
Computational Genomics in the Era of Precision Medicine: Applications to Variant Analysis and Gene Therapy

 , ,
, ,  , , , and
, , , and
Abstract
:
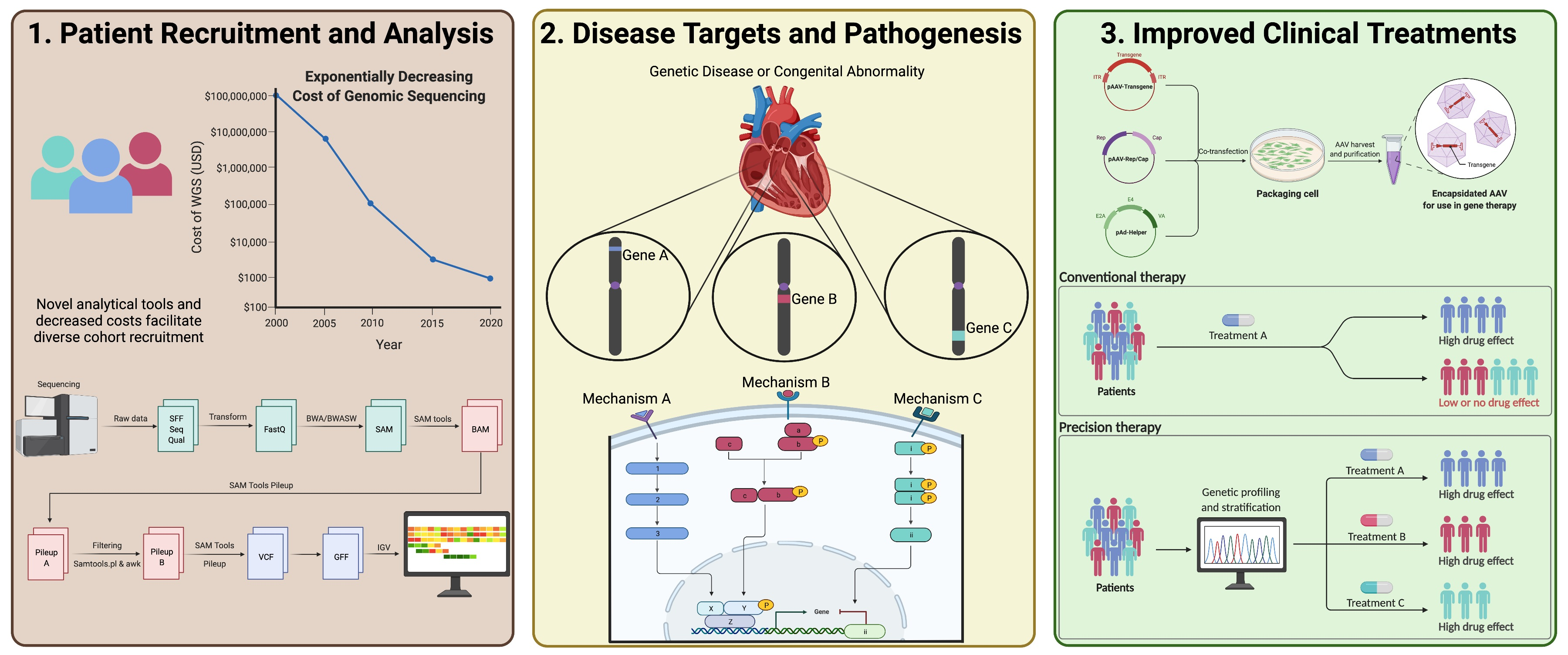
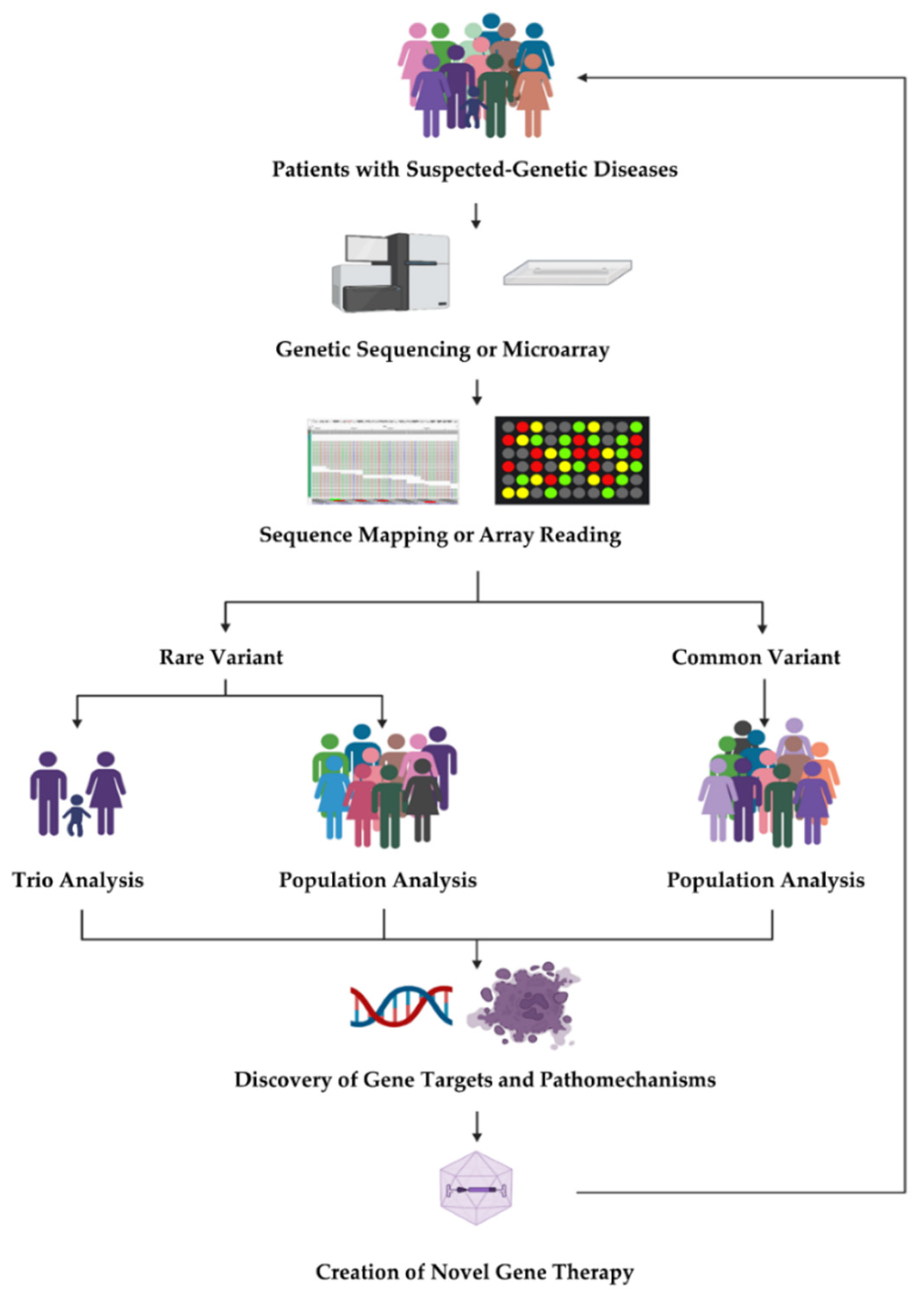
1. Introduction
2. Rare Variant Analysis in Unrelated Individuals

{kind=link}
{kind=link}
| Type | Methods | Strengths | Weaknesses | Ref. |
|---|---|---|---|---|
| Rare variant analysis in unrelated individuals | Combined Multivariate and Collapsing (CMC) test |
|
| [14] |
| Variable Threshold (VT) |
|
| [15] | |
| Sequence kernel association test (SKAT) |
|
| [16] | |
| Cohort allelic sums test (CAST) |
|
| [17] | |
| Weighted sum test (WST) |
|
| [18] | |
| Kernel-based adaptive clustering method (KBAC) |
|
| [19] | |
| Versatile gene-based association study (VEGAS) |
|
| [20] | |
| Gene-based association test that uses extended Simes procedure (GATES) |
|
| [21] | |
| Multivariate Association Analysis using Score Statistics (MAAUSS) |
|
| [22] | |
| Multi-trait analysis of rare-variant associations (MTAR) |
|
| [23] | |
| De novo variants analysis | DeNovoWEST |
|
| [4] |
| Chimpanzee–human divergence model |
|
| [25] | |
| denovolyzeR |
|
| [26] | |
| Autosomal recessive variant analysis | Resampling-based statistical framework |
|
| [27] |
| Sampling the observed genotypes and phenotypes by chance |
|
| [28] | |
| The phased haplotypes-based framework |
|
| [29] | |
| Joint analysis of transmitted variants and DNVs | Transmission and de novo association test (TADA), extTADA |
|
| [30,31] |
| TADA-Annotations (TADA-A) |
|
| [32] | |
| TADA-Recessive (TADA-R) |
|
| [33] | |
| Multi-trait TADA (M-TADA) |
|
| [34] | |
| X-linked variant analysis | Various XCI modes integrated statistical approach |
|
| [35] |
| 1 and 2 degree-of-freedom tests for association |
|
| [36] | |
| Distinct XCI processes combined using a modified Fisher’s method |
|
| [37] | |
| Sex-specific burden analyses |
|
| [38] | |
| Digenic variant analysis | The genetic linkage method |
|
| [39] |
| The candidate gene approach |
|
| [40] | |
| Case-only study design |
|
| [41] | |
| Random forests |
|
| [42] |
3. Rare Variant Analysis for Family-Based Studies
3.1. De Novo Variant
3.2. Autosomal Recessive Variant Analysis
3.3. Joint Analysis of Transmitted Variants and DNVs
4. X-Linked Variant Analysis
5. Digenic Variant Analysis
5.1. Case-Only Approach
5.2. Machine Learning
6. Common Variant Association Analysis
7. Disease Risk Prediction
8. Gene Therapy
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium ITP-CAoWG. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplanis, J.; Samocha, K.E.; Wiel, L.; Zhang, Z.; Arvai, K.J.; Eberhardt, R.Y.; Gallone, G.; Lelieveld, S.H.; Martin, H.C.; McRae, J.F.; et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature 2020, 586, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.-Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef]
- Samani, N.J.; Erdmann, J.; Hall, A.S.; Hengstenberg, C.; Mangino, M.; Mayer, B.; Dixon, R.J.; Meitinger, T.; Braund, P.; Wichmann, H.-E.; et al. Genomewide association analysis of coronary artery disease. N. Engl. J. Med. 2007, 357, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Frayling, T.M.; Timpson, N.J.; Weedon, M.N.; Zeggini, E.; Freathy, R.M.; Lindgren, C.M.; Perry, J.R.B.; Elliott, K.S.; Lango, H.; Rayner, N.W.; et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 2007, 316, 889–894. [Google Scholar] [CrossRef] [Green Version]
- Herbert, A.; Gerry, N.P.; McQueen, M.B.; Heid, I.M.; Pfeufer, A.; Illig, T.; Wichmann, H.-E.; Meitinger, T.; Hunter, D.; Hu, F.B.; et al. A common genetic variant is associated with adult and childhood obesity. Science 2006, 312, 279–283. [Google Scholar] [CrossRef] [Green Version]
- Saxena, R.; Voight, B.F.; Lyssenko, V.; Burtt, N.P.; de Bakker, P.I.W.; Chen, H.; Roix, J.J.; Kathiresan, S.; Hirschhorn, J.N.; Daly, M.J.; et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007, 316, 1331–1336. [Google Scholar] [CrossRef]
- Stefansson, H.; Ophoff, R.A.; Steinberg, S.; Andreassen, O.A.; Cichon, S.; Rujescu, D.; Werge, T.; Pietilainen, O.P.; Mors, O.; Mortensen, P.B.; et al. Common variants conferring risk of schizophrenia. Nature 2009, 460, 744–747. [Google Scholar] [CrossRef] [Green Version]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [Green Version]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.H.; Dauber, A.; Lippincott, M.; Chan, Y.-M.; Salem, R.; Hirschhorn, J.N. Determinants of Power in Gene-Based Burden Testing for Monogenic Disorders. Am. J. Hum. Genet. 2016, 99, 527–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Leal, S.M. Methods for detecting associations with rare variants for common diseases: Application to analysis of sequence data. Am. J. Hum. Genet. 2008, 83, 311–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, A.L.; Kryukov, G.; de Bakker, P.I.; Purcell, S.M.; Staples, J.; Wei, L.-J.; Sunyaev, S.R. Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet. 2010, 86, 832–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.C.; Lee, S.; Cai, T.; Li, Y.; Boehnke, M.; Lin, X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011, 89, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgenthaler, S.; Thilly, W.G. A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: A cohort allelic sums test (CAST). Mutat. Res. 2007, 615, 28–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Elston, R.C. Improved power by use of a weighted score test for linkage disequilibrium mapping. Am. J. Hum. Genet. 2007, 80, 353–360. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.J.; Leal, S.M. A novel adaptive method for the analysis of next-generation sequencing data to detect complex trait associations with rare variants due to gene main effects and interactions. PLoS Genet. 2010, 6, e1001156. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Z.; Mcrae, A.F.; Nyholt, D.R.; Medland, S.E.; Wray, N.R.; Brown, K.M.; Hayward, N.K.; Montgomery, G.; Visscher, P.; Martin, N.; et al. A versatile gene-based test for genome-wide association studies. Am. J. Hum. Genet. 2010, 87, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Li, M.-X.; Gui, H.-S.; Kwan, J.S.; Sham, P.C. GATES: A rapid and powerful gene-based association test using extended Simes procedure. Am. J Hum Genet. 2011, 88, 283–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Won, S.; Kim, Y.J.; Kim, Y.; Consortium, T.D.-G.; Kim, B.J.; Park, T. Rare variant association test with multiple phenotypes. Genet. Epidemiol. 2017, 41, 198–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Shen, J.; Zhang, H.; Chhibber, A.; Mehrotra, D.V.; Tang, Z.-Z. Multi-trait analysis of rare-variant association summary statistics using MTAR. Nat. Commun. 2020, 11, 2850. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Abecasis, G.R.; Boehnke, M.; Lin, X. Rare-variant association analysis: Study designs and statistical tests. Am. J. Hum. Genet. 2014, 95, 5–23. [Google Scholar] [CrossRef] [Green Version]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Ware, J.; Samocha, K.; Homsy, J.; Daly, M.J. Interpreting de novo Variation in Human Disease Using denovolyzeR. Curr. Protoc. Hum. Genet. 2015, 87, 7.25.1–7.25.15. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of rare inherited and de novo variants in 2871 congenital heart disease probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef] [Green Version]
- Akawi, N.; McRae, J.; Ansari, M.; Balasubramanian, M.; Blyth, M.; Brady, A.F.; Clayton, S.; Cole, T.; Deshpande, C.; Fitzgerald, T.W.; et al. Discovery of four recessive developmental disorders using probabilistic genotype and phenotype matching among 4125 families. Nat. Genet. 2015, 47, 1363–1369. [Google Scholar] [CrossRef]
- Martin, H.C.; Jones, W.D.; McIntyre, R.; Sanchez-Andrade, G.; Sanderson, M.; Stephenson, J.D.; Jones, C.P.; Handsaker, J.; Gallone, G.; Bruntraeger, M.; et al. Quantifying the contribution of recessive coding variation to developmental disorders. Science 2018, 362, 1161–1164. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Sanders, S.; Liu, L.; De Rubeis, S.; Lim, T.T.; Sutcliffe, J.S.; Schellenberg, G.D.; Gibbs, R.A.; Daly, M.J.; Buxbaum, J.; et al. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 2013, 9, e1003671. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.T.; Bryois, J.; Kim, A.; Dobbyn, A.; Huckins, L.M.; Munoz-Manchado, A.B.; Ruderfer, D.M.; Genovese, G.; Fromer, M.; Xu, X.; et al. Integrated Bayesian analysis of rare exonic variants to identify risk genes for schizophrenia and neurodevelopmental disorders. Genome Med. 2017, 9, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Liang, Y.; Cicek, A.E.; Li, Z.; Li, J.; Muhle, R.A.; Krenzer, M.; Mei, Y.; Wang, Y.; Knoblauch, N.; et al. A Statistical Framework for Mapping Risk Genes from De Novo Mutations in Whole-Genome-Sequencing Studies. Am. J. Hum. Genet. 2018, 102, 1031–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Zeng, X.; Jin, C.; Jin, S.C.; Dong, W.; Brueckner, M.; Lifton, R.; Lu, Q.; Zhao, H. Integrative modeling of transmitted and de novo variants identifies novel risk genes for congenital heart disease. Quant. Biol. 2021, 9, 216–227. [Google Scholar] [CrossRef]
- Nguyen, T.-H.; Dobbyn, A.; Brown, R.C.; Riley, B.P.; Buxbaum, J.; Pinto, D.; Purcell, S.M.; Sullivan, P.F.; He, X.; Stahl, E.A. mTADA is a framework for identifying risk genes from de novo mutations in multiple traits. Nat. Commun. 2020, 11, 2929. [Google Scholar] [CrossRef]
- Wang, J.; Yu, R.; Shete, S. X-chromosome genetic association test accounting for X-inactivation, skewed X-inactivation, and escape from X-inactivation. Genet. Epidemiol. 2014, 38, 483–493. [Google Scholar] [CrossRef]
- Clayton, D. Testing for association on the X chromosome. Biostatistics 2008, 9, 593–600. [Google Scholar] [CrossRef]
- Jin, H.; Park, T.; Won, S. Efficient Statistical Method for Association Analysis of X-Linked Variants. Hum. Hered. 2016, 82, 50–63. [Google Scholar] [CrossRef] [Green Version]
- Martin, H.C.; Gardner, E.J.; Samocha, K.E.; Kaplanis, J.; Akawi, N.; Sifrim, A.; Eberhardt, R.Y.; Tavares, A.L.T.; Neville, M.D.C.; Niemi, M.E.K.; et al. The contribution of X-linked coding variation to severe developmental disorders. Nat. Commun. 2021, 12, 627. [Google Scholar] [CrossRef]
- March, R.E. Gene mapping by linkage and association analysis. Mol. Biotechnol. 1999, 13, 113–122. [Google Scholar] [CrossRef]
- Tabor, H.K.; Risch, N.J.; Myers, R.M. Candidate-gene approaches for studying complex genetic traits: Practical considerations. Nat. Rev. Genet. 2002, 3, 391–397. [Google Scholar] [CrossRef]
- Kerner, G.; Bouaziz, M.; Cobat, A.; Bigio, B.; Timberlake, A.T.; Bustamante, J.; Lifton, R.P.; Casanova, J.-L.; Abel, L. A genome-wide case-only test for the detection of digenic inheritance in human exomes. Proc. Natl. Acad. Sci. USA 2020, 117, 19367–19375. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ishwaran, H. Random forests for genomic data analysis. Genomics 2012, 99, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, D.C.; Yang, Z.; Yang, F. Two-phase and family-based designs for next-generation sequencing studies. Front. Genet. 2013, 4, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013, 498, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Feng, T.; Zhang, S.; Sha, Q. Two-stage association tests for genome-wide association studies based on family data with arbitrary family structure. Eur. J. Hum. Genet. 2007, 15, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; DeMeo, D.; Silverman, E.K.; Weiss, S.T.; Laird, N.M. Using the noninformative families in family-based association tests: A powerful new testing strategy. Am. J. Hum. Genet. 2003, 73, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Murphy, A.; Weiss, S.T.; Lange, C. Screening and replication using the same data set: Testing strategies for family-based studies in which all probands are affected. PLoS Genet. 2008, 4, e1000197. [Google Scholar] [CrossRef]
- Van Steen, K.; McQueen, M.B.; Herbert, A.; Raby, B.; Lyon, H.; DeMeo, D.L.; Murphy, A.; Su, J.; Datta, S.; Rosenow, C.; et al. Genomic screening and replication using the same data set in family-based association testing. Nat. Genet. 2005, 37, 683–691. [Google Scholar] [CrossRef]
- Homsy, J.; Zaidi, S.; Shen, Y.; Ware, J.S.; Samocha, K.E.; Karczewski, K.J.; DePalma, S.R.; McKean, D.; Wakimoto, H.; Gorham, J.; et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 2015, 350, 1262–1266. [Google Scholar] [CrossRef] [Green Version]
- Sifrim, A.; Hitz, M.-P.; Wilsdon, A.; Breckpot, J.; Al Turki, S.H.; Thienpont, B.; McRae, J.; Fitzgerald, T.W.; Singh, T.; Swaminathan, G.J.; et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 2016, 48, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Conrad, D.F.; Keebler, J.E.; De Pristo, M.A.; Lindsay, S.J.; Zhang, Y.; Cassals, F.; Idaghdour, Y.; Hartl, C.L.; Torroja, C.; Garimella, K.V.; et al. Variation in genome-wide mutation rates within and between human families. Nat. Genet. 2011, 43, 712–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. USA 2010, 107, 961–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samocha, K.; Robinson, E.; Sanders, S.; Stevens, C.; Sabo, A.; McGrath, L.; Kosmicki, J.A.; Rehnström, K.; Mallick, S.; Kirby, A.; et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 2014, 46, 944–950. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- An, J.-Y.; Lin, K.; Zhu, L.; Werling, D.M.; Dong, S.; Brand, H.; Wang, H.Z.; Zhao, X.; Schwartz, G.B.; Collins, R.L.; et al. Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 2018, 362, eaat6576. [Google Scholar] [CrossRef] [Green Version]
- Sayres, M.A.W. Genetic Diversity on the Sex Chromosomes. Genome Biol. Evol. 2018, 10, 1064–1078. [Google Scholar] [CrossRef]
- Peeters, S.B.; Cotton, A.M.; Brown, C.J. Variable escape from X-chromosome inactivation: Identifying factors that tip the scales towards expression. Bioessays 2014, 36, 746–756. [Google Scholar] [CrossRef]
- Heard, E.; Chaumeil, J.; Masui, O.; Okamoto, I. Mammalian X-chromosome inactivation: An epigenetics paradigm. Cold Spring Harb. Symp. Quant. Biol. 2004, 69, 89–102. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.; Caspi, A.; Williams, B.; Houts, R.; Craig, I.W.; Mill, J. A longitudinal twin study of skewed X chromosome-inactivation. PLoS ONE 2011, 6, e17873. [Google Scholar] [CrossRef]
- Wang, J.; Talluri, R.; Shete, S. Selection of X-chromosome Inactivation Model. Cancer Inform. 2017, 16, 1176935117747272. [Google Scholar] [CrossRef] [PubMed]
- Busque, L.; Paquette, Y.; Provost, S.; Roy, D.-C.; Levine, R.L.; Mollica, L.; Gilliland, D.G. Skewing of X-inactivation ratios in blood cells of aging women is confirmed by independent methodologies. Blood 2009, 113, 3472–3474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, G.; Pedersen, J.; Klingenberg, O.; Lygren, I.; Ørstavik, K. Increased skewing of X chromosome inactivation with age in both blood and buccal cells. Cytogenet. Genome Res. 2007, 116, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, A.A. Digenic inheritance in medical genetics. J. Med. Genet. 2013, 50, 641–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasche, B.; Yi, N. Candidate gene association studies: Successes and failures. Curr. Opin. Genet. Dev. 2010, 20, 257–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Khoury, M.J.; Sun, F.; Flanders, W.D. Case-only design to measure gene-gene interaction. Epidemiology 1999, 10, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Begg, C.B.; Zhang, Z.F. Statistical analysis of molecular epidemiology studies employing case-series. Cancer Epidemiol. Biomark. Prev. 1994, 3, 173–175. [Google Scholar]
- Piegorsch, W.W.; Weinberg, C.R.; Taylor, J.A. Non-hierarchical logistic models and case-only designs for assessing susceptibility in population-based case-control studies. Stat. Med. 1994, 13, 153–162. [Google Scholar] [CrossRef]
- McKinney, B.A.; Reif, D.; Ritchie, M.D.; Moore, J.H. Machine learning for detecting gene-gene interactions: A review. Appl. Bioinform. 2006, 5, 77–88. [Google Scholar] [CrossRef]
- Byrjalsen, A.; Hansen, T.V.O.; Stoltze, U.K.; Mehrjouy, M.M.; Barnkob, N.M.; Hjalgrim, L.L.; Mathiasen, R.; Lautrup, C.K.; Gregersen, P.A.; Hasle, H.; et al. Nationwide germline whole genome sequencing of 198 consecutive pediatric cancer patients reveals a high incidence of cancer prone syndromes. PLoS Genet. 2020, 16, e1009231. [Google Scholar] [CrossRef]
- Costantini, A.; Valta, H.; Suomi, A.-M.; Mäkitie, O.; Taylan, F. Oligogenic Inheritance of Monoallelic TRIP11, FKBP10, NEK1, TBX5, and NBAS Variants Leading to a Phenotype Similar to Odontochondrodysplasia. Front. Genet. 2021, 12, 680838. [Google Scholar] [CrossRef] [PubMed]
- Dallali, H.; Kheriji, N.; Kammoun, W.; Mrad, M.; Soltani, M.; Trabelsi, H.; Hamdi, W.; Bahlous, A.; Ben Ahmed, M.; Mahjoub, F.; et al. Multiallelic Rare Variants in BBS Genes Support an Oligogenic Ciliopathy in a Non-obese Juvenile-Onset Syndromic Diabetic Patient: A Case Report. Front. Genet. 2021, 12, 664963. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Ma, Y.; Zhang, Z.; Xian, J.; Geng, X.; Wang, F.; Huang, J.; Yang, Z.; Luo, Y.; Lin, Y. Young and early-onset dilated cardiomyopathy with malignant ventricular arrhythmia and sudden cardiac death induced by the heterozygous LDB3, MYH6, and SYNE1 missense mutations. Ann. Noninvasive Electrocardiol. 2021, 26, e12840. [Google Scholar] [CrossRef] [PubMed]
- Libbrecht, M.; Noble, W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, H.L.; John, C.R.; Watson, D.; Munroe, P.B.; Barnes, M.R.; Cabrera, C.P. Reaching the End-Game for GWAS: Machine Learning Approaches for the Prioritization of Complex Disease Loci. Front. Genet. 2020, 11, 350. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yagi, H.; Onuoha, E.O.; Damerla, R.R.; Francis, R.; Furutani, Y.; Tariq, M.; King, S.M.; Hendricks, G.; Cui, C.; et al. DNAH6 and Its Interactions with PCD Genes in Heterotaxy and Primary Ciliary Dyskinesia. PLoS Genet. 2016, 12, e1005821. [Google Scholar] [CrossRef] [Green Version]
- Gifford, C.A.; Ranade, S.S.; Samarakoon, R.; Salunga, H.T.; de Soysa, T.Y.; Huang, Y.; Zhou, P.; Elfenbein, A.; Wyman, S.K.; Bui, Y.K.; et al. Oligogenic inheritance of a human heart disease involving a genetic modifier. Science 2019, 364, 865–870. [Google Scholar] [CrossRef]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [Green Version]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 years of GWAS discovery: Biology, function, and translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [Green Version]
- Willer, C.; Li, Y.; Abecasis, G.R. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010, 26, 2190–2191. [Google Scholar] [CrossRef]
- Uffelmann, E.; Huang, Q.Q.; Munung, N.S.; de Vries, J.; Okada, Y.; Martin, A.R.; Martin, H.C.; Lappalainen, T.; Posthuma, D. Genome-wide association studies. Nat. Rev. Methods Primers 2021, 1, 59. [Google Scholar] [CrossRef]
- Chatterjee, N.; Shi, J.; García-Closas, M. Developing and evaluating polygenic risk prediction models for stratified disease prevention. Nat. Rev. Genet. 2016, 17, 392–406. [Google Scholar] [CrossRef]
- Kong, A.; Thorleifsson, G.; Frigge, M.L.; Vilhjalmsson, B.J.; Young, A.I.; Thorgeirsson, T.E.; Benonisdottir, S.; Oddsson, A.; Halldorsson, B.V.; Masson, G.; et al. The nature of nurture: Effects of parental genotypes. Science 2018, 359, 424–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.I.; Benonisdottir, S.; Przeworski, M.; Kong, A. Deconstructing the sources of genotype-phenotype associations in humans. Science 2019, 365, 1396–1400. [Google Scholar] [CrossRef]
- Howe, L.J.; Nivard, M.G.; Morris, T.T.; Hansen, A.F.; Rasheed, H.; Cho, Y.; Chittoor, G.; Lind, P.A.; Palviainen, T.; van der Zee, M.D.; et al. Within-sibship GWAS improve estimates of direct genetic effects. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wu, Y.; Zhong, X.; Lin, Y.; Zhao, Z.; Chen, J.; Zheng, B.; Li, J.J.; Fletcher, J.M.; Lu, Q. Estimating genetic nurture with summary statistics of multigenerational genome-wide association studies. Proc. Natl. Acad. Sci. USA 2021, 118, e2023184118. [Google Scholar] [CrossRef]
- Cooper, D.N.; Krawczak, M.; Polychronakos, C.; Tyler-Smith, C.; Kehrer-Sawatzki, H. Where genotype is not predictive of phenotype: Towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum. Genet. 2013, 132, 1077–1130. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Hemani, G.; Haley, C. Detecting epistasis in human complex traits. Nat. Rev. Genet. 2014, 15, 722–733. [Google Scholar] [CrossRef]
- Sinnott-Armstrong, N.; Naqvi, S.; Rivas, M.; Pritchard, J.K. GWAS of three molecular traits highlights core genes and pathways alongside a highly polygenic background. eLife 2021, 10, e58615. [Google Scholar] [CrossRef]
- Wainschtein, P.; Jain, D.; Zheng, Z.; TOPMed Anthropometry Working Group; NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium; Cupples, L.A.; Shadyab, A.H.; McKnight, B.; Shoemaker, B.M.; Mitchell, B.D.; et al. Recovery of trait heritability from whole genome sequence data. bioRxiv 2021. [Google Scholar] [CrossRef]
- Crowley, J.J.; Szatkiewicz, J.; Kähler, A.K.; Giusti-Rodríguez, P.; Ancalade, N.; Booker, J.K.; Carr, J.L.; Crawford, G.E.; Losh, M.; Stockmeier, C.A.; et al. Common-variant associations with fragile X syndrome. Mol. Psychiatry 2019, 24, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Claussnitzer, M.; Cho, J.H.; Collins, R.; Cox, N.J.; Dermitzakis, E.T.; Hurles, M.E.; Kathiresan, S.; Kenny, E.E.; Lindgren, C.M.; MacArthur, D.G.; et al. A brief history of human disease genetics. Nature 2020, 577, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordell, H.J.; Bentham, J.; Topf, A.; Zelenika, D.; Heath, S.; Mamasoula, C.; Cosgrove, C.; Blue, G.M.; Granados-Riveron, J.T.; Setchfield, K.; et al. Genome-wide association study of multiple congenital heart disease phenotypes identifies a susceptibility locus for atrial septal defect at chromosome 4p16. Nat. Genet. 2013, 45, 822. [Google Scholar] [CrossRef] [PubMed]
- Agopian, A.J.; Goldmuntz, E.; Hakonarson, H.; Sewda, A.; Taylor, D.; Mitchell, L.E.; Pediatric Cardiac Genomics Consortium. Genome-wide association studies and meta-analyses for congenital heart defects. Circ. Cardiovasc. Genet. 2017, 10, e001449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiner, D.J.; iPSYCH-Broad Autism Group; Wigdor, E.M.; Ripke, S.; Walters, R.K.; Kosmicki, J.A.; Grove, J.; Samocha, K.E.; Goldstein, J.I.; Okbay, A.; et al. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat. Genet. 2017, 49, 978–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.-Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 2020, 180, 568–584. [Google Scholar] [CrossRef]
- Timberlake, A.T.; Choi, J.; Zaidi, S.; Lu, Q.; Nelson-Williams, C.; Brooks, E.D.; Bilguvar, K.; Tikhonova, I.; Mane, S.; Yang, J.F.; et al. Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. eLife 2016, 5, e20125. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Wu, Y.; Shin, J.; Zheng, Y.; Siahpirani, A.F.; Lin, Y.; Ni, Z.; Chen, J.; You, J.; Keles, S.; et al. Transcriptome-wide transmission disequilibrium analysis identifies novel risk genes for autism spectrum disorder. PLoS Genet. 2021, 17, e1009309. [Google Scholar] [CrossRef]
- Halldorsson, B.V.; Eