Structure
The IMM is a complex structure that can be subdivided into two further regions: the inner boundary membrane (IBM) and the cristae (Figure 3b). The IBM runs parallel to the OMM and houses proteins responsible for localising and importing proteins into the matrix, as well as inserting and assembling proteins into the IMM [182,183]. The IBM is divided at regions where the IMM is folded inwards to form cristae structures. Cristae are functionally active structures that contain the respiratory chain complexes and the ATP synthase [184]. At points where the cristae joins the IBM, the cristae narrows to form a cristae junction (approximately 20–40 nm in diameter) which allows the separation of the intracristal space from the intermembrane space between the IMM and OMM [185,186,187]. The mitochondrial contact site and cristae organising system (MICOS) complex is located at these junctions and plays a key role in maintaining their structure.
It has long been appreciated that mtDNA is located close to the IMM and to the cristae structure. It has been recently demonstrated using correlative 3D super-resolution fluorescence and electron microscopy that nucleoids are found within cristae regions of the mitochondria [68]. Sophisticated super-resolution imaging has now revealed that nucleoids are typically located in the voids that form between groups of cristae rather than being embedded within the cristae structure itself [188]. This is in line with the observation that the size of the nucleoid, at approximately 100 nm, is greater than the gaps between cristae, which are found tightly packed together [53,54,188]. It is postulated that these voids where nucleoids are located may function to provide space for transcription, replication and segregation to occur [68,188]. In this section we will summarise current data on cristae structure and modulation, and its relationship with mtDNA maintenance and segregation.
4.1. The Importance of Lipids in the IMM and Their Role in mtDNA Tethering
The capacity for mtDNA to freely diffuse around the mitochondrial network is limited, due in part to the relatively large size of the nucleoid and to the high density of proteins located within the matrix and cristae regions [53,69,189]. There is also evidence that has led to the suggestion that mtDNA is tethered to the IMM [67,69,70,190,191]. However, at this stage the factors responsible remain poorly understood.
Several studies have highlighted the importance of the lipid composition of the IMM to mtDNA attachment, replication and organisation. The composition of phospholipids in the IMM is tightly regulated. Phospholipids have a hydrophobic lipid domain tail which is larger than their charged hydrophilic head groups; this configuration forms a rough conical shape which pulls head groups together, inducing membrane tension and facilitating the bending of the IMM to form cristae invaginations [192,193]. Cardiolipin has an important role in stabilising IMM proteins such as the respiratory complexes [194]. Alterations in the level of cardiolipin have been associated with mitochondrial dysfunction, mtDNA loss and the formation of abnormal cristae structures [121,123]. Similarly, in mammalian cells the removal of the IMM phospholipid phosphatidylethanolamine has comparable effects; mitochondrial function is reduced coupled with obvious swelling of mitochondria which lack distinct cristae structures [195].
It has been demonstrated in both yeast and mammalian cells that cardiolipin is physically associated with the nucleoid [120]. Furthermore, as discussed earlier cardiolipin is a key constituent of the IMM and has a role in mitochondrial fission and fusion. As such, the role of cardiolipin in mtDNA maintenance is multi-faceted. The loss of cardiolipin is associated with a reduced capacity to correctly segregate and guide nucleoids to daughter mitochondria. In addition, this defective IMM-nucleoid association coupled with defects in mitochondrial dynamics leads to a lack of mtDNA inheritance between replicating cells resulting in a dysfunctional respiratory phenotype in daughter cells [120,121,122].
The composition of the mitochondrial IMM is subject to a relatively low ratio of cholesterol to phospholipids [196]. Cholesterol has been implicated in nucleoid organisation as its modulation either through pharmacological inhibition or supplementation prompts the formation of aggregated nucleoids in human fibroblasts [171]. The majority of mitochondrial cholesterol is clustered in specialised structures that span the IMM and OMM [197]. TWINKLE-containing nucleoids were shown to be associated with these cholesterol structures in a complex that also contains ATAD3. ATAD3 is an essential protein that is anchored into the IMM, with its C-terminal AAA ATPase domain located in the mitochondrial matrix while its N-terminus interacts with the OMM where it is present at ER-mitochondria contact sites [62,169,197,198,199]. It has been proposed that these cholesterol-rich sites provide a platform for mtDNA replication, and act as an attachment site at ER-mitochondria junctions to allow for the coordinated distribution of mtDNA. As discussed earlier, ATAD3 has been demonstrated to have a regulatory role in mitochondrial morphology and dynamics and has been implicated in nucleoid organisation [167,168,169,170]. ATAD3 also has an important role in maintaining IMM architecture as the disruption of ATAD3 leads to impaired transfer of cholesterol from the ER into the mitochondria and lipid metabolism [171,200]. The knockout of ATAD3 in skeletal muscle has been associated with alterations in cholesterol metabolism and the progressive formation of mtDNA deletions and copy number depletion [173]. As discussed earlier, the formation of these mtDNA rearrangements has been associated with stalling of the replication machinery and a loss of cristae structure. It has been suggested that ATAD3 may interact with OPA1 or the MICOS complex to stabilise cristae structure [173]. Collectively, it is clear that ATAD3 is an important regulator of mitochondrial dynamics, mtDNA attachment, maintenance and organisation. However, at this stage it is not entirely clear whether these effects are directly associated with the loss of ATAD3 or due to cholesterol-mediated alterations in the IMM structure as a consequence of the role of ATAD3 in the uptake of cholesterol to the mitochondria [200].
In addition to ATAD3, OPA1 has also been implicated as having a potential role in the attachment of nucleoids to the IMM [135]. OPA1 is embedded in the IMM, co-immunoprecipitates with nucleoids and as discussed earlier, its deletion has profound effects on nucleoid organisation [77,135,136]. Thus, it has been suggested that OPA1 may have a role in membrane attachment of the nucleoid (Figure 3b). Loss of either ATAD3 or OPA1 has been shown to cause significant aberrations in cristae structure [161,197,201].
The structure of proteins and lipids within the IMM is also supported by a group of evolutionarily conserved scaffolding proteins termed prohibitins. Prohibitin 1 and 2 are essential proteins that form high molecular weight ring-shaped heterooligomers in the IMM. They have also been found to associate with nucleoids which prompts speculation that they may be involved in tethering of nucleoids to the IMM [57]. It has been demonstrated that the prohibitins can influence mtDNA copy number through their interactions with TFAM [202]. Cultured cells lacking the prohibitins display impaired cardiolipin maturation, loss of cristae structure and disorganisation of nucleoids [203,204,205]. It has also been demonstrated that the effects of prohibitin 2 depletion upon cristae structure are dependent upon OPA1, a known modulator of cristae structure [204].
4.2. Modulators of Cristae Structure
Mitochondrial cristae are dynamic structures that can modulate their shape in response to various physiological conditions. As they are the primary site of the OXPHOS machinery, it is important that the folding of the IMM into cristae structures facilitates the most efficient manner of producing ATP. Recent work has found that individual cristae act as autonomous bioenergetic units, highlighting the importance of their structural integrity [206]. Cristae structure is dictated by a variety of “cristae-shaping proteins” (Figure 3b). A key mediator of cristae architecture is the MICOS complex. The MICOS complex consists of at least the subunits MIC10, MIC12, MIC13, MIC19, MIC25, MIC26, MIC27 and MIC60 [207]. Each of these proteins has a specific role in shaping the cristae structure. MIC60 is a core component of the complex and is associated with the formation of cristae junctions and contact sites with the OMM [208,209,210]. MIC10 can also bend membranes and is known to be responsible for forming hairpin structures in the IMM [211,212]. MIC13 has also been recently recognised as being essential for maintaining the stability of the MICOS complex and cristae junction formation [212]. MIC26 has a role in cristae junction formation, and both MIC26 and MIC27 are necessary to maintain normal cristae architecture [213,214].
The F1F0 ATP synthase is embedded in the IMM and is localised to the cristae tips, where it exists in a dimer confirmation. These dimers are also assembled into oligomers. The presence of these dimers prompts a bending of the surrounding IMM lipid bilayer, highlighting their importance as mediators of cristae structure [215,216]. As such, the loss of the F1F0 ATP synthase is associated with a loss of cristae invaginations [216]. However, the F1F0 ATP synthase may also act indirectly to maintain cristae morphology, as it has also been recognized that the F1F0 ATP synthase interacts with the MICOS complex [217]. It has also been reported that oligomerisation of F1F0 ATP synthase dimers is promoted by OPA1, which also has a role in cristae shaping [218].
The role of OPA1 in cristae shaping is reflected in its ability to define the diameter and width of cristae junctions [209]. It has been demonstrated in yeast that the OPA1 homologue MGM1 is required to maintain cristae structure by tethering to other OPA1 molecules on the opposing IMM [219]. Indeed, it has been observed that high molecular weight multimers of OPA1 stabilise and induce the formation of tight cristae junctions, whereas lower-order OPA1 oligomers are associated with increased cristae junction and lumen width [209]. The remodeling of cristae is mediated by the integral membrane protease PARL. Processing by PARL produces a short, intramembrane-soluble form of OPA1 that binds with the membrane-bound form to maintain tight cristae junctions [220]. There is some evidence that OPA1 has a broader regulatory role and can adjust mitochondrial respiration by modulating cristae shape. It has been reported that in response to hypoxia there is an increase in the abundance of OPA1 oligomers that prevent cristae remodeling, thereby acting to enhance mitochondrial respiration [221]. It has also been shown that OPA1 can interact with metabolic sensors in response to starvation, acting to adjust the cristae shape to maintain ATP production [222]. High molecular weight OPA1 multimers have also found to be associated with MIC60 and F1F0 ATP synthase [209,218,223]. Further work will be required to understand the extent to which OPA1 dictates cristae shape alone, and how it functions as part of an interactive network with other cristae shaping proteins.
A number of other proteins have been observed to have an impact on cristae structure, but for which the precise mechanisms are less well understood. MCL1 has been found to have a role in preserving cristae ultrastructure and the maintenance of oligomeric ATP synthase [224]. The loss of proteins that interact with cardiolipin have also been found to disrupt cristae architecture. Specifically, the prohibitins and UQCC3 have all been demonstrated to bind to cardiolipin, and their loss is associated with alterations in cristae structure [204,225,226].
4.3. The Relationship between Cristae Structure and mtDNA
A number of studies indicate that there is an interesting but poorly-understood link between mtDNA and cristae ultrastructure. Dissecting this relationship is problematic because of the role that mtDNA-encoded proteins, such as components of the ATP synthase, play in maintaining cristae structure. For example Rho-0 (ρ0) fibroblasts, which are devoid of mtDNA, display sparse cristae structures and swollen mitochondria compared to mtDNA-containing cells [227,228,229]. Embryos lacking TFAM also lack mtDNA and display abnormal cristae structure [230]. Similarly, tissue-specific knockout of TFAM in developed animals has also been shown to ablate cristae structure [231,232]. In cultured cells, the transient depletion of mtDNA using the replication inhibitor 2′-3′-dideoxycytidine also leads to a loss of cristae structure [77]. Other data has suggested a direct role for mtDNA in maintaining cristae structure. The loss of DRP1 is associated with the formation of structures filled with very densely packed cristae and clustered nucleoids termed “mito-bulbs” (Figure 3c) [77]. The formation of these cristae-rich mito-bulbs in DRP1 knockdown cells was not impacted when mitochondrial translation was inhibited using chloramphenicol. However, the depletion of mtDNA prior to DRP1 knockdown prevented the formation of mito-bulbs. This suggests that mtDNA itself may be important for determining cristae architecture rather than its gene products. These data highlight the need for further study to understand how mtDNA contributes to the maintenance of normal cristae structure.
The loss of OPA1 has been associated with a reduced mtDNA copy number, both in model systems and in patients harbouring OPA1 mutations [135,161,233]. OPA1-deficient cultured cells also show a distinct loss of cristae structure (Figure 3d) [201,234]. Similarly, in mice, both the mutation of Opa1 in the heart and deletion of Opa1 in the skeletal muscle was found to cause the loss of cristae structures [161,235,236]. Other in vivo models in which mtDNA content was reduced, such as the deletion of Mfn2 in Purkinje cells or the dual knockout of Mfn1 & 2 in skeletal muscle, have also been shown to be associated with sparse cristae structures. Whilst these observations support the idea that the loss of mtDNA is linked with the disruption of cristae structure, it is likely that the relationship is not this simple. For example, one study noted that the loss of cristae structure in Opa1 mutant mice occurred prior to the reduction in mtDNA content [235], although this is not surprising because of the key role of OPA1 in maintaining cristae junctions [209].
Analysis of the relationship between mtDNA and cristae structure is also complicated by the fact that a number of interventions which alter mtDNA content also impact on the balance of fission and fusion rates of mitochondria, and therefore the proper organisation of nucleoids around the network. There is also evidence to suggest that normal cristae structure is required to facilitate fission and fusion [210]. The MICOS complex can be destabilized by the loss of the core protein MIC60. This results in a striking phenotype of enlarged mitochondria that display abnormal circular cristae, commonly described as “concentric rings” or an “onion-like structure”, alongside an almost complete loss of cristae junctions (Figure 3e) [210,237]. It has also been reported that the loss of MIC60 causes a reduction in the copy number of mtDNA (139). The downregulation of other MICOS subunits such as MIC19 and MIC10 also leads to a disruption of cristae structure [210]. The silencing of MIC19 and MIC60 led to the formation of enlarged and disorganised nucleoids in both mammalian cells and yeast, accompanied by a reduction in mtDNA transcription [210,238]. Similarly, it has been noted that mutations in CHCHD10, a constituent of the MICOS complex, leads to a loss of cristae junctions and structure. Furthermore, this was also shown to be linked with the formation of mtDNA deletions, as well as a reduction in nucleoid number without an overall effect on copy number [239].
It is worth noting that it is often difficult to determine whether apparently enlarged nucleoids represent one single enlarged nucleoid consisting of multiple physically joined (catenated) mtDNA molecules, or a group of nucleoids clustered in close proximity. An interesting aspect of the enlargement or clustering of nucleoids associated with the loss of MIC60 is that it was not reported to lead to the formation of cristae-enriched mito-bulb structures as are seen following DRP1 knockdown [77]. However, it is likely that these cells are unable to form dense cristae regions as a consequence of the fundamental role of MIC60 and the MICOS complex in shaping cristae [207]. Interestingly, MIC60 knockdown cells showed a normal balance of fission and fusion compared to wild-type cells, but this occurred at a reduced rate with DRP1 being one of the mitochondrial dynamics proteins observed to be downregulated. When DRP1 was overexpressed to promote mitochondrial fragmentation, the presence of enlarged nucleoids associated with the loss of MIC60 was partially reverted [210]. Furthermore, work in yeast has found that when MIC60 (FCJ1) is knocked out in conjunction with DRP1 (DNM1) the nucleoids are further enlarged compared to the loss of MIC60 alone [238].
Cristae junctions have also been implicated in enabling nucleoid distribution. Unlike MIC60 deletion, the knockout of F1F0 ATP synthase dimerization factors does not promote nucleoid aggregation or affect the number of cristae junctions. However, its knockout does reduce the number of cristae tips. The dual knockout of MIC60 (FCJ1) and dimerization partners of F1F0 ATP synthase results in an increased number of ring-like cristae structures that rescues the nucleoid aggregation phenotype associated with MIC60 deletion alone [238]. This finding indicates that cristae structure is likely important for partitioning nucleoids to prevent their aggregation.
Addressed collectively, these studies support the idea that normal cristae structure is required to support the even distribution of mtDNA nucleoids around the mitochondrial network (Figure 3). Firstly, dynamic cristae structure is required to facilitate normal rates of fission and fusion, which as discussed earlier are key mediators of nucleoid dispersal. Secondly, cristae can also prevent the aggregation of nucleoids by forming partitioning structures. This is in logical agreement with the observation that nucleoids are generally located in voids between groups of cristae structures [188]. At this stage, it is unclear whether nucleoid partitioning is a precursor to the distribution of nucleoids around the network via fission. Therefore, further study will be necessary to understand the intricate interplay between these two events.
4.4. Cristae Remodelling by Independent Fission and Fusion of Cristae Membranes
It has been speculated for some time that cristae are dynamic structures that can undergo remodelling of their membranes in response to various physiological conditions. However, studying these remodelling events in real-time has been particularly challenging until recently. A number of groups have developed methods that allow for the visualisation of cristae structures in live cells using super-resolution microscopy [188,206,240]. Recent work has established that cristae within the same mitochondrion can have different membrane potentials, thus demonstrating that cristae can behave as independent bioenergetic units [206]. This work suggests that within a mitochondrion cristae can functionally isolate themselves from one another, and it is postulated that this mechanism can prevent individual dysfunctional cristae from disrupting membrane potential in the broader mitochondrial network. Further work has revealed that cristae membranes within a mitochondrion undergo fission and fusion events in a MICOS-dependent manner [241]. Specifically, this study provides evidence for a model in which MIC60 is evenly distributed along the IBM, acting as a docking or scaffolding platform to facilitate the formation of cristae junctions following the recruitment of MIC10. The recruitment of other subunits to complete the MICOS complex allows cristae junctions to fully form. Both cristae junctions and cristae membranes are dynamic, and it was observed that cristae junctions have the capacity to split and merge. Cristae membranes can detach from one cristae junction, and fuse with either the same cristae junction or another cristae junction, such as one on the opposing IBM. It was demonstrated that the merging of cristae membranes is associated with changes in membrane potential at that region, and that these events can facilitate content mixing between distinct cristae compartments [241].
The fact that cristae continuously undergo remodelling and have the capacity to function independently raises the question of how these processes are involved in maintaining the integrity of mtDNA, as well as its segregation and normal distribution. This novel ability to study cristae dynamics in live cells allows an unprecedented opportunity for the field to address some of these experimentally difficult questions. For example, what is the importance of dynamic cristae membranes in maintaining mtDNA integrity by content mixing? Other directions may include understanding the interplay between overall mitochondrial dynamics and cristae dynamics, as well as addressing how the modelling of cristae membranes impacts the segregation and movement of mtDNA around the mitochondrial network.
4.5. Genetic Defects Associated with Perturbed Mitochondrial Cristae Structure and Diseases
Mammalian MICOS comprises seven protein subunits of which defects in two encoding genes have been linked to human disease so far. Mutations in MICOS13 (aka QIL1) cause severe infantile mitochondrial disease characterised by failure to thrive, microcephaly, truncal hypotonia, spasticity and cerebellar atrophy, lactic acidosis and 3-methylglutoconic aciduria (3-MGA) [242,243,244]. Subclinical hepatic involvement manifesting with persistently abnormal liver function tests was also reported. Altered mitochondrial cristae morphology was evident in liver tissue and fibroblasts, but the mitochondrial ultrastructure in muscle appeared normal. Multiple mitochondrial respiratory chain deficiencies were identified in skeletal muscle and liver biopsies, and isolated complex IV deficiency was present in fibroblasts. However, there was no evidence of mtDNA deletions or depletion in muscles and fibroblasts [242,243,244]. More recently, a mutation in APOO, encoding MIC26, has been reported to cause an X-linked disease associated with developmental delay, hypotonia, autistic spectrum disorder, gastrointestinal symptoms, lactic acidosis and abnormal carnitine profile [245]. The pathogenicity of the APOO variant was supported by work with fibroblasts and a fly model.
Homozygous mutations in the PINK1 gene, encoding for PTEN-induced kinase 1, were first identified in three consanguineous families with early-onset Parkinsonism through linkage analysis in 2004 [246]. Overall, PINK1 mutations account for less than 10% of autosomal recessive Parkinson’s disease (PD) [247]. Both Parkin and PINK1 have been shown to play crucial roles in mediating the mitophagy process [248]. More recently, PINK1 was found to maintain cristae junctions by the phosphorylation of the MICOS subunit MIC60 in both Drosophila melanogaster and human neurons [249]. Furthermore, mutations within the mitochondrial targeting sequence of MIC60 were evident in a subset of PD patients. The introduction of these patient mutations into a Drosophila melanogaster model disrupted the ability for MIC60 to localise to mitochondria and prompted the formation of abnormal cristae junctions. Furthermore, the overexpression of MIC60 in a PINK1−/− model of PD compensated for the loss of PINK1-mediated phosphorylation and rescued both cristae defects and mitochondrial function [249]. In addition, mutations in the mitochondrial targeting sequence of CHCHD2 have been identified as a risk factor for PD and Lewy body disease [250].
CHCHD10 is a protein located in the mitochondrial inter-membrane space and it has been demonstrated to play a role in the maintenance of cristae integrity [239]. Heterozygous mutations in CHCHD10 were first linked to motor neuron disease and frontotemporal dementia, myopathy and hyperkalemia, impaired respiratory chain function and multiple mtDNA deletions in the skeletal muscle in a large family of French origin, and in a Spanish family (none of the family members underwent muscle or skin biopsy) in 2014 [251]. Whilst mutations of CHCHD10 display a mitochondrial disease that resembles motor neuron disease, a large consortium of motor neuron disease (amyotrophic lateral sclerosis) patients (n = 4365) and healthy controls (n = 1832) from seven countries subsequently demonstrated that pathogenic CHCHD10 variants are exceptionally rare [252]. Therefore, in pure forms of amyotrophic lateral sclerosis it is not necessarily associated with CHCHD10 mutations.
Prominent extra-neurological disease has been observed following the disruption of several proteins implicated in the maintenance of cristae structure, for example, hypertrophic cardiomyopathy occurs in patients harbouring mutated forms of ATAD3A and TAZ (Barth syndrome) [253,254], and 3-MGA in ATAD3A, TAZ, MICOS13 and ATP5F1E [243,253,255,256].
5. Conclusions
The structure and dynamics of the mitochondrial membranes are required to maintain both the integrity of mtDNA and well as its distribution within the mitochondrial network. Mitochondrial membranes make a poorly understood contribution to mtDNA replication, the impairment of which manifests either as an inability to maintain a sufficient number of copies of mtDNA, or as rearrangements of the genome. Following replication, the dynamics of mitochondria are also required for the segregation of mtDNA, and the disruption of mitochondrial dynamics can lead to the clustering of nucleoids within cells. The direct association between mtDNA and the respiratory chain means that an uneven distribution of nucleoids can lead to a mosaic pattern of respiratory activity within cells, which may represent an under-appreciated molecular contributor to mitochondrial pathologies. Observations from the clinic highlight the importance of these mechanisms, with mutations in genes associated with fusion, fission and cristae structure manifesting mainly in severe neurological disorders frequently associated with mtDNA depletion or rearrangements.
Work from the laboratory puts forward the notion that mitochondrial fusion is necessary to maintain adequate levels of mtDNA replication, likely highlighting the importance of content mixing for maintaining a proper stoichiometry of replisome components between mitochondria. Indeed, it is plausible that this protein heterogeneity may prompt replication stalling resulting in the formation of deletions and rearrangements of mtDNA. The reduction in mitochondrial DNA copy number observed in conjunction with the disruption of fusion therefore likely occurs due to reduced rates of replication.
Defects in mitochondrial fission on the other hand have less of an association with the onset of mtDNA rearrangements or a reduction in mtDNA copy number in the laboratory. However, mutations of fission genes still result in severe neurological disorders in the clinic. As mitochondrial function is often observed to be normal when fission is impaired, the mechanism underlying these clinical phenotypes is not entirely clear at this stage. It is understood that mitochondrial fission is necessary for mitochondrial quality control which can facilitate the accumulation of dysfunctional mitochondria. Furthermore, fission is essential for the even allocation of nucleoids around the mitochondrial network and to subsequent daughter cells. Disruption of fission leads to the clustering of nucleoids. Indeed, the presence of mtDNA is intricately linked to the formation of normal cristae architecture. The clustering of nucleoids is associated with the formation of dense cristae regions in the form of mito-bulb structures, and the loss of mtDNA is often correlated with the loss of cristae structure. The data available suggests that dynamic cristae are necessary to facilitate normal rates of mitochondrial fission and fusion, as well as allow for the partitioning of mtDNA. As such the interactions between dynamics, cristae and mtDNA organisation are tightly interwoven and interdependent. It is therefore likely that if each of these processes is not tightly regulated then there are subsequent downstream effects that disrupt the delicate balance and prompt the onset of mitochondrial dysfunction.
Author Contributions
Conceptualization, J.C. and T.J.N.; writing—original draft preparation, J.C. and Y.S.N.; writing—review and editing, T.J.N.; supervision, T.J.N.; funding acquisition, T.J.N. All authors have read and agreed to the published version of the manuscript.
Funding
T.J.N. is the recipient of a Sir Henry Dale Fellowship (213464/Z/18/Z), jointly funded by the Wellcome Trust and Royal Society, and a Rosetrees and Stoneygate Trust fellowship (M811). Y.S.N. holds an NIHR Clinical Lectureship in Neurology (CL-2016-01-003).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| 3-MGA | 3-methylglutoconic aciduria |
| AD | autosomal dominant |
| AR | autosomal recessive |
| ATAD3 | ATPase family AAA domain-containing protein 3 |
| ATP | adenosine triphosphate |
| CK | creatine kinase |
| CMT | Charcot-Marie-Tooth |
| COX1 | cytochrome c oxidase 1 subunit |
| CPEO | chronic progressive external ophthalmoplegia |
| CSF | cerebrospinal fluid |
| DOA | dominant optic atrophy |
| DD | developmental delay |
| DRP1 | dynamin-related protein 1 |
| ER | endoplasmic reticulum |
| ESRF | end-stage renal failure |
| FIS1 | mitochondrial fission 1 protein |
| FSGS | focal segmental glomerulosclerosis |
| GTP | guanine triphosphate |
| HSP | heavy strand promoter |
| IBM | inner boundary membrane |
| IMM | inner mitochondrial membrane |
| IMS | intermembrane space |
| LA | lactic acidosis |
| LS | Leigh syndrome |
| LSP | light strand promoter |
| MCL1 | induced myeloid leukemia cell differentiation protein |
| MEF | mouse embryonic fibroblast |
| MERRF | myoclonic epilepsy and ragged-red fibres |
| MFN | Mitofusin |
| MICOS | mitochondrial contact site and cristae organising system |
| MID49 | mitochondrial dynamics protein of 49kDa |
| MitoPLD | mitochondria-localised phospholipase D |
| MRC | mitochondrial respiratory chain |
| mtDNA | mitochondrial DNA |
| MTERF1 | mitochondrial transcription termination factor 1 |
| mtSSB | mitochondrial single stranded DNA-binding protein |
| NCR | noncoding region |
| OA | optic atrophy |
| OMA1 | overlapping with the M-AAA protease 1 homolog |
| OMM | outer mitochondrial membrane |
| OPA1 | optic atrophy 1 |
| OriH | origin of H-strand replication |
| OriL | origin of L-strand replication |
| OXPHOS | oxidative phosphorylation |
| PA | phosphatidic acid |
| PARL | presenilin-associated rhomboid-like |
| PD | Parkinson’s disease |
| POLγ | DNA polymerase-γ |
| POLRMT | mitochondrial RNA polymerase |
| rRNA | ribosomal RNA |
| RRFs | ragged-red fibres |
| RTA | renal tubular acidosis |
| SCA28 | spinocerebellar ataxia type 28 |
| SNHL | sensorineural hearing loss |
| TEFM | mitochondrial transcription elongation factor |
| TFAM | mitochondrial transcription factor A |
| TFB2M | mitochondrial transcription factor B2 |
| tRNA | transfer RNA |
| VLCFA | very long chain fatty acid |
| WM | white matter |
| YME1L1 | YME1 like 1 ATPase |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table A1.
Summary of clinical phenotype, extra-neurological involvement and integrity of mitochondrial DNA associated with the genetic defects of mitochondrial dynamics.
Table A1.
Summary of clinical phenotype, extra-neurological involvement and integrity of mitochondrial DNA associated with the genetic defects of mitochondrial dynamics.
| Gene | Protein | Protein Function | Inheritance | Clinical Phenotype | Extra-Neurological | mtDNA Integrity | References | |
|---|---|---|---|---|---|---|---|---|
| Regulators of Mitochondrial Dynamics | DRP1 | Dynamin-related protein 1 | Mitochondrial fission | AD | Microcephaly, abnormal brain development, OA, LA, elevated VLCFA; DD, refractory epilepsy; isolated DOA (OPA5) | None | EM & confocal microscopy study showing concentric cristae structure in patient-derived fibroblasts [97] | [92,97,257,258] |
| CIV deficiency in muscle [257] | ||||||||
| Elongated mitochondria in optic nerve and RGC layer without axonal degeneration in Dnm1l+/− mice [258] | ||||||||
| No reports of mtDNA deletions or depletion | ||||||||
| DNM2 | Dynamin 2 | Mitochondrial fission | AD | Centronuclear myopathy; dominant intermediate CMT neuropathy type B associated with neutropenia and cataract; CPEO, facial weakness, neck flexor weakness; severe cardiomyopathy and centronuclear myopathy | Cardiac, neutropenia | COX negative fibres and multiple mtDNA deletions in skeletal muscle [103] | [100,101,102,103,259,260] | |
| GDAP1 | Ganglioside Induced Differentiation Associated Protein 1 | Mitochondrial fission | AR | CMT4A (early onset with rapid progression demyelinating neuropathy) | None | No reports of mtDNA deletions or depletion | [93,106,108,109] | |
| AD | CMT2RV (axonal neuropathy with hoarse voice and diaphragmatic weakness) | |||||||
| INF2 | Inverted Formin, FH2 And WH2 Domain Containing | Mitochondrial fission | AD | CMT, dominant, intermediate type, E (CMTDIE) with SNHL and renal phenotype (FSGS, proteinuria and ESRF) | Renal | No reports of mtDNA deletions or depletion | [94,104,105,261] | |
| MFF | Mitochondrial Fission Factor | Mitochondrial fission | AR | DD, pyramidal signs and neuropathy; Leigh-like disease with infantile spasm, OA and neuropathy | None | Normal mitochondrial respiratory chain activities in muscle; significant branching of mitochondria in patient-derived fibroblasts [98] | [95,98,107] | |
| SLC25A46 | Solute Carrier Family 25 Member 46 | Mitochondrial fission | AD | DOA and CMT2; hereditary motor and sensory neuropathy, Type VIB (HMSN6B) | None | Abnormal mitochondrial cristae following knockdown in fibroblasts [99] | [96,99,262,263] | |
| Mitochondrial elongation following knockdown in Zebrafish, mtDNA integrity not assessed [262] | ||||||||
| AR | Progressive myoclonic ataxia, OA and neuropathy; LS; pontocerebellar hypoplasia and apnoea | |||||||
| No reports of mtDNA deletions or depletion | ||||||||
| OPA1 | OPA1 Mitochondrial Dynamin Like GTPase | IM fusion/cristae shaping | AD | DOA (most common genetic cause); DOA plus syndrome (CPEO, ataxia, axonal neuropathy, deafness); syndromic Parkinsonism and dementia | Cardiac | Abnormal cristae in OPA1+/− mouse model [264] | [141,143,174,265,266,267,268,269,270] | |
| AR | Ataxia, early-onset OA, gastrointestinal dysmotility; Behr syndrome (OA and ataxia); Infantile encephalomyopathy, HCM and OA; Leigh-like imaging changes | COX negative fibres and multiple mtDNA deletions in cases of heterozygous mutation [141,174] | ||||||
| Multiple respiratory chain deficiencies and mtDNA depletion in muscle with recessive inheritance [265] | ||||||||
| MFN2 | Mitofusin 2 | OM fusion | AD | CMT2A; DOA plus; DD, progressive weakness, failure to thrive, axonal sensorimotor neuropathy, chorea and elevated CSF lactate | Lipoma | Multiple mtDNA deletions and mtDNA depletion detected in muscles and fibroblasts [154] | [154,157,176,271,272,273,274,275] | |
| AR | Severe, early-onset axonal neuropathy, OA; multiple symmetric lipomatosis, LA and axonal neuropathy | RRFs, COX deficient fibres, altered mitochondrial morphology and multiple mtDNA deletion detected in muscle [176] | ||||||
| COX deficient fibres, ultrastructural changes and mtDNA depletion | ||||||||
| FBLX4 | F-Box And Leucine Rich Repeat Protein 4 | OM fusion | AR | Encephalopathy, microcephaly and persistent LA; encephalomyopathy, abnormal WM changes, LA, dysmorphism, RTA, seizures | Renal (RTA) | Multiple respiratory chain deficiencies and mtDNA depletion in patient-derived fibroblasts [155] | [155,276,277,278,279] | |
| Normal MRC enzymes but mtDNA depletion in muscle [276] | ||||||||
| mtDNA depletion, reduced respiratory function and ultrastructure abnormalities in muscle, reduced respiratory function in fibroblasts [277] | ||||||||
| MSTO1 | Misato Mitochondrial Distribution And Morphology Regulator 1 | Interacts with mitochondrial fusion machinery | AR | Myopathy, elevated CK (1200–4500 IU/L), pigmentary retinopathy, scoliosis and cerebellar ataxia and atrophy | None | Normal MRC enzymes, reduced mtDNA copy number in muscle; mitochondrial fragmentation in patient-derived fibroblasts [280] | [280,281,282] | |
| AD | Myopathy, cerebellar ataxia and non-specific neuropsychiatric symptoms | |||||||
| YME1L1 (YME1) | YME1 like 1 ATPase | OPA1 processing | AR | Intellectual disability, DD, OA, hearing impairment, ataxia, LA, leukoencephalopathic changes on MRI, | None | Altered cristae ultrastructure in muscle, mitochondrial fragmentation in patient-derived fibroblasts [283] | [283] | |
| SPG7 | SPG7 Matrix AAA Peptidase Subunit, Paraplegin | OPA1 processing | AR | Spastic paraplegia 7; spastic ataxia; cerebellar ataxia; CPEO and myopathy; | None | Altered mitochondrial ultrastructure; multiple respiratory chain deficiencies in muscle [147,284] | [147,148,153,284,285] | |
| RRFs, COX-deficient fibres and multiple mtDNA deletions; normal mtDNA copy number in muscle; reduced network size in patient-derived fibroblasts [153] | ||||||||
| AFG3L2 | AFG3 Like Matrix AAA Peptidase Subunit 2 | OPA1 processing | AD | Spinocerebellar ataxia 28 (SCA28); CPEO | None | RRFs, COX deficient fibres and multiple mtDNA deletions in muscle [152] | [152,286,287,288] | |
| Regulators of Cristae Structure | MICOS13 (MIC13; QIL1) | Mitochondrial Contact Site And Cristae Organizing System Subunit 13 | Stabilises MIC60 and MIC10 in the MICOS Complex | AR | Failure to thrive, acquired microcephaly, truncal hypotonia, limb spasticity cerebellar atrophy, LA, 3-MGA, elevated liver transaminases and myoclonic seizures | Liver | Normal histology and MRC activities in muscle; low MRC enzymatic activities and abnormal cristae in liver [243] | [243,244] |
| Multiple respiratory chain deficiencies in muscle and liver [244] | ||||||||
| APOO (MIC26) | Apolipoprotein O | Regulatory role in MICOS complex | X-linked | DD, myopathy, LA, cognitive impairment, autistic features, abnormal acylcarnitidine profile | None | Abnormal ultrastructure in fibroblasts and a Drosophila Melanogaster model [245] | [245] | |
| CHCHD10 | Coiled-Coil-Helix-Coiled-Coil-Helix Domain Containing 10 | Stabilises MICOS complex | AD | Frontotemporal dementia and amyotrophic lateral sclerosis; late onset spinal motor neuronopathy and elevated CK; myopathy, elevated CK and lactate; CMT2 | None | RRFs, COX deficient fibres, multiple respiratory chain deficiencies, abnormal assembly of CV, multiple mtDNA deletions in muscle, reduced fusion and loss of cristae structure in patient-derived fibroblast [251] | [239,251,252,289,290,291,292] | |
| Mildly increased COX deficient fibres and occasional RRFs [290] and multiple mtDNA deletions [290] | ||||||||
| Multiple respiratory chain deficiencies (worst with CIV), RRFs, lipid accumulation and abnormal circular cristae ultrastructure [291] | ||||||||
| Loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis in patient-derived fibroblasts [239] | ||||||||
| PINK1 | PTEN Induced Kinase 1 | Mitophagy/Interacts with MIC60 to regulate CJs | AR | Parkinson disease 6 (PARK6) | None | Complex I deficiency in Drosophila Melanogaster model [293] | [246,293,294,295,296,297] | |
| ATP5F1E F1Fo-ATP synthase (sub unit E) | ATP Synthase F1 Subunit Epsilon | Cristae shaping/ATP Production | AR | LA, 3-MGA, mild mental retardation and development of peripheral neuropathy | None | Complex V deficiency in patient-derived fibroblasts [256] | [256] | |
| ATP5F1A F1Fo-ATP synthase (sub unit a) | ATP Synthase F1 Subunit Alpha | Cristae shaping/ATP production | AR | Intrauterine growth retardation, microcephaly, hypotonia, pulmonary hypertension, failure to thrive, encephalopathy, and heart failure; neonatal-onset encephalopathy with intractable seizures, extensive signal changes involving WM, basal ganglia and thalamus | Cardiac | Multiple respiratory chain deficiency and mtDNA depletion in muscle [298] | [298,299] | |
| Complex V deficiency in muscle [299] | ||||||||
| IMM Structure | ATAD3A | ATPase Family AAA Domain Containing 3A | mtDNA and cristae organisation | De novo & AR | Global DD, hypotonia, OA, axonal neuropathy, mildly elevated lactate, seizures, 3-MGA and hypertrophic cardiomyopathy; fatal congenital pontocerebellar hypoplasia with a simplified gyral pattern; cerebellar atrophy with dystonia and ataxia | Cardiac | Abnormal cristae in Drosophila Melanogaster and patient-derived fibroblasts [253] | [171,253] |
| mtDNA abnormalities in patient-derived fibroblasts [171] | ||||||||
| TAZ | Tafazzin | Organisation of cardiolipin in the IM | X-linked | Severe infantile cardiomyopathy (left ventricular noncompaction, Barth syndrome with dilated cardiomyopathy), sudden cardiac death, myopathy, neutropenia, growth failure, 3-MGA, facial dysmorphism | Cardiac, neutropenia | Altered mitochondrial ultrastructure, stacked and circular cristae, in patient-derived lymphoblasts [300] | [254,255,300,301,302,303,304] |
References
- Gray, M.W. Mitochondrial Evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403. [Google Scholar] [CrossRef] [Green Version]
- Gualberto, J.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotfi, F.; Dietrich, A. The plant mitochondrial genome: Dynamics and maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef]
- Foury, F.; Roganti, T.; Lecrenier, N.; Purnelle, B. The complete sequence of the mitochondrial genome of Saccharomyces cerevisiae. FEBS Lett. 1998, 440, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Chrzanowska-Lightowlers, Z.M.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef] [PubMed]
- Chaban, Y.; Boekema, E.J.; Dudkina, N.V. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim. Biophys. Acta 2014, 1837, 418–426. [Google Scholar] [CrossRef] [Green Version]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2015, 44, D1251–D1257. [Google Scholar] [CrossRef]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [Green Version]
- Falkenberg, M.; Larsson, N.-G.; Gustafsson, C.M. DNA Replication and Transcription in Mammalian Mitochondria. Annu. Rev. Biochem. 2007, 76, 679–699. [Google Scholar] [CrossRef]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.-G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef]
- Falkenberg, M. Mitochondrial DNA replication in mammalian cells: Overview of the pathway. Essays Biochem. 2018, 62, 287–296. [Google Scholar]
- Berk, A.J.; Clayton, D.A. Mechanism of mitochondrial DNA replication in mouse L-cells: Asynchronous replication of strands, segregation of circular daughter molecules, aspects of topology and turnover of an initiation sequence. J. Mol. Biol. 1974, 86, 801–824. [Google Scholar] [CrossRef]
- Falkenberg, M.; Gaspari, M.; Rantanen, A.; Trifunovic, A.; Larsson, N.-G.; Gustafsson, C.M. Mitochondrial transcription factors B1 and B2 activate transcription of human mtDNA. Nat. Genet. 2002, 31, 289–294. [Google Scholar] [CrossRef]
- Hillen, H.S.; Morozov, Y.I.; Sarfallah, A.; Temiakov, D.; Cramer, P. Structural Basis of Mitochondrial Transcription Initiation. Cell 2017, 171, 1072–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minczuk, M.; He, J.; Duch, A.M.; Ettema, T.J.; Chlebowski, A.; Dzionek, K.; Nijtmans, L.G.; Huynen, M.A.; Holt, I.J. TEFM (c17orf42) is necessary for transcription of human mtDNA. Nucleic Acids Res. 2011, 39, 4284–4299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posse, V.; Shahzad, S.; Falkenberg, M.; Hallberg, B.M.; Gustafsson, C.M. TEFM is a potent stimulator of mitochondrial transcription elongation in vitro. Nucleic Acids Res. 2015, 43, 2615–2624. [Google Scholar] [CrossRef] [PubMed]
- Posse, V.; Al-Behadili, A.; Uhler, J.P.; Clausen, A.R.; Reyes, A.; Zeviani, M.; Falkenberg, M.; Gustafsson, C.M. RNase H1 directs origin-specific initiation of DNA replication in human mitochondria. PLoS Genet. 2019, 15, e1007781. [Google Scholar] [CrossRef] [Green Version]
- Wanrooij, P.H.; Uhler, J.P.; Simonsson, T.; Falkenberg, M.; Gustafsson, C.M. G-quadruplex structures in RNA stimulate mitochondrial transcription termination and primer formation. Proc. Natl. Acad. Sci. USA 2010, 107, 16072–16077. [Google Scholar] [CrossRef] [Green Version]
- Wanrooij, P.H.; Uhler, J.P.; Shi, Y.; Westerlund, F.; Falkenberg, M.; Gustafsson, C.M. A hybrid G-quadruplex structure formed between RNA and DNA explains the extraordinary stability of the mitochondrial R-loop. Nucleic Acids Res. 2012, 40, 10334–10344. [Google Scholar] [CrossRef] [Green Version]
- Gray, H.; Wong, T.W. Purification and identification of subunit structure of the human mitochondrial DNA polymerase. J. Biol. Chem. 1992, 267, 5835–5841. [Google Scholar]
- Yakubovskaya, E.; Chen, Z.; Carrodeguas, J.A.; Kisker, C.; Bogenhagen, D.F. Functional human mitochondrial dna polymerase forms a heterotrimer. J. Biol. Chem. 2005, 281, 374–382. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-S.; Kennedy, W.D.; Yin, Y.W. Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell 2009, 139, 312–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrodeguas, J.A.; Pinz, K.G.; Bogenhagen, D.F. DNA binding properties of human pol, γB. J. Biol. Chem. 2002, 277, 50008–50014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korhonen, J.A.; Pham, X.H.; Pellegrini, M.; Falkenberg, M. Reconstitution of a minimal mtDNA replisome in vitro. EMBO J. 2004, 23, 2423–2429. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, J.A.; Gaspari, M.; Falkenberg, M. TWINKLE Has 5′ -> 3′ DNA Helicase Activity and Is Specifically Stimulated by Mitochondrial Single-stranded DNA-binding Protein. J. Biol. Chem. 2003, 278, 48627–48632. [Google Scholar] [CrossRef] [Green Version]
- Martens, P.A.; Clayton, D.A. Mechanism of mitochondrial DNA replication in mouse L-cells: Localization and sequence of the light-strand origin of replication. J. Mol. Biol. 1979, 135, 327–351. [Google Scholar] [CrossRef]
- Fusté, J.M.; Wanrooij, S.; Jemt, E.; Granycome, C.E.; Cluett, T.J.; Shi, Y.; Atanassova, N.; Holt, I.J.; Gustafsson, C.M.; Falkenberg, M. Mitochondrial RNA Polymerase Is Needed for Activation of the Origin of Light-Strand DNA Replication. Mol. Cell 2010, 37, 67–78. [Google Scholar] [CrossRef]
- Wanrooij, S.; Fuste, J.M.; Farge, G.; Shi, Y.; Gustafsson, C.M.; Falkenberg, M. Human mitochondrial RNA polymerase primes lagging-strand DNA synthesis in vitro. Proc. Natl. Acad. Sci. USA 2008, 105, 11122–11127. [Google Scholar] [CrossRef] [Green Version]
- Reyes, A.; Kazak, L.; Wood, S.R.; Yasukawa, T.; Jacobs, H.T.; Holt, I.J. Mitochondrial DNA replication proceeds via a ‘bootlace’ mechanism involving the incorporation of processed transcripts. Nucleic Acids Res. 2013, 41, 5837–5850. [Google Scholar] [CrossRef] [Green Version]
- Yasukawa, T.; Kang, N. An overview of mammalian mitochondrial DNA replication mechanisms. J. Biochem. 2018, 164, 183–193. [Google Scholar] [CrossRef]
- Holt, I.J.; Jacobs, H.T. Unique features of DNA replication in mitochondria: A functional and evolutionary perspective. BioEssays 2014, 36, 1024–1031. [Google Scholar] [CrossRef]
- Holt, I.; Lorimer, H.E.; Jacobs, H.T. Coupled Leading- and Lagging-Strand Synthesis of Mammalian Mitochondrial DNA. Cell 2000, 100, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Holmes, J.B.; Akman, G.; Wood, S.R.; Sakhuja, K.; Cerritelli, S.M.; Moss, C.; Bowmaker, M.R.; Jacobs, H.T.; Crouch, R.J.; Holt, I.J.; et al. Primer retention owing to the absence of RNase H1 is catastrophic for mitochondrial DNA replication. Proc. Natl. Acad. Sci. USA 2015, 112, 9334–9339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Behadili, A.; Uhler, J.P.; Berglund, A.K.; Peter, B.; Doimo, M.; Reyes, A.; Wanrooij, S.; Zeviani, M.; Falkenberg, M. A two-nuclease pathway involving RNase H1 is required for primer removal at human mitochondrial, O.r.i.L. Nucleic Acids Res. 2018, 46, 9471–9483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerritelli, S.M.; Frolova, E.G.; Feng, C.; Grinberg, A.; Love, P.E.; Crouch, R.J. Failure to Produce Mitochondrial DNA Results in Embryonic Lethality in Rnaseh1 Null Mice. Mol. Cell 2003, 11, 807–815. [Google Scholar] [CrossRef]
- Lakshmipathy, U.; Campbell, C. The Human DNA Ligase III Gene Encodes Nuclear and Mitochondrial Proteins. Mol. Cell. Biol. 1999, 19, 3869–3876. [Google Scholar] [CrossRef] [Green Version]
- Uhler, J.P.; Thörn, C.; Nicholls, T.J.; Matic, S.; Milenkovic, D.; Gustafsson, C.M.; Falkenberg, M. MGME1 processes flaps into ligatable nicks in concert with DNA polymerase gamma during mtDNA replication. Nucleic Acids Res. 2016, 44, 5861–5871. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, T.J.; Zsurka, G.; Peeva, V.; Schöler, S.; Szczesny, R.J.; Cysewski, D.; Reyes, A.; Kornblum, C.; Sciacco, M.; Moggio, M.; et al. Linear mtDNA fragments and unusual mtDNA rearrangements associated with pathological deficiency of MGME1 exonuclease. Hum. Mol. Genet. 2014, 23, 6147–6162. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, T.J.; Nadalutti, C.A.; Motori, E.; Sommerville, E.W.; Gorman, G.S.; Basu, S.; Hoberg, E.; Turnbull, D.M.; Chinnery, P.F.; Larsson, N.G.; et al. Topoisomerase 3alpha Is Required for Decatenation Segregation of Human mtDNA. Mol. Cell 2018, 69, 9–23.e6. [Google Scholar] [CrossRef] [Green Version]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef]
- Schon, E.A.; Rizzuto, R.; Moraes, C.T.; Nakase, H.; Zeviani, M.; DiMauro, S. A direct repeat is a hotspot for large-scale deletion of human mitochondrial DNA. Science 1989, 244, 346–349. [Google Scholar] [CrossRef]
- Viscomi, C.; Zeviani, M. MtDNA-maintenance defects: Syndromes and genes. J. Inherit. Metab. Dis. 2017, 40, 587–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Lawless, C.; Greaves, L.C.; Reeve, A.K.; Turnbull, D.M.; Vincent, A.E. The rise and rise of mitochondrial DNA mutations. Open Biol. 2020, 10, 200061. [Google Scholar] [CrossRef] [PubMed]
- Nissanka, N.; Minczuk, M.; Moraes, C.T. Mechanisms of Mitochondrial DNA Deletion Formation. Trends Genet. 2019, 35, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Shoffner, J.M.; Lott, M.T.; Voljavec, A.S.; Soueidan, S.A.; Costigan, D.A.; Wallace, D.C. Spontaneous Kearns-Sayre/chronic external ophthalmoplegia plus syndrome associated with a mitochondrial DNA deletion: A slip-replication model and metabolic therapy. Proc. Natl. Acad. Sci. USA 1989, 86, 7952–7956. [Google Scholar] [CrossRef] [Green Version]
- Persson, Ö.; Muthukumar, Y.; Basu, S.; Jenninger, L.; Uhler, J.P.; Berglund, A.-K.; McFarland, R.; Taylor, R.W.; Gustafsson, C.M.; Larsson, E. Copy-choice recombination during mitochondrial L-strand synthesis causes DNA deletions. Nat. Commun. 2019, 10, 1–10. [Google Scholar]
- Nissanka, N.; Bacman, S.R.; Plastini, M.; Moraes, C.T. The mitochondrial DNA polymerase gamma degrades linear DNA fragments precluding the formation of deletions. Nat. Commun. 2018, 9, 2491. [Google Scholar] [CrossRef]
- Srivastava, S.; Moraes, C.T. Double-strand breaks of mouse muscle mtDNA promote large deletions similar to multiple mtDNA deletions in humans. Hum. Mol. Genet. 2005, 14, 893–902. [Google Scholar] [CrossRef] [Green Version]
- Bacman, S.R.; Williams, S.L.; Moraes, C.T. Intra-and inter-molecular recombination of mitochondrial DNA after in vivo induction of multiple double-strand breaks. Nucleic Acids Res. 2009, 37, 4218–4226. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, K.J.; Reeve, A.K.; Samuels, D.C.; Chinnery, P.F.; Blackwood, J.K.; Taylor, R.W.; Wanrooij, S.; Spelbrink, J.N.; Chrzanowska-Lightowlers, Z.M.; Turnbull, D.M. What causes mitochondrial DNA deletions in human cells? Nat. Genet. 2008, 40, 275–279. [Google Scholar] [CrossRef] [PubMed]
- Farge, G.; Mehmedovic, M.; Baclayon, M.; Wildenberg, S.M.V.D.; Roos, W.H.; Gustafsson, C.M.; Wuite, G.J.; Falkenberg, M. In Vitro-Reconstituted Nucleoids Can Block Mitochondrial DNA Replication and Transcription. Cell Rep. 2014, 8, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, T.A.; Tkachuk, A.N.; Shtengel, G.; Kopek, B.G.; Bogenhagen, D.F.; Hess, H.F.; Clayton, D.A. Superresolution fluorescence imaging of mitochondrial nucleoids reveals their spatial range, limits, and membrane interaction. Mol. Cell. Biol. 2011, 31, 4994–5010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukat, C.; Wurm, C.A.; Spahr, H.; Falkenberg, M.; Larsson, N.G.; Jakobs, S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Natl. Acad. Sci. USA 2011, 108, 13534–13539. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, B.A.; Durisic, N.; Mativetsky, J.M.; Costantino, S.; Hancock, M.A.; Grutter, P.; Shoubridge, E.A. The Mitochondrial Transcription Factor TFAM Coordinates the Assembly of Multiple DNA Molecules into Nucleoid-like Structures. Mol. Biol. Cell 2007, 18, 3225–3236. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.-G. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 2004, 13, 935–944. [Google Scholar] [CrossRef]
- Bogenhagen, D.F. Mitochondrial DNA nucleoid structure. Biochim. Biophys. Acta 2012, 1819, 914–920. [Google Scholar] [CrossRef]
- Takamatsu, C.; Umeda, S.; Ohsato, T.; Ohno, T.; Abe, Y.; Fukuoh, A.; Shinagawa, H.; Hamasaki, N.; Kang, D. Regulation of mitochondrial D-loops by transcription factor A and single-stranded DNA-binding protein. EMBO Rep. 2002, 3, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Bogenhagen, D.F. Human Mitochondrial DNA Nucleoids Are Linked to Protein Folding Machinery and Metabolic Enzymes at the Mitochondrial Inner Membrane. J. Biol. Chem. 2006, 281, 25791–25802. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Cooper, H.M.; Reyes, A.; Di Re, M.; Sembongi, H.; Litwin, T.R.; Gao, J.; Neuman, K.C.; Fearnley, I.M.; Spinazzola, A. Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res. 2012, 40, 6109–6121. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Udeshi, N.D.; Deerinck, T.J.; Svinkina, T.; Ellisman, M.H.; Carr, S.A.; Ting, A.Y. Proximity Biotinylation as a Method for Mapping Proteins Associated with mtDNA in Living Cells. Cell Chem. Biol. 2017, 24, 404–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogenhagen, D.F.; Rousseau, D.; Burke, S. The Layered Structure of Human Mitochondrial DNA Nucleoids. J. Biol. Chem. 2007, 283, 3665–3675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couvillion, M.T.; Soto, I.C.; Shipkovenska, G.; Churchman, L.S. Synchronized mitochondrial and cytosolic translation programs. Nature 2016, 533, 499–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter-Dennerlein, R.; Oeljeklaus, S.; Lorenzi, I.; Ronsör, C.; Bareth, B.; Schendzielorz, A.B.; Wang, C.; Warscheid, B.; Rehling, P.; Dennerlein, S. Mitochondrial Protein Synthesis Adapts to Influx of Nuclear-Encoded Protein. Cell 2016, 167, 471–483.e10. [Google Scholar] [CrossRef] [Green Version]
- Gilkerson, R.W.; Schon, E.A.; Hernandez, E.; Davidson, M.M. Mitochondrial nucleoids maintain genetic autonomy but allow for functional complementation. J. Cell Biol. 2008, 181, 1117–1128. [Google Scholar] [CrossRef]
- Ishihara, T.; Ban-Ishihara, R.; Maeda, M.; Matsunaga, Y.; Ichimura, A.; Kyogoku, S.; Aoki, H.; Katada, S.; Nakada, K.; Nomura, M. Dynamics of Mitochondrial DNA Nucleoids Regulated by Mitochondrial Fission Is Essential for Maintenance of Homogeneously Active Mitochondria during Neonatal Heart Development. Mol. Cell. Biol. 2014, 35, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Nass, M.M. Mitochondrial DNA. I. Intramitochondrial distribution and structural relations of single- \and double-length circular DNA. J. Mol. Biol. 1969, 42, 521–528. [Google Scholar] [CrossRef]
- Kopek, B.G.; Shtengel, G.; Xu, C.S.; Clayton, D.A.; Hess, H.F. Correlative 3D superresolution fluorescence and electron microscopy reveal the relationship of mitochondrial nucleoids to membranes. Proc. Natl. Acad. Sci. USA 2012, 109, 6136–6141. [Google Scholar] [CrossRef] [Green Version]
- Iborra, F.J.; Kimura, H.; Cook, P.R. The functional organisation of mitochondrial genomes in human cells. BMC Biol. 2004, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Albring, M.; Griffith, J.; Attardi, G. Association of a protein structure of probable membrane derivation with HeLa cell mitochondrial DNA near its origin of replication. Proc. Natl. Acad. Sci. USA 1977, 74, 1348–1352. [Google Scholar] [CrossRef] [Green Version]
- Collins, T.J.; Berridge, M.J.; Lipp, P.; Bootman, M.D. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 2002, 21, 1616–1627. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.G.; Scorrano, L. Mitochondrial morphology in mitophagy and macroautophagy. Biochim. Biophys. Acta 2013, 1833, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial dysfunction and cell senescence: Deciphering a complex relationship. FEBS Lett. 2019, 593, 1566–1579. [Google Scholar] [CrossRef] [Green Version]
- Lewis, S.C.; Uchiyama, L.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016, 353, aaf5549. [Google Scholar] [CrossRef] [Green Version]
- Murley, A.; Lackner, L.L.; Osman, C.; West, M.; Voeltz, G.K.; Walter, P.; Nunnari, J.; Youle, R.J. ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. ELife 2013, 2, e00422. [Google Scholar] [CrossRef] [PubMed]
- Ban-Ishihara, R.; Ishihara, T.; Sasaki, N.; Mihara, K.; Ishihara, N. Dynamics of nucleoid structure regulated by mitochondrial fission contributes to cristae reformation and release of cytochrome c. Proc. Natl. Acad. Sci. USA 2013, 110, 11863–11868. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An Actin-Dependent Step in Mitochondrial Fission Mediated by the ER-Associated Formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [Green Version]
- Manor, U.; Bartholomew, S.; Golani, G.; Christenson, E.; Kozlov, M.; Higgs, H.N.; Spudich, J.; Lippincott-Schwartz, J. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. ELife 2015, 4. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.-L.; Van Der Bliek, A.M. Dynamin-related Protein Drp1 Is Required for Mitochondrial Division in Mammalian Cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, D.I.; Parone, P.A.; Mattenberger, Y.; Martinou, J.-C. hFis1, a Novel Component of the Mammalian Mitochondrial Fission Machinery. J. Biol. Chem. 2003, 278, 36373–36379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Kalia, R.; Wang, R.Y.-R.; Yusuf, A.; Thomas, P.V.; Agard, D.A.; Shaw, J.M.; Frost, A. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 2018, 558, 401–405. [Google Scholar] [CrossRef]
- Kamerkar, S.; Kraus, F.; Sharpe, A.; Pucadyil, T.J.; Ryan, M.T. Dynamin-related protein 1 has membrane constricting and severing abilities sufficient for mitochondrial and peroxisomal fission. Nat. Commun. 2018, 9, 5239. [Google Scholar] [CrossRef]
- Fonseca, T.B.; Sánchez-Guerrero, Á.; Milosevic, I.; Raimundo, N. Mitochondrial fission requires DRP1 but not dynamins. Nature 2019, 570, E34–E42. [Google Scholar] [CrossRef]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria–lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef]
- Nagashima, S.; Tábara, L.-C.; Tilokani, L.; Paupe, V.; Anand, H.; Pogson, J.H.; Zunino, R.; McBride, H.M.; Prudent, J. Golgi-derived PI(4)P-containing vesicles drive late steps of mitochondrial division. Science 2020, 367, 1366–1371. [Google Scholar] [CrossRef]
- Garrido, N.; Griparic, L.; Jokitalo, E.; Wartiovaara, J.; Van Der Bliek, A.M.; Spelbrink, J.N. Composition and Dynamics of Human Mitochondrial Nucleoids. Mol. Biol. Cell 2003, 14, 1583–1596. [Google Scholar] [CrossRef] [Green Version]
- Waterham, H.R.; Koster, J.; Van Roermund, C.W.; Mooyer, P.A.; Wanders, R.J.; Leonard, J.V. A Lethal Defect of Mitochondrial and Peroxisomal Fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.V.; Ben Othmane, K.; Rochelle, J.M.; Stajich, J.E.; Hulette, C.; Dew-Knight, S.; Hentati, F.; Ben Hamida, M.; Bel, S.; Stenger, J.E. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat. Genet. 2001, 30, 21–22. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Schlöndorff, J.S.; Becker, D.J.; Tsukaguchi, H.; Tonna, S.J.; Uscinski, A.L.; Higgs, H.N.; Henderson, J.M.; Pollak, M.R. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat. Genet. 2010, 42, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Shamseldin, H.E.; Alshammari, M.; Al-Sheddi, T.; Salih, M.A.M.; Alkhalidi, H.; Kentab, A.; Repetto, G.M.; Hashem, M.O.; Alkuraya, F.S. Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J. Med. Genet. 2012, 49, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Abrams, A.J.; Hufnagel, R.B.; Rebelo, A.; Zanna, C.; Patel, N.; Gonzalez, M.A.; Campeanu, I.J.; Griffin, L.B.; Groenewald, S.; Strickland, A.V.; et al. Mutations in SLC25A46, encoding a UGO1-like protein, cause an optic atrophy spectrum disorder. Nat. Genet. 2015, 47, 926–932. [Google Scholar] [CrossRef] [Green Version]
- Vanstone, J.R.; Smith, A.M.; McBride, S.; Naas, T.; Holčík, M.; Antoun, G.; Harper, M.-E.; Michaud, J.; Sell, E.; Chakraborty, P.; et al. DNM1L-related mitochondrial fission defect presenting as refractory epilepsy. Eur. J. Hum. Genet. 2015, 24, 1084–1088. [Google Scholar] [CrossRef] [Green Version]
- Koch, J.; Feichtinger, R.G.; Freisinger, P.; Pies, M.; Schrödl, F.; Iuso, A.; Sperl, W.; Mayr, J.A.; Prokisch, H.; Haack, T.B. Disturbed mitochondrial and peroxisomal dynamics due to loss of MFF causes Leigh-like encephalopathy, optic atrophy and peripheral neuropathy. J. Med. Genet. 2016, 53, 270–278. [Google Scholar] [CrossRef]
- Janer, A.; Prudent, J.; Paupe, V.; Fahiminiya, S.; Majewski, J.; Sgarioto, N.; Rosiers, C.D.; Forest, A.; Lin, Z.; Gingras, A.-C.; et al. SLC 25A46 is required for mitochondrial lipid homeostasis and cristae maintenance and is responsible for Leigh syndrome. EMBO Mol. Med. 2016, 8, 1019–1038. [Google Scholar] [CrossRef]
- Bitoun, M.; Maugenre, S.; Jeannet, P.Y.; Lacène, E.; Ferrer, X.; Laforêt, P.; Martin, J.J.; Laporte, J.; Lochmüller, H.; Beggs, A.H.; et al. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat. Genet. 2005, 37, 1207–1209. [Google Scholar] [CrossRef]
- Claeys, K.G.; Züchner, S.; Kennerson, M.; Berciano, J.; Garcia, A.; Verhoeven, K.; Storey, E.; Merory, J.R.; Bienfait, H.M.E.; Lammens, M.; et al. Phenotypic spectrum of dynamin 2 mutations in Charcot-Marie-Tooth neuropathy. Brain 2009, 132 Pt 7, 1741–1752. [Google Scholar] [CrossRef]
- Böhm, J.; Biancalana, V.; Dechene, E.T.; Bitoun, M.; Pierson, C.R.; Schaefer, E.; Karasoy, H.; Dempsey, M.A.; Klein, F.; Dondaine, N.; et al. Mutation spectrum in the large GTPase dynamin 2, and genotype-phenotype correlation in autosomal dominant centronuclear myopathy. Hum. Mutat. 2012, 33, 949–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gal, A.; Inczedy-Farkas, G.; Pál, E.; Remenyi, V.; Bereznai, B.; Gellér, L.; Szelid, Z.; Merkely, B.; Molnar, M.J. The coexistence of dynamin 2 mutation and multiple mitochondrial DNA (mtDNA) deletions in the background of severe cardiomyopathy and centronuclear myopathy. Clin. Neuropathol. 2015, 34, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, O.; Nevo, F.; Plaisier, E.; Funalot, B.; Gribouval, O.; Benoit, G.; Huynh Cong, E.; Arrondel, C.; Tête, M.J.; Montjean, R.; et al. INF2Mutations in Charcot–Marie–Tooth Disease with Glomerulopathy. N. Engl. J. Med. 2011, 365, 2377–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caridi, G.; Lugani, F.; Dagnino, M.; Gigante, M.; Iolascon, A.; Falco, M.; Graziano, C.; Benetti, E.; Dugo, M.; Del Prete, D.; et al. Novel INF2 mutations in an Italian cohort of patients with focal segmental glomerulosclerosis, renal failure and Charcot-Marie-Tooth neuropathy. Nephrol. Dial. Transpl. 2014, 29 (Suppl. S4), iv80–iv86. [Google Scholar] [CrossRef] [Green Version]
- Crimella, C.; Tonelli, A.; Airoldi, G.; Baschirotto, C.; D’Angelo, M.G.; Bonato, S.; Losito, L.; Trabacca, A.; Bresolin, N.; Bassi, M.T.; et al. The GST domain of GDAP1 is a frequent target of mutations in the dominant form of axonal Charcot Marie Tooth type, 2.K. J. Med. Genet. 2010, 47, 712–716. [Google Scholar] [CrossRef] [Green Version]
- Nasca, A.; Nardecchia, F.; Commone, A.; Semeraro, M.; Legati, A.; Garavaglia, B.; Ghezzi, D.; Leuzzi, V. Clinical and Biochemical Features in a Patient With Mitochondrial Fission Factor Gene Alteration. Front. Genet. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Pedrola, L.; Espert, A.; Wu, X.; Claramunt, R.; Shy, M.E.; Palau, F. GDAP1, the protein causing Charcot-Marie-Tooth disease type 4A, is expressed in neurons and is associated with mitochondria. Hum. Mol. Genet. 2005, 14, 1087–1094. [Google Scholar] [CrossRef]
- Pezzini, I.; Geroldi, A.; Capponi, S.; Gulli, R.; Schenone, A.; Grandis, M.; Doria-Lamba, L.; La Piana, C.; Cremonte, M.; Pisciotta, C.; et al. GDAP1 mutations in Italian axonal Charcot–Marie–Tooth patients: Phenotypic features and clinical course. Neuromuscul. Disord. 2016, 26, 26–32. [Google Scholar] [CrossRef]
- Parone, P.A.; Da Cruz, S.; Tondera, D.; Mattenberger, Y.; James, D.I.; Maechler, P.; Barja, F.; Martinou, J.C. Preventing Mitochondrial Fission Impairs Mitochondrial Function and Leads to Loss of Mitochondrial DNA. PLoS ONE 2008, 3, e3257. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.; et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nature 2009, 11, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, J.; Zhang, Z.; Wakabayashi, N.; Tamura, Y.; Fukaya, M.; Kensler, T.W.; Iijima, M.; Sesaki, H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 2009, 186, 805–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Ren, S.; Clish, C.; Jain, M.; Mootha, V.; McCaffery, J.M.; Chan, D.C. Titration of mitochondrial fusion rescues Mff-deficient cardiomyopathy. J. Cell Biol. 2015, 211, 795–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kameoka, S.; Adachi, Y.; Okamoto, K.; Iijima, M.; Sesaki, H. Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics. Trends Cell Biol. 2017, 28, 67–76. [Google Scholar] [CrossRef]
- Macdonald, P.J.; Stepanyants, N.; Mehrotra, N.; Mears, J.A.; Qi, X.; Sesaki, H.; Ramachandran, R. A dimeric equilibrium intermediate nucleates Drp1 reassembly on mitochondrial membranes for fission. Mol. Biol. Cell 2014, 25, 1905–1915. [Google Scholar] [CrossRef]
- Francy, C.A.; Alvarez, F.J.D.; Zhou, L.; Ramachandran, R.; Mears, J.A. The Mechanoenzymatic Core of Dynamin-related Protein 1 Comprises the Minimal Machinery Required for Membrane Constriction. J. Biol. Chem. 2015, 290, 11692–11703. [Google Scholar] [CrossRef] [Green Version]
- Francy, C.A.; Clinton, R.W.; Fröhlich, C.; Murphy, C.; Mears, J.A. Cryo-EM Studies of Drp1 Reveal Cardiolipin Interactions that Activate the Helical Oligomer. Sci. Rep. 2017, 7, 10744. [Google Scholar] [CrossRef]
- Stepanyants, N.; Macdonald, P.J.; Francy, C.A.; Mears, J.A.; Qi, X.; Ramachandran, R. Cardiolipin’s propensity for phase transition and its reorganisation by dynamin-related protein 1 form a basis for mitochondrial membrane fission. Mol. Biol. Cell 2015, 26, 3104–3116. [Google Scholar] [CrossRef]
- dachi, Y.; Itoh, K.; Yamada, T.; Cerveny, K.L.; Suzuki, T.L.; Macdonald, P.; Frohman, M.A.; Ramachandran, R.; Iijima, M.; Sesaki, H. Coincident Phosphatidic Acid Interaction Restrains Drp1 in Mitochondrial Division. Mol. Cell 2016, 63, 1034–1043. [Google Scholar]
- Luévano-Martínez, L.A.; Forni, M.F.; Dos Santos, V.T.; Souza-Pinto, N.C.; Kowaltowski, A.J. Cardiolipin is a key determinant for mtDNA stability and segregation during mitochondrial stress. Biochim. Biophys. Acta 2015, 1847, 587–598. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Q.; Gohil, V.M.; Ma, L.; Greenberg, M.L. Absence of Cardiolipin Results in Temperature Sensitivity, Respiratory Defects, and Mitochondrial DNA Instability Independent of pet56. J. Biol. Chem. 2004, 279, 32294–32300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Liu, D.; Finley, R.L.; Greenberg, M.L., Jr. Loss of mitochondrial DNA in the yeast cardiolipin synthase crd1 mutant leads to up-regulation of the protein kinase Swe1p that regulates the G2/M transition. J. Biol. Chem. 2010, 285, 10397–10407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acehan, D.; Malhotra, A.; Xu, Y.; Ren, M.; Stokes, D.L.; Schlame, M. Cardiolipin Affects the Supramolecular Organisation of ATP Synthase in Mitochondria. Biophys. J. 2011, 100, 2184–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, T.; Isobe, K.; Nakada, K.; Hayashi, J.-I. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat. Genet. 2001, 28, 272–275. [Google Scholar] [CrossRef]
- Cao, Y.L.; Meng, S.; Chen, Y.; Feng, J.X.; Gu, D.D.; Yu, B.; Li, Y.J.; Yang, J.Y.; Liao, S.; Chan, D.C.; et al. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 2017, 542, 372–376. [Google Scholar] [CrossRef]
- Yan, L.; Qi, Y.; Huang, X.; Yu, C.; Lan, L.; Guo, X.; Rao, Z.; Hu, J.; Lou, Z. Structural basis for GTP hydrolysis and conformational change of MFN1 in mediating membrane fusion. Nat. Struct. Mol. Biol. 2018, 25, 233–243. [Google Scholar] [CrossRef]
- Qi, Y.; Yan, L.; Yu, C.; Guo, X.; Zhou, X.; Hu, X.; Huang, X.; Rao, Z.; Lou, Z.; Hu, J. Structures of human mitofusin 1 provide insight into mitochondrial tethering. J. Cell Biol. 2016, 215, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef] [Green Version]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [Google Scholar] [CrossRef] [Green Version]
- Wai, T.; Garcia-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Ruperez, F.J.; Barbas, C.; Ibanez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef] [PubMed]
- Griparic, L.; Kanazawa, T.; Van Der Bliek, A.M. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 2007, 178, 757–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.S.; Motori, E.; Bruser, C.; Kuhl, I.; Yeroslaviz, A.; Ruzzenente, B.; Kauppila, J.H.K.; Busch, J.D.; Hultenby, K.; Habermann, B.H.; et al. Mitochondrial fusion is required for regulation of mitochondrial DNA replication. PLoS Genet. 2019, 15, e1008085. [Google Scholar] [CrossRef] [Green Version]
- Elachouri, G.; Vidoni, S.; Zanna, C.; Pattyn, A.; Boukhaddaoui, H.; Gaget, K.; Yu-Wai-Man, P.; Gasparre, G.; Sarzi, E.; Delettre, C.; et al. OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution. Genome Res. 2010, 21, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial Fusion Is Required for mtDNA Stability in Skeletal Muscle and Tolerance of mtDNA Mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, A.; Nagashima, S.; Tokuyama, T.; Amo, T.; Matsuki, Y.; Ishido, S.; Kudo, Y.; McBride, H.M.; Fukuda, T.; Matsushita, N.; et al. MITOL Regulates Endoplasmic Reticulum-Mitochondria Contacts via Mitofusin2. Mol. Cell 2013, 51, 20–34. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Csordás, G.; Jowdy, C.; Schneider, T.G.; Csordás, N.; Wang, W.; Liu, Y.; Kohlhaas, M.; Meiser, M.; Bergem, S.; et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ. Res. 2012, 111, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Yu-Wai-Man, P.; Griffiths, P.G.; Burke, A.; Sellar, P.W.; Clarke, M.P.; Gnanaraj, L.; Ah-Kine, D.; Hudson, G.; Czermin, B.; Taylor, R.W.; et al. The prevalence and natural history of dominant optic atrophy due to OPA1 mutations. Ophthalmology 2010, 117, 1538–1546. [Google Scholar] [CrossRef] [Green Version]
- Yu-Wai-Man, P.; Chinnery, P.F. Dominant optic atrophy: Novel OPA1 mutations and revised prevalence estimates. Ophthalmology 2013, 120, 1712. [Google Scholar] [CrossRef]
- Hudson, G.; Amati-Bonneau, P.; Blakely, E.L.; Stewart, J.D.; He, L.; Schaefer, A.M.; Griffiths, P.G.; Ahlqvist, K.; Suomalainen, A.; Reynier, P.; et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: A novel disorder of mtDNA maintenance. Brain 2008, 131 Pt 2, 329–337. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain 2010, 133 Pt 3, 771–786. [Google Scholar] [CrossRef]
- Carelli, V.; Musumeci, O.; Caporali, L.; Zanna, C.; La Morgia, C.; Del Dotto, V.; Porcelli, A.M.; Rugolo, M.; Valentino, M.L.; Iommarini, L.; et al. Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann. Neurol. 2015, 78, 21–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braathen, G.J.; Sand, J.C.; Lobato, A.; Høyer, H.; Russell, M.B. Genetic epidemiology of Charcot–Marie–Tooth in the general population. Eur. J. Neurol. 2010, 18, 39–48. [Google Scholar] [CrossRef]
- Braathen, G.J.; Sand, J.C.; Lobato, A.; Høyer, H.; Russell, M.B. MFN2 point mutations occur in 3.4% of Charcot-Marie-Tooth families. An investigation of 232 Norwegian CMT families. BMC Med. Genet. 2010, 11, 48. [Google Scholar] [CrossRef] [Green Version]
- Feely, S.M.; Laura, M.; Siskind, C.E.; Sottile, S.; Davis, M.; Gibbons, V.S.; Reilly, M.M.; Shy, M.E. MFN2 mutations cause severe phenotypes in most patients with, C.M.T.2.A. Neurology 2011, 76, 1690–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casari, G.; De Fusco, M.; Ciarmatori, S.; Zeviani, M.; Mora, M.; Fernandez, P.; De Michele, G.; Filla, A.; Cocozza, S.; Marconi, R.; et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 1998, 93, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, G.; Pyle, A.; Griffin, H.; Miller, J.; Wilson, V.; Turnbull, L.; Fawcett, K.; Sims, D.; Eglon, G.; Hadjivassiliou, M.; et al. SPG7 mutations are a common cause of undiagnosed ataxia. Neurology 2015, 84, 1174–1176. [Google Scholar] [CrossRef] [Green Version]
- Hewamadduma, C.A.; Hoggard, N.; O’Malley, R.; Robinson, M.K.; Beauchamp, N.J.; Segamogaite, R.; Martindale, J.; Rodgers, T.; Rao, G.; Sarrigiannis, P.; et al. Novel genotype-phenotype and MRI correlations in a large cohort of patients with SPG7 mutations. Neurol. Genet. 2018, 4, e279. [Google Scholar] [CrossRef] [Green Version]
- De la Casa-Fages, B.; Fernández-Eulate, G.; Gamez, J.; Barahona-Hernando, R.; Morís, G.; García-Barcina, M.; Infante, J.; Zulaica, M.; Fernández-Pelayo, U.; Muñoz-Oreja, M.; et al. Parkinsonism and spastic paraplegia type 7: Expanding the spectrum of mitochondrial Parkinsonism. Mov. Disord. 2019, 34, 1547–1561. [Google Scholar] [CrossRef]
- Di Bella, D.; Lazzaro, F.; Brusco, A.; Plumari, M.; Battaglia, G.; Pastore, A.; Finardi, A.; Cagnoli, C.; Tempia, F.; Frontali, M.; et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat. Genet. 2010, 42, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Pfeffer, G.; Griffin, H.; Blakely, E.L.; Kurzawa-Akanbi, M.; Gabriel, J.; Sitarz, K.; Roberts, M.; Schoser, B.; Pyle, A.; et al. Clonal expansion of secondary mitochondrial DNA deletions associated with spinocerebellar ataxia type 28. JAMA Neurol. 2015, 72, 106–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, G.; Gorman, G.S.; Griffin, H.; Kurzawa-Akanbi, M.; Blakely, E.L.; Wilson, I.; Sitarz, K.; Moore, D.; Murphy, J.L.; Alston, C.L.; et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 2014, 137 Pt 5, 1323–1336. [Google Scholar] [CrossRef] [Green Version]
- Vielhaber, S.; Debska-Vielhaber, G.; Peeva, V.; Schoeler, S.; Kudin, A.P.; Minin, I.; Schreiber, S.; Dengler, R.; Kollewe, K.; Zuschratter, W.; et al. Mitofusin 2 mutations affect mitochondrial function by mitochondrial DNA depletion. Acta Neuropathol. 2013, 125, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Bonnen, P.E.; Yarham, J.W.; Besse, A.; Wu, P.; Faqeih, E.A.; Al-Asmari, A.M.; Saleh, M.A.; Eyaid, W.; Hadeel, A.; He, L.; et al. Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am. J. Hum. Genet. 2013, 93, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Di Mauro, S.; Hirano, M.M.E.R.R.F. GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1520/ (accessed on 24 August 2020).
- Sawyer, S.L.; Cheuk-Him Ng, A.; Innes, A.M.; Wagner, J.D.; Dyment, D.A.; Tetreault, M.; Majewski, J.; Boycott, K.M.; Screaton, R.A.; Nicholson, G. Homozygous mutations in MFN2 cause multiple symmetric lipomatosis associated with neuropathy. Hum. Mol. Genet. 2015, 24, 5109–5114. [Google Scholar] [CrossRef] [Green Version]
- Capel, E.; Vatier, C.; Cervera, P.; Stojkovic, T.; Disse, E.; Cottereau, A.S.; Auclair, M.; Verpont, M.C.; Mosbah, H.; Gourdy, P.; et al. MFN2-associated lipomatosis: Clinical spectrum and impact on adipose tissue. J. Clin. Lipidol. 2018, 12, 1420–1435. [Google Scholar] [CrossRef]
- Guan, K.; Farh, L.; Marshall, T.K.; Deschenes, R.J. Normal mitochondrial structure and genome maintenance in yeast requires the dynamin-like product of the MGM1 gene. Curr. Genet. 1993, 24, 141–148. [Google Scholar] [CrossRef]
- Rapaport, D.; Brunner, M.; Neupert, W.; Westermann, B. Fzo1p is a mitochondrial outer membrane protein essential for the biogenesis of functional mitochondria in Saccharomyces cerevisiae. J. Biol. Chem. 1998, 273, 20150–20155. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liu, T.; Tran, A.; Lu, X.; Tomilov, A.A.; Davies, V.; Cortopassi, G.; Chiamvimonvat, N.; Bers, D.M.; Votruba, M.; et al. OPA1 mutation and late-onset cardiomyopathy: Mitochondrial dysfunction and mtDNA instability. J. Am. Heart Assoc. 2012, 1, e003012. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef] [PubMed]
- DeVay, R.M.; Dominguez-Ramirez, L.; Lackner, L.L.; Hoppins, S.; Stahlberg, H.; Nunnari, J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J. Cell Biol. 2009, 186, 793–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ban, T.; Heymann, J.A.; Song, Z.; Hinshaw, J.E.; Chan, D.C. OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum. Mol. Genet. 2010, 19, 2113–2122. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.Y.; Huang, P.; Jenkins, G.M.; Chan, D.C.; Schiller, J.; Frohman, M.A. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat. Cell Biol. 2006, 8, 1255–1262. [Google Scholar] [CrossRef] [Green Version]
- Ban-Ishihara, R.; Tomohiro-Takamiya, S.; Tani, M.; Baudier, J.; Ishihara, N.; Kuge, O. COX assembly factor ccdc56 regulates mitochondrial morphology by affecting mitochondrial recruitment of Drp1. FEBS Lett. 2015, 589 Pt B, 3126–3132. [Google Scholar] [CrossRef]
- Cooper, H.M.; Yang, Y.; Ylikallio, E.; Khairullin, R.; Woldegebriel, R.; Lin, K.-L.; Euro, L.; Palin, E.; Wolf, A.; Trokovic, R.; et al. ATPase-deficient mitochondrial inner membrane protein ATAD3A disturbs mitochondrial dynamics in dominant hereditary spastic paraplegia. Hum. Mol. Genet. 2017, 26, 1432–1443. [Google Scholar] [CrossRef]
- Gilquin, B.; Taillebourg, E.; Cherradi, N.; Hubstenberger, A.; Gay, O.; Merle, N.; Assard, N.; Fauvarque, M.O.; Tomohiro, S.; Kuge, O.; et al. The AAA+ ATPase ATAD3A controls mitochondrial dynamics at the interface of the inner and outer membranes. Mol. Cell Biol. 2010, 30, 1984–1996. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Sun, X.; Hu, D.; Prosdocimo, D.A.; Hoppel, C.; Jain, M.K.; Ramachandran, R.; Qi, X. ATAD3A oligomerization causes neurodegeneration by coupling mitochondrial fragmentation and bioenergetics defects. Nat. Commun. 2019, 10, 1317. [Google Scholar] [CrossRef] [Green Version]
- Desai, R.; Frazier, A.E.; Durigon, R.; Patel, H.; Jones, A.W.; Dalla Rosa, I.; Lake, N.J.; Compton, A.G.; Mountford, H.S.; Tucker, E.J.; et al. ATAD3 gene cluster deletions cause cerebellar dysfunction associated with altered mitochondrial DNA and cholesterol metabolism. Brain 2017, 140, 1595–1610. [Google Scholar] [CrossRef]
- Gunning, A.C.; Strucinska, K.; Muñoz Oreja, M.; Parrish, A.; Caswell, R.; Stals, K.L.; Durigon, R.; Durlacher-Betzer, K.; Cunningham, M.H.; Grochowski, C.M.; et al. Recurrent De Novo NAHR Reciprocal Duplications in the ATAD3 Gene Cluster Cause a Neurogenetic Trait with Perturbed Cholesterol and Mitochondrial Metabolism. Am. J. Hum. Genet. 2020, 106, 272–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peralta, S.; Goffart, S.; Williams, S.L.; Diaz, F.; Garcia, S.; Nissanka, N.; Area-Gomez, E.; Pohjoismaki, J.; Moraes, C.T. ATAD3 controls mitochondrial cristae structure in mouse muscle, influencing mtDNA replication and cholesterol levels. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amati-Bonneau, P.; Valentino, M.L.; Reynier, P.; Gallardo, M.E.; Bornstein, B.; Boissiere, A.; Campos, Y.; Rivera, H.; de la Aleja, J.G.; Carroccia, R.; et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain 2008, 131 Pt 2, 338–351. [Google Scholar] [CrossRef] [Green Version]
- Lodi, R.; Tonon, C.; Valentino, M.L.; Manners, D.; Testa, C.; Malucelli, E.; La Morgia, C.; Barboni, P.; Carbonelli, M.; Schimpf, S.; et al. Defective mitochondrial adenosine triphosphate production in skeletal muscle from patients with dominant optic atrophy due to OPA1 mutations. Arch. Neurol. 2011, 68, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Rouzier, C.; Bannwarth, S.; Chaussenot, A.; Chevrollier, A.; Verschueren, A.; Bonello-Palot, N.; Bonello-Palot, N.; Fragaki, K.; Cano, A.; Pouget, J.; et al. The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy ‘plus’ phenotype. Brain 2012, 135 Pt 1, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [Green Version]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlof, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef]
- Legros, F.; Malka, F.; Frachon, P.; Lombes, A.; Rojo, M. Organisation and dynamics of human mitochondrial DNA. J. Cell Sci. 2004, 117 Pt 13, 2653–2662. [Google Scholar] [CrossRef] [Green Version]
- Kandul, N.P.; Zhang, T.; Hay, B.A.; Guo, M. Selective removal of deletion-bearing mitochondrial DNA in heteroplasmic Drosophila. Nat. Commun. 2016, 7, 13100. [Google Scholar] [CrossRef]
- Malena, A.; Loro, E.; Di Re, M.; Holt, I.J.; Vergani, L. Inhibition of mitochondrial fission favours mutant over wild-type mitochondrial DNA. Hum. Mol. Genet. 2009, 18, 3407–3416. [Google Scholar] [CrossRef] [Green Version]
- Pfanner, N.; Meijer, M. The Tom and Tim machine. Curr. Biol. 1997, 7, R100–R103. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, J.M.; Neupert, W. Protein insertion into the inner membrane of mitochondria. IUBMB Life 2003, 55, 219–225. [Google Scholar] [CrossRef]
- Wilkens, V.; Kohl, W.; Busch, K. Restricted diffusion of OXPHOS complexes in dynamic mitochondria delays their exchange between cristae and engenders a transitory mosaic distribution. J. Cell Sci. 2013, 126 Pt 1, 103–116. [Google Scholar] [CrossRef] [Green Version]
- Perkins, G.; Renken, C.; Martone, M.E.; Young, S.J.; Ellisman, M.; Frey, T. Electron tomography of neuronal mitochondria: Three-dimensional structure and organisation of cristae and membrane contacts. J. Struct. Biol. 1997, 119, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Perkins, G.; Song, J.Y.; Tarsa, L.; Deerinck, T.J.; Ellisman, M.H.; Frey, T.G. Electron tomography of mitochondria from brown adipocytes reveals crista junctions. J. Bioenergy Biomembr. 1998, 30, 431–442. [Google Scholar] [CrossRef]
- Gilkerson, R.W.; Selker, J.M.; Capaldi, R.A. The cristal membrane of mitochondria is the principal site of oxidative phosphorylation. FEBS Lett. 2003, 546, 355–358. [Google Scholar] [CrossRef] [Green Version]
- Stephan, T.; Roesch, A.; Riedel, D.; Jakobs, S. Live-cell STED nanoscopy of mitochondrial cristae. Sci. Rep. 2019, 9, 12419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jajoo, R.; Jung, Y.; Huh, D.; Viana, M.P.; Rafelski, S.M.; Springer, M.; Paulsson, J. Accurate concentration control of mitochondria and nucleoids. Science 2016, 351, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Prachar, J. Ultrastructure of mitochondrial nucleoid and its surroundings. Gen. Physiol. Biophys. 2016, 35, 273–286. [Google Scholar] [CrossRef] [Green Version]
- Rajala, N.; Gerhold, J.; Martinsson, P.; Klymov, A.; Spelbrink, J.N. Replication factors transiently associate with mtDNA at the mitochondrial inner membrane to facilitate replication. Nucleic Acids Res. 2013, 42, 952–967. [Google Scholar] [CrossRef] [Green Version]
- Khalifat, N.; Puff, N.; Bonneau, S.; Fournier, J.-B.; Angelova, M.I. Membrane Deformation under Local pH Gradient: Mimicking Mitochondrial Cristae Dynamics. Biophys. J. 2008, 95, 4924–4933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, A.; Barriere, S.; Broseus, L.; Brooke, J.; Lorenzi, C.; Villemin, J.P.; Beurier, G.; Sabatier, R.; Reynes, C.; Mancheron, A.; et al. GECKO is a genetic algorithm to classify and explore high throughput sequencing data. Commun. Biol. 2019, 2, 222. [Google Scholar] [CrossRef]
- Pfeiffer, K.; Gohil, V.; Stuart, R.A.; Hunte, C.; Brandt, U.; Greenberg, M.L.; Schägger, H. Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 2003, 278, 52873–52880. [Google Scholar] [CrossRef] [Green Version]
- Tasseva, G.; Bai, H.D.; Davidescu, M.; Haromy, A.; Michelakis, E.; Vance, J.E. Phosphatidylethanolamine Deficiency in Mammalian Mitochondria Impairs Oxidative Phosphorylation and Alters Mitochondrial Morphology. J. Biol. Chem. 2012, 288, 4158–4173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleischer, S.; Rouser, G.; Fleischer, B.; Casu, A.; Kritchevsky, G. Lipid composition of mitochondria from bovine heart, liver, and kidney. J. Lipid Res. 1967, 8, 170–180. [Google Scholar] [PubMed]
- Gerhold, J.M.; Cansiz-Arda, S.; Lohmus, M.; Engberg, O.; Reyes, A.; van Rennes, H.; Sanz, A.; Holt, I.J.; Cooper, H.M.; Spelbrink, J.N. Human Mitochondrial DNA-Protein Complexes Attach to a Cholesterol-Rich Membrane Structure. Sci. Rep. 2015, 5, 15292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, V.; Lam, S.S.; Udeshi, N.D.; Svinkina, T.; Guzman, G.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife 2017, 6, 1206. [Google Scholar] [CrossRef] [Green Version]
- Hubstenberger, A.; Merle, N.; Charton, R.; Brandolin, G.; Rousseau, D. Topological analysis of ATAD3A insertion in purified human mitochondria. J. Bioenergy Biomembr. 2010, 42, 143–150. [Google Scholar] [CrossRef]
- Issop, L.; Fan, J.; Lee, S.; Rone, M.B.; Basu, K.; Mui, J.; Papadopoulos, V.; Fan, J. Mitochondria-Associated Membrane Formation in Hormone-Stimulated Leydig Cell Steroidogenesis: Role of ATAD3. Endocrinology 2015, 156, 334–345. [Google Scholar] [CrossRef] [Green Version]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [Green Version]
- Kasashima, K.; Sumitani, M.; Satoh, M.; Endo, H. Human prohibitin 1 maintains the organisation and stability of the mitochondrial nucleoids. Exp. Cell Res. 2008, 314, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Osman, C.; Haag, M.; Potting, C.; Rodenfels, J.; Dip, P.V.; Wieland, F.T.; Brügger, B.; Westermann, B.; Langer, T. The genetic interactome of prohibitins: Coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J. Cell Biol. 2009, 184, 583–596. [Google Scholar] [CrossRef] [Green Version]
- Merkwirth, C.; Dargazanli, S.; Tatsuta, T.; Geimer, S.; Löwer, B.; Wunderlich, F.T.; von Kleist-Retzow, J.C.; Waisman, A.; Westermann, B.; Langer, T. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008, 22, 476–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter-Dennerlein, R.; Korwitz, A.; Haag, M.; Tatsuta, T.; Dargazanli, S.; Baker, M.; Decker, T.; Lamkemeyer, T.; Rugarli, E.I.; Langer, T. DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell Metab. 2014, 20, 158–171. [Google Scholar] [CrossRef] [Green Version]
- Wolf, D.M.; Segawa, M.; Kondadi, A.K.; Anand, R.; Bailey, S.T.; Reichert, A.S.; van der Bliek, A.M.; Shackelford, D.B.; Liesa, M.; Shirihai, O.S. Individual cristae within the same mitochondrion display different membrane potentials and are functionally independent. EMBO J. 2019, 38, e101056. [Google Scholar] [CrossRef]
- Friedman, J.R.; Mourier, A.; Yamada, J.; McCaffery, J.M.; Nunnari, J. MICOS coordinates with respiratory complexes and lipids to establish mitochondrial inner membrane architecture. Elife 2015, 4, e07739. [Google Scholar] [CrossRef] [PubMed]
- Korner, C.; Barrera, M.; Dukanovic, J.; Eydt, K.; Harner, M.; Rabl, R.; Vogel, F.; Rapaport, D.; Neupert, W.; Reichert, A.S. The C-terminal domain of Fcj1 is required for formation of crista junctions and interacts with the TOB/SAM complex in mitochondria. Mol. Biol. Cell 2012, 23, 2143–2155. [Google Scholar] [CrossRef] [PubMed]
- Glytsou, C.; Calvo, E.; Cogliati, S.; Mehrotra, A.; Anastasia, I.; Rigoni, G.; Raimondi, A.; Shintani, N.; Loureiro, M.; Vazquez, J.; et al. Optic Atrophy 1 Is Epistatic to the Core MICOS Component MIC60 in Mitochondrial Cristae Shape Control. Cell Rep. 2016, 17, 3024–3034. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Ruan, Y.; Zhang, K.; Jian, F.; Hu, C.; Miao, L.; Gong, L.; Sun, L.; Zhang, X.; Chen, S.; et al. Mic60/Mitofilin determines MICOS assembly essential for mitochondrial dynamics and mtDNA nucleoid organisation. Cell Death Differ. 2016, 23, 380–392. [Google Scholar] [CrossRef] [Green Version]
- Barbot, M.; Jans, D.C.; Schulz, C.; Denkert, N.; Kroppen, B.; Hoppert, M.; Jakobs, S.; Meinecke, M. Mic10 oligomerizes to bend mitochondrial inner membranes at cristae junctions. Cell Metab. 2015, 21, 756–763. [Google Scholar] [CrossRef] [Green Version]
- Bohnert, M.; Zerbes, R.M.; Davies, K.M.; Muhleip, A.W.; Rampelt, H.; Horvath, S.E.; Boenke, T.; Kram, A.; Perschil, I.; Veenhuis, M.; et al. Central role of Mic10 in the mitochondrial contact site and cristae organizing system. Cell Metab. 2015, 21, 747–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koob, S.; Barrera, M.; Anand, R.; Reichert, A.S. The non-glycosylated isoform of MIC26 is a constituent of the mammalian MICOS complex and promotes formation of crista junctions. Biochim. Biophys. Acta 2015, 1853, 1551–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, T.A.; Koob, S.; Heide, H.; Wittig, I.; Head, B.; van der Bliek, A.; Brandt, U.; Mittelbronn, M.; Reichert, A.S. APOOL is a cardiolipin-binding constituent of the Mitofilin/MINOS protein complex determining cristae morphology in mammalian mitochondria. PLoS ONE 2013, 8, e63683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, K.M.; Anselmi, C.; Wittig, I.; Faraldo-Gómez, J.D.; Kühlbrandt, W. Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2012, 109, 13602–13607. [Google Scholar] [CrossRef] [Green Version]
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brethes, D.; di Rago, J.P.; Velours, J. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 2002, 21, 221–230. [Google Scholar] [CrossRef]
- Eydt, K.; Davies, K.J.; Behrendt, C.; Wittig, I.; Reichert, A.S. Cristae architecture is determined by an interplay of the MICOS complex and the F1Fo ATP synthase via Mic27 and Mic10. Microb. Cell 2017, 4, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Quintana-Cabrera, R.; Quirin, C.; Glytsou, C.; Corrado, M.; Urbani, A.; Pellattiero, A.; Calvo, E.; Vazquez, J.; Enriquez, J.A.; Gerle, C.; et al. The cristae modulator Optic atrophy 1 requires mitochondrial ATP synthase oligomers to safeguard mitochondrial function. Nat. Commun. 2018, 9, 3399. [Google Scholar] [CrossRef]
- Meeusen, S.; DeVay, R.; Block, J.; Cassidy-Stone, A.; Wayson, S.; McCaffery, J.M.; Nunnari, J. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell 2006, 127, 383–395. [Google Scholar] [CrossRef] [Green Version]
- Cipolat, S.; Rudka, T.; Hartmann, D.; Costa, V.; Serneels, L.; Craessaerts, K.; Metzger, K.; Frezza, C.; Annaert, W.; D’Adamio, L.; et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell 2006, 126, 163–175. [Google Scholar] [CrossRef]
- Khacho, M.; Tarabay, M.; Patten, D.; Khacho, P.; MacLaurin, J.G.; Guadagno, J.; Bergeron, R.; Cregan, S.P.; Harper, M.E.; Park, D.S.; et al. Acidosis overrides oxygen deprivation to maintain mitochondrial function and cell survival. Nat. Commun. 2014, 5, 3550. [Google Scholar] [CrossRef] [Green Version]
- Patten, D.A.; Wong, J.; Khacho, M.; Soubannier, V.; Mailloux, R.J.; Pilon-Larose, K.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Trinkle-Mulcahy, L.; et al. OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 2014, 33, 2676–2691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darshi, M.; Mendiola, V.L.; Mackey, M.R.; Murphy, A.N.; Koller, A.; Perkins, G.A.; Ellisman, M.H.; Taylor, S.S. ChChd3, an inner mitochondrial membrane protein, is essential for maintaining crista integrity and mitochondrial function. J. Biol. Chem. 2011, 286, 2918–2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perciavalle, R.M.; Stewart, D.P.; Koss, B.; Lynch, J.; Milasta, S.; Bathina, M.; Temirov, J.; Cleland, M.M.; Pelletier, S.; Schuetz, J.D.; et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol. 2012, 14, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Böttinger, L.; Horvath, S.E.; Kleinschroth, T.; Hunte, C.; Daum, G.; Pfanner, N.; Becker, T. Phosphatidylethanolamine and cardiolipin differentially affect the stability of mitochondrial respiratory chain supercomplexes. J. Mol. Biol. 2012, 423, 677–686. [Google Scholar] [CrossRef] [Green Version]
- Desmurs, M.; Foti, M.; Raemy, E.; Vaz, F.M.; Martinou, J.C.; Bairoch, A.; Lane, L. C11orf83, a mitochondrial cardiolipin-binding protein involved in bc1 complex assembly and supercomplex stabilization. Mol. Cell Biol. 2015, 35, 1139–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marusich, M.F.; Robinson, B.H.; Taanman, J.W.; Kim, S.J.; Schillace, R.; Smith, J.L.; Capaldi, R.A. Expression of mtDNA and nDNA encoded respiratory chain proteins in chemically and genetically-derived Rho0 human fibroblasts: A comparison of subunit proteins in normal fibroblasts treated with ethidium bromide and fibroblasts from a patient with mtDNA depletion syndrome. Biochim. Biophys. Acta 1997, 1362, 145–159. [Google Scholar]
- Fukuyama, R.; Nakayama, A.; Nakase, T.; Toba, H.; Mukainaka, T.; Sakaguchi, H.; Saiwaki, T.; Sakurai, H.; Wada, M.; Fushiki, S. A newly established neuronal rho-0 cell line highly susceptible to oxidative stress accumulates iron and other metals. Relevance to the origin of metal ion deposits in brains with neurodegenerative disorders. J. Biol. Chem. 2002, 277, 41455–41462. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta Bioenergy 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Larsson, N.G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef]
- Wang, J.; Wilhelmsson, H.; Graff, C.; Li, H.; Oldfors, A.; Rustin, P.; Bruning, J.C.; Kahn, C.R.; Clayton, D.A.; Barsh, G.S.; et al. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat. Genet. 1999, 21, 133–137. [Google Scholar] [CrossRef]
- Wredenberg, A.; Wibom, R.; Wilhelmsson, H.; Graff, C.; Wiener, H.H.; Burden, S.J.; Oldfors, A.; Westerblad, H.; Larsson, N.G. Increased mitochondrial mass in mitochondrial myopathy mice. Proc. Natl. Acad. Sci. USA 2002, 99, 15066–15071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Hwang, J.M.; Ko, H.S.; Seong, M.W.; Park, B.J.; Park, S.S. Mitochondrial DNA content is decreased in autosomal dominant optic atrophy. Neurology 2005, 64, 966–972. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tezze, C.; Romanello, V.; Desbats, M.A.; Fadini, G.P.; Albiero, M.; Favaro, G.; Ciciliot, S.; Soriano, M.E.; Morbidoni, V.; Cerqua, C.; et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab. 2017, 25, 1374–1389.e6. [Google Scholar] [CrossRef] [PubMed]
- Alavi, M.V.; Bette, S.; Schimpf, S.; Schuettauf, F.; Schraermeyer, U.; Wehrl, H.F.; Ruttiger, L.; Beck, S.C.; Tonagel, F.; Pichler, B.J.; et al. A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain 2007, 130 Pt 4, 1029–1042. [Google Scholar] [CrossRef] [Green Version]
- Rabl, R.; Soubannier, V.; Scholz, R.; Vogel, F.; Mendl, N.; Vasiljev-Neumeyer, A.; Korner, C.; Jagasia, R.; Keil, T.; Baumeister, W.; et al. Formation of cristae and crista junctions in mitochondria depends on antagonism between Fcj1 and Su e/g. J. Cell Biol. 2009, 185, 1047–1063. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Tamura, Y.; Iijima, M.; Sesaki, H. Effects of Fcj1-Mos1 and mitochondrial division on aggregation of mitochondrial DNA nucleoids and organelle morphology. Mol. Biol. Cell 2013, 24, 1842–1851. [Google Scholar] [CrossRef]
- Genin, E.C.; Plutino, M.; Bannwarth, S.; Villa, E.; Cisneros-Barroso, E.; Roy, M.; Ortega-Vila, B.; Fragaki, K.; Lespinasse, F.; Pinero-Martos, E.; et al. CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO Mol. Med. 2016, 8, 58–72. [Google Scholar] [CrossRef]
- Wang, C.; Taki, M.; Sato, Y.; Tamura, Y.; Yaginuma, H.; Okada, Y.; Yamaguchi, S. A photostable fluorescent marker for the superresolution live imaging of the dynamic structure of the mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2019, 116, 15817–15822. [Google Scholar] [CrossRef] [Green Version]
- Kondadi, A.K.; Anand, R.; Hänsch, S.; Urbach, J.; Zobel, T.; Wolf, D.M.; Segawa, M.; Liesa, M.; Shirihai, O.S.; Weidtkamp-Peters, S.; et al. Cristae undergo continuous cycles of membrane remodelling in a MICOS-dependent manner. EMBO Rep. 2020, 21, e49776. [Google Scholar] [CrossRef]
- Guarani, V.; Jardel, C.; Chrétien, D.; Lombès, A.; Bénit, P.; Labasse, C.; Lacène, E.; Bourillon, A.; Imbard, A.; Benoist, J.F.; et al. QIL1 mutation causes MICOS disassembly and early onset fatal mitochondrial encephalopathy with liver disease. Elife 2016, 5, e17163. [Google Scholar] [CrossRef] [PubMed]
- Zeharia, A.; Friedman, J.R.; Tobar, A.; Saada, A.; Konen, O.; Fellig, Y.; Shaag, A.; Nunnari, J.; Elpeleg, O. Mitochondrial hepato-encephalopathy due to deficiency of QIL1/MIC13 (C19orf70), a MICOS complex subunit. Eur. J. Hum. Genet. 2016, 24, 1778–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gödiker, J.; Grüneberg, M.; DuChesne, I.; Reunert, J.; Rust, S.; Westermann, C.; Wada, Y.; Classen, G.; Langhans, C.D.; Schlingmann, K.P.; et al. QIL1-dependent assembly of MICOS complex-lethal mutation in C19ORF70 resulting in liver disease and severe neurological retardation. J. Hum. Genet. 2018, 63, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Benincá, C.; Zanette, V.; Brischigliaro, M.; Johnson, M.; Reyes, A.; Valle, D.A.D.; Robinson, A.R.; Degiorgi, A.; Yeates, A.; Telles, B.A.; et al. Mutation in the MICOS subunit gene APOO (MIC26) associated with an X-linked recessive mitochondrial myopathy, lactic acidosis, cognitive impairment and autistic features. J. Med. Genet. 2020. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Pankratz, N.; Foroud, T. Genetics of Parkinson disease. Genet. Med. 2007, 9, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Tsai, P.I.; Lin, C.H.; Hsieh, C.H.; Papakyrikos, A.M.; Kim, M.J.; Napolioni, V.; Schoor, C.; Couthouis, J.; Wu, R.M.; Wszolek, Z.K.; et al. PINK1 Phosphorylates MIC60/Mitofilin to Control Structural Plasticity of Mitochondrial Crista Junctions. Mol. Cell 2018, 69, 744–756.e6. [Google Scholar] [CrossRef] [Green Version]
- Ogaki, K.; Koga, S.; Heckman, M.G.; Fiesel, F.C.; Ando, M.; Labbé, C.; Lorenzo-Betancor, O.; Moussaud-Lamodière, E.L.; Soto-Ortolaza, A.I.; Walton, R.L.; et al. Mitochondrial targeting sequence variants of the CHCHD2 gene are a risk for Lewy body disorders. Neurology 2015, 85, 2016–2025. [Google Scholar] [CrossRef] [Green Version]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137 Pt 8, 2329–2345. [Google Scholar] [CrossRef] [Green Version]
- Project MinE ALS Sequencing Consortium. CHCHD10 variants in amyotrophic lateral sclerosis: Where is the evidence? Ann. Neurol. 2018, 84, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Harel, T.; Yoon, W.H.; Garone, C.; Gu, S.; Coban-Akdemir, Z.; Eldomery, M.K.; Posey, J.E.; Jhangiani, S.N.; Rosenfeld, J.A.; Cho, M.T.; et al. Recurrent De Novo and Biallelic Variation of ATAD3A, Encoding a Mitochondrial Membrane Protein, Results in Distinct Neurological Syndromes. Am. J. Hum. Genet. 2016, 99, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Bione, S.; D’Adamo, P.; Maestrini, E.; Gedeon, A.K.; Bolhuis, P.A.; Toniolo, D. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 1996, 12, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.L.; Bowron, A.; Gonzalez, I.L.; Groves, S.J.; Newbury-Ecob, R.; Clayton, N.; Martin, R.P.; Tsai-Goodman, B.; Garratt, V.; Ashworth, M.; et al. Barth syndrome. Orphanet J. Rare Dis. 2013, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Mayr, J.A.; Havlícková, V.; Zimmermann, F.; Magler, I.; Kaplanová, V.; Jesina, P.; Pecinová, A.; Nusková, H.; Koch, J.; Sperl, W.; et al. Mitochondrial ATP synthase deficiency due to a mutation in the ATP5E gene for the F1 epsilon subunit. Hum. Mol. Genet. 2010, 19, 3430–3439. [Google Scholar] [CrossRef] [Green Version]
- Sheffer, R.; Douiev, L.; Edvardson, S.; Shaag, A.; Tamimi, K.; Soiferman, D.; Meiner, V.; Saada, A. Postnatal microcephaly and pain insensitivity due to a de novo heterozygous DNM1L mutation causing impaired mitochondrial fission and function. Am. J. Med. Genet. A 2016, 170, 1603–1607. [Google Scholar] [CrossRef]
- Gerber, S.; Charif, M.; Chevrollier, A.; Chaumette, T.; Angebault, C.; Kane, M.S.; Paris, A.; Alban, J.; Quiles, M.; Delettre, C.; et al. Mutations in DNM1L, as in OPA1, result in dominant optic atrophy despite opposite effects on mitochondrial fusion and fission. Brain 2017, 140, 2586–2596. [Google Scholar] [CrossRef]
- Jungbluth, H.; Cullup, T.; Lillis, S.; Zhou, H.; Abbs, S.; Sewry, C.; Muntoni, F. Centronuclear myopathy with cataracts due to a novel dynamin 2 (DNM2) mutation. Neuromuscul. Disord. 2010, 20, 49–52. [Google Scholar] [CrossRef]
- Susman, R.D.; Quijano-Roy, S.; Yang, N.; Webster, R.; Clarke, N.F.; Dowling, J.; Kennerson, M.; Nicholson, G.; Biancalana, V.; Ilkovski, B.; et al. Expanding the clinical, pathological and MRI phenotype of DNM2-related centronuclear myopathy. Neuromuscul. Disord. 2010, 20, 229–237. [Google Scholar] [CrossRef]
- Mademan, I.; Deconinck, T.; Dinopoulos, A.; Voit, T.; Schara, U.; Devriendt, K.; Meijers, B.; Lerut, E.; De Jonghe, P.; Baets, J. De novo INF2 mutations expand the genetic spectrum of hereditary neuropathy with glomerulopathy. Neurology 2013, 81, 1953–1958. [Google Scholar] [CrossRef]
- Wan, J.; Steffen, J.; Yourshaw, M.; Mamsa, H.; Andersen, E.; Rudnik-Schöneborn, S.; Pope, K.; Howell, K.B.; McLean, C.A.; Kornberg, A.J.; et al. Loss of function of SLC25A46 causes lethal congenital pontocerebellar hypoplasia. Brain 2016, 139, 2877–2890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, M.B.; Ding, J.; Mochel, F.; Eleuch-Fayache, G.; Charles, P.; Coutelier, M.; Gibbs, J.R.; Arepalli, S.K.; Chong, S.B.; Hernandez, D.G.; et al. SLC25A46 Mutations Associated with Autosomal Recessive Cerebellar Ataxia in North African Families. Neurodegener Dis. 2017, 17, 208–212. [Google Scholar] [CrossRef] [PubMed]
- White, K.E.; Davies, V.J.; Hogan, V.E.; Piechota, M.J.; Nichols, P.P.; Turnbull, D.M.; Votruba, M. OPA1 deficiency associated with increased autophagy in retinal ganglion cells in a murine model of dominant optic atrophy. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2567–2571. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, R.; Saada, A.; Flannery, P.J.; Burté, F.; Soiferman, D.; Khayat, M.; Eisner, V.; Vladovski, E.; Taylor, R.W.; Bindoff, L.A.; et al. Fatal infantile mitochondrial encephalomyopathy, hypertrophic cardiomyopathy and optic atrophy associated with a homozygous OPA1 mutation. J. Med. Genet. 2016, 53, 127–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Bonneau, D.; Colin, E.; Oca, F.; Ferré, M.; Chevrollier, A.; Guéguen, N.; Desquiret-Dumas, V.; N’Guyen, S.; Barth, M.; Zanlonghi, X.; et al. Early-onset Behr syndrome due to compound heterozygous mutations in OPA1. Brain 2014, 137, e301. [Google Scholar] [CrossRef] [Green Version]
- Schaaf, C.P.; Blazo, M.; Lewis, R.A.; Tonini, R.E.; Takei, H.; Wang, J.; Wong, L.J.; Scaglia, F. Early-onset severe neuromuscular phenotype associated with compound heterozygosity for OPA1 mutations. Mol. Genet. Metab. 2011, 103, 383–387. [Google Scholar] [CrossRef]
- Rubegni, A.; Pisano, T.; Bacci, G.; Tessa, A.; Battini, R.; Procopio, E.; Giglio, S.; Pasquariello, R.; Santorelli, F.M.; Guerrini, R.; et al. Leigh-like neuroimaging features associated with new biallelic mutations in OPA1. Eur. J. Paediatr. Neurol. 2017, 21, 671–677. [Google Scholar] [CrossRef]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type, 2.A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Kijima, K.; Numakura, C.; Izumino, H.; Umetsu, K.; Nezu, A.; Shiiki, T.; Ogawa, M.; Ishizaki, Y.; Kitamura, T.; Shozawa, Y.; et al. Mitochondrial GTPase mitofusin 2 mutation in Charcot-Marie-Tooth neuropathy type, 2.A. Hum. Genet. 2005, 116, 23–27. [Google Scholar] [CrossRef]
- Nicholson, G.A.; Magdelaine, C.; Zhu, D.; Grew, S.; Ryan, M.M.; Sturtz, F.; Vallat, J.M.; Ouvrier, R.A. Severe early-onset axonal neuropathy with homozygous and compound heterozygous MFN2 mutations. Neurology 2008, 70, 1678–1681. [Google Scholar] [CrossRef] [PubMed]
- Polke, J.M.; Laurá, M.; Pareyson, D.; Taroni, F.; Milani, M.; Bergamin, G.; Gibbons, V.S.; Houlden, H.; Chamley, S.C.; Blake, J.; et al. Recessive axonal Charcot-Marie-Tooth disease due to compound heterozygous mitofusin 2 mutations. Neurology 2011, 77, 168–173. [Google Scholar] [CrossRef] [Green Version]
- Renaldo, F.; Amati-Bonneau, P.; Slama, A.; Romana, C.; Forin, V.; Doummar, D.; Barnerias, C.; Bursztyn, J.; Mayer, M.; Khouri, N.; et al. MFN2, a new gene responsible for mitochondrial DNA depletion. Brain 2012, 135 Pt 8, e223, 1–4; Author reply e4, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Barøy, T.; Pedurupillay, C.R.; Bliksrud, Y.T.; Rasmussen, M.; Holmgren, A.; Vigeland, M.D.; Hughes, T.; Brink, M.; Rodenburg, R.; Nedregaard, B.; et al. A novel mutation in FBXL4 in a Norwegian child with encephalomyopathic mitochondrial DNA depletion syndrome 13. Eur. J. Med. Genet. 2016, 59, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Antoun, G.; McBride, S.; Vanstone, J.R.; Naas, T.; Michaud, J.; Redpath, S.; McMillan, H.J.; Brophy, J.; Daoud, H.; Chakraborty, P.; et al. Detailed Biochemical and Bioenergetic Characterization of FBXL4-Related Encephalomyopathic Mitochondrial DNA Depletion. JIMD Rep. 2016, 27, 1–9. [Google Scholar] [PubMed] [Green Version]
- Gai, X.; Ghezzi, D.; Johnson, M.A.; Biagosch, C.A.; Shamseldin, H.E.; Haack, T.B.; Reyes, A.; Tsukikawa, M.; Sheldon, C.A.; Srinivasan, S.; et al. Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am. J. Hum. Genet. 2013, 93, 482–495. [Google Scholar] [CrossRef] [Green Version]
- Huemer, M.; Karall, D.; Schossig, A.; Abdenur, J.E.; Al Jasmi, F.; Biagosch, C.; Distelmaier, F.; Freisinger, P.; Graham, B.H.; Haack, T.B.; et al. Clinical, morphological, biochemical, imaging and outcome parameters in 21 individuals with mitochondrial maintenance defect related to FBXL4 mutations. J. Inherit. Metab. Dis. 2015, 38, 905–914. [Google Scholar] [CrossRef]
- Nasca, A.; Scotton, C.; Zaharieva, I.; Neri, M.; Selvatici, R.; Magnusson, O.T.; Gal, A.; Weaver, D.; Rossi, R.; Armaroli, A.; et al. Recessive mutations in MSTO1 cause mitochondrial dynamics impairment, leading to myopathy and ataxia. Hum. Mutat. 2017, 38, 970–977. [Google Scholar] [CrossRef] [Green Version]
- Gal, A.; Balicza, P.; Weaver, D.; Naghdi, S.; Joseph, S.K.; Várnai, P.; Gyuris, T.; Horváth, A.; Nagy, L.; Seifert, E.L.; et al. MSTO1 is a cytoplasmic pro-mitochondrial fusion protein, whose mutation induces myopathy and ataxia in humans. EMBO Mol. Med. 2017, 9, 967–984. [Google Scholar] [CrossRef]
- Iwama, K.; Takaori, T.; Fukushima, A.; Tohyama, J.; Ishiyama, A.; Ohba, C.; Mitsuhashi, S.; Miyatake, S.; Takata, A.; Miyake, N.; et al. Novel recessive mutations in MSTO1 cause cerebellar atrophy with pigmentary retinopathy. J. Hum. Genet. 2018, 63, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, B.; Wai, T.; Hu, H.; MacVicar, T.; Musante, L.; Fischer-Zirnsak, B.; Stenzel, W.; Gräf, R.; van den Heuvel, L.; Ropers, H.H.; et al. Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation. Elife 2016, 5, 5. [Google Scholar] [CrossRef]
- Wilkinson, P.A.; Crosby, A.H.; Turner, C.; Bradley, L.J.; Ginsberg, L.; Wood, N.W.; Schapira, A.H.; Warner, T.T. A clinical, genetic and biochemical study of SPG7 mutations in hereditary spastic paraplegia. Brain 2004, 127 Pt 5, 973–980. [Google Scholar] [CrossRef] [Green Version]
- Elleuch, N.; Depienne, C.; Benomar, A.; Hernandez, A.M.; Ferrer, X.; Fontaine, B.; Grid, D.; Tallaksen, C.M.; Zemmouri, R.; Stevanin, G.; et al. Mutation analysis of the paraplegin gene (SPG7) in patients with hereditary spastic paraplegia. Neurology 2006, 66, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Banfi, S.; Bassi, M.T.; Andolfi, G.; Marchitiello, A.; Zanotta, S.; Ballabio, A.; Casari, G.; Franco, B. Identification and characterization of AFG3L2, a novel paraplegin-related gene. Genomics 1999, 59, 51–58. [Google Scholar] [CrossRef]
- Edener, U.; Wöllner, J.; Hehr, U.; Kohl, Z.; Schilling, S.; Kreuz, F.; Bauer, P.; Bernard, V.; Gillessen-Kaesbach, G.; Zühlke, C. Early onset and slow progression of SCA28, a rare dominant ataxia in a large four-generation family with a novel AFG3L2 mutation. Eur. J. Hum. Genet. 2010, 18, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, C.; Brusco, A.; Di Bella, D.; Cagnoli, C.; Seri, M.; Gellera, C.; Di Donato, S.; Taroni, F. Spinocerebellar ataxia type 28: A novel autosomal dominant cerebellar ataxia characterized by slow progression and ophthalmoparesis. Cerebellum 2008, 7, 184–188. [Google Scholar] [CrossRef]
- Penttilä, S.; Jokela, M.; Bouquin, H.; Saukkonen, A.M.; Toivanen, J.; Udd, B. Late onset spinal motor neuronopathy is caused by mutation in CHCHD10. Ann. Neurol. 2015, 77, 163–172. [Google Scholar] [CrossRef]
- Auranen, M.; Ylikallio, E.; Shcherbii, M.; Paetau, A.; Kiuru-Enari, S.; Toppila, J.P.; Tyynismaa, H. CHCHD10 variant p.(Gly66Val) causes axonal Charcot-Marie-Tooth disease. Neurol. Genet. 2015, 1, e1. [Google Scholar] [CrossRef] [Green Version]
- Ajroud-Driss, S.; Fecto, F.; Ajroud, K.; Lalani, I.; Calvo, S.E.; Mootha, V.K.; Deng, H.X.; Siddique, N.; Tahmoush, A.J.; Heiman-Patterson, T.D.; et al. Mutation in the novel nuclear-encoded mitochondrial protein CHCHD10 in a family with autosomal dominant mitochondrial myopathy. Neurogenetics 2015, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chaussenot, A.; Le Ber, I.; Ait-El-Mkadem, S.; Camuzat, A.; de Septenville, A.; Bannwarth, S.; Genin, E.C.; Serre, V.; Augé, G.; Brice, A.; et al. Screening of CHCHD10 in a French cohort confirms the involvement of this gene in frontotemporal dementia with amyotrophic lateral sclerosis patients. Neurobiol. Aging 2014, 35, 2884.e1–e4. [Google Scholar] [CrossRef]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Chishti, M.A.; Bohlega, S.; Ahmed, M.; Loualich, A.; Carroll, P.; Sato, C.; St George-Hyslop, P.; Westaway, D.; Rogaeva, E. T313M PINK1 mutation in an extended highly consanguineous Saudi family with early-onset Parkinson disease. Arch. Neurol. 2006, 63, 1483–1485. [Google Scholar] [CrossRef] [Green Version]
- Ibáñez, P.; Lesage, S.; Lohmann, E.; Thobois, S.; De Michele, G.; Borg, M.; Agid, Y.; Dürr, A.; Brice, A.; The French Parkinson’s Disease Genetics Study Group. Mutational analysis of the PINK1 gene in early-onset parkinsonism in Europe and North Africa. Brain 2006, 129 Pt 3, 686–694. [Google Scholar]
- Silvestri, L.; Caputo, V.; Bellacchio, E.; Atorino, L.; Dallapiccola, B.; Valente, E.M.; Casari, G. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum. Mol. Genet. 2005, 14, 3477–3492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogaeva, E.; Johnson, J.; Lang, A.E.; Gulick, C.; Gwinn-Hardy, K.; Kawarai, T.; Sato, C.; Morgan, A.; Werner, J.; Nussbaum, R.; et al. Analysis of the PINK1 gene in a large cohort of cases with Parkinson disease. Arch. Neurol. 2004, 61, 1898–1904. [Google Scholar] [CrossRef] [PubMed]
- Lieber, D.S.; Calvo, S.E.; Shanahan, K.; Slate, N.G.; Liu, S.; Hershman, S.G.; Gold, N.B.; Chapman, B.A.; Thorburn, D.R.; Berry, G.T.; et al. Targeted exome sequencing of suspected mitochondrial disorders. Neurology 2013, 80, 1762–1770. [Google Scholar] [CrossRef] [Green Version]
- Jonckheere, A.I.; Renkema, G.H.; Bras, M.; van den Heuvel, L.P.; Hoischen, A.; Gilissen, C.; Nabuurs, S.B.; Huynen, M.A.; de Vries, M.C.; Smeitink, J.A.; et al. A complex V ATP5A1 defect causes fatal neonatal mitochondrial encephalopathy. Brain 2013, 136 Pt 5, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Saric, A.; Andreau, K.; Armand, A.S.; Møller, I.M.; Petit, P.X. Barth Syndrome: From Mitochondrial Dysfunctions Associated with Aberrant Production of Reactive Oxygen Species to Pluripotent Stem Cell Studies. Front. Genet. 2015, 6, 359. [Google Scholar] [CrossRef] [Green Version]
- Barth, P.G.; Scholte, H.R.; Berden, J.A.; Van der Klei-Van Moorsel, J.M.; Luyt-Houwen, I.E.; Van’t Veer-Korthof, E.T.; Van der Harten, J.J.; Sobotka-Plojhar, M.A. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 1983, 62, 327–355. [Google Scholar] [CrossRef]
- D’Adamo, P.; Fassone, L.; Gedeon, A.; Janssen, E.A.; Bione, S.; Bolhuis, P.A.; Barth, P.G.; Wilson, M.; Haan, E.; Orstavik, K.H.; et al. The X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. Am. J. Hum. Genet. 1997, 61, 862–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichida, F.; Tsubata, S.; Bowles, K.R.; Haneda, N.; Uese, K.; Miyawaki, T.; Dreyer, W.J.; Messina, J.; Li, H.; Bowles, N.E.; et al. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation 2001, 103, 1256–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, C.T.; Bryant, R.M.; Day, J.; Gonzalez, I.L.; Colan, S.D.; Thompson, W.R.; Berthy, J.; Redfearn, S.P.; Byrne, B.J. Cardiac and clinical phenotype in Barth syndrome. Pediatrics 2006, 118, e337–e346. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic overview of the human mtDNA genome, its replication and the formation of deletions. (a) The structure of mtDNA highlighting the arrangement of protein coding genes, rRNAs (orange) and tRNAs (yellow). The replication origins of the heavy and light strand (OriH and OriL, respectively) are highlighted. (b) Enlargement of the mtDNA non-coding region (NCR) depicting the arrangement of the heavy strand promoter (HSP) and light strand promoter (LSP), the three conserved sequence boxes (CSB), OriH and the termination-associated sequence (TAS). The premature termination of the DNA synthesis of the H-strand at TAS results in the formation of a triple-stranded displacement-loop structure termed the D-loop. The short double stranded product formed within the D-loop is termed 7S DNA. (c) mtDNA replication is initiated at OriH and proceeds unidirectionally until OriL is reached. At this point, DNA synthesis of the light strand is initiated, and both strands are synthesised simultaneously until two completely replicated genomes are produced. The two replicated genomes are physically interlinked by a single-stranded overlap structure, termed a hemicatenane. This structure is resolved by topoisomerase 3α (TOP3α) to produce two separate mtDNA molecules. (d) Copy choice recombination model for the formation of mtDNA deletions. mtDNA deletions generally occur in the major arc. The replication of a repeat sequence in the template heavy strand (yellow boxes) can lead to stalling of POLγ which results in its dissociation from the newly synthesised DNA-end. When Polγ reanneals, it may associate at another repeat sequence further along the template. Following the completion of replication, this slippage event produces two mtDNA genomes; one full length molecule and a second heteroduplex molecule (which has a full-length heavy strand alongside a deletion-containing light strand). The subsequent replication of the heteroduplex molecule culminates in the formation of mtDNA harbouring the deletion.
Figure 1.
Schematic overview of the human mtDNA genome, its replication and the formation of deletions. (a) The structure of mtDNA highlighting the arrangement of protein coding genes, rRNAs (orange) and tRNAs (yellow). The replication origins of the heavy and light strand (OriH and OriL, respectively) are highlighted. (b) Enlargement of the mtDNA non-coding region (NCR) depicting the arrangement of the heavy strand promoter (HSP) and light strand promoter (LSP), the three conserved sequence boxes (CSB), OriH and the termination-associated sequence (TAS). The premature termination of the DNA synthesis of the H-strand at TAS results in the formation of a triple-stranded displacement-loop structure termed the D-loop. The short double stranded product formed within the D-loop is termed 7S DNA. (c) mtDNA replication is initiated at OriH and proceeds unidirectionally until OriL is reached. At this point, DNA synthesis of the light strand is initiated, and both strands are synthesised simultaneously until two completely replicated genomes are produced. The two replicated genomes are physically interlinked by a single-stranded overlap structure, termed a hemicatenane. This structure is resolved by topoisomerase 3α (TOP3α) to produce two separate mtDNA molecules. (d) Copy choice recombination model for the formation of mtDNA deletions. mtDNA deletions generally occur in the major arc. The replication of a repeat sequence in the template heavy strand (yellow boxes) can lead to stalling of POLγ which results in its dissociation from the newly synthesised DNA-end. When Polγ reanneals, it may associate at another repeat sequence further along the template. Following the completion of replication, this slippage event produces two mtDNA genomes; one full length molecule and a second heteroduplex molecule (which has a full-length heavy strand alongside a deletion-containing light strand). The subsequent replication of the heteroduplex molecule culminates in the formation of mtDNA harbouring the deletion.

Figure 2.
Mitochondrial dynamics and its role in mtDNA distribution. (a) Schematic overview of mitochondrial fission. Constriction of the mitochondrion occurs by the endoplasmic reticulum (ER) and dynamin related protein 1 (DRP1). The final scission step is performed by DRP1 to produce multiple independent mitochondria. (b) Mitochondrial fusion outline. The mitofusins (MFNs) are responsible for tethering neighbouring mitochondria and fusing the outer membranes together. Fusion of the inner mitochondrial membrane is mediated by optic atrophy 1 (OPA1). (c) Summary of mtDNA segregation and distribution. First, newly replicated mtDNA molecules that are joined by a hemicatenane must be physically separated by topoisomerase 3α. Failure of this process results in the formation of multiple physically joined mtDNA genomes, termed mtDNA catenanes. Following separation, mitochondrial fission is required to distribute the replicated nucleoids into separate mitochondria and facilitate distribution of the genomes around the mitochondrial network. An impairment of fission results in a clustered phenotype whereby many replicated nucleoids are observed in close proximity to each other but are not physically linked together. (d) A lack of mitochondrial fusion is associated with mtDNA instability as observed by the progressive onset of mtDNA deletions and point mutations. In addition, the lack of fusion leads to a depletion in mtDNA copy number.
Figure 2.
Mitochondrial dynamics and its role in mtDNA distribution. (a) Schematic overview of mitochondrial fission. Constriction of the mitochondrion occurs by the endoplasmic reticulum (ER) and dynamin related protein 1 (DRP1). The final scission step is performed by DRP1 to produce multiple independent mitochondria. (b) Mitochondrial fusion outline. The mitofusins (MFNs) are responsible for tethering neighbouring mitochondria and fusing the outer membranes together. Fusion of the inner mitochondrial membrane is mediated by optic atrophy 1 (OPA1). (c) Summary of mtDNA segregation and distribution. First, newly replicated mtDNA molecules that are joined by a hemicatenane must be physically separated by topoisomerase 3α. Failure of this process results in the formation of multiple physically joined mtDNA genomes, termed mtDNA catenanes. Following separation, mitochondrial fission is required to distribute the replicated nucleoids into separate mitochondria and facilitate distribution of the genomes around the mitochondrial network. An impairment of fission results in a clustered phenotype whereby many replicated nucleoids are observed in close proximity to each other but are not physically linked together. (d) A lack of mitochondrial fusion is associated with mtDNA instability as observed by the progressive onset of mtDNA deletions and point mutations. In addition, the lack of fusion leads to a depletion in mtDNA copy number.

Figure 3.
The relationship between cristae modulators and mtDNA organisation. (a) Schematic overview of a normal mitochondrion. (b) Enlarged cristae region depicting the spatial arrangement of key proteins that are necessary for dictating cristae structure and shape, as well as the organisation of the respiratory chain complexes. Proteins that are potentially responsible for the tethering of mtDNA to the IBM are also highlighted. (c) Loss of the fission factor DRP1 has been associated with the formation of mito-bulb structures. These are regions of dense cristae structure which harbour a number of clustered mtDNA molecules. (d) The loss of IMM fusion and cristae shaping protein OPA1 results in mitochondria that display enlarged cristae junctions and a perturbed cristae structure. In addition, mtDNA copy number is reduced. (e) The loss of MICOS components (MIC60, MIC10 and MIC19) has been associated with a complete loss of cristae junctions and the formation of cristae in concentric circles. Nucleoid clustering is evident following the loss of MICOS components.
Figure 3.
The relationship between cristae modulators and mtDNA organisation. (a) Schematic overview of a normal mitochondrion. (b) Enlarged cristae region depicting the spatial arrangement of key proteins that are necessary for dictating cristae structure and shape, as well as the organisation of the respiratory chain complexes. Proteins that are potentially responsible for the tethering of mtDNA to the IBM are also highlighted. (c) Loss of the fission factor DRP1 has been associated with the formation of mito-bulb structures. These are regions of dense cristae structure which harbour a number of clustered mtDNA molecules. (d) The loss of IMM fusion and cristae shaping protein OPA1 results in mitochondria that display enlarged cristae junctions and a perturbed cristae structure. In addition, mtDNA copy number is reduced. (e) The loss of MICOS components (MIC60, MIC10 and MIC19) has been associated with a complete loss of cristae junctions and the formation of cristae in concentric circles. Nucleoid clustering is evident following the loss of MICOS components.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chapman, J.; Ng, Y.S.; Nicholls, T.J. The Maintenance of Mitochondrial DNA Integrity and Dynamics by Mitochondrial Membranes. Life 2020, 10, 164. https://doi.org/10.3390/life10090164
AMA Style
Chapman J, Ng YS, Nicholls TJ. The Maintenance of Mitochondrial DNA Integrity and Dynamics by Mitochondrial Membranes. Life. 2020; 10(9):164. https://doi.org/10.3390/life10090164
Chicago/Turabian StyleChapman, James, Yi Shiau Ng, and Thomas J. Nicholls. 2020. "The Maintenance of Mitochondrial DNA Integrity and Dynamics by Mitochondrial Membranes" Life 10, no. 9: 164. https://doi.org/10.3390/life10090164
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.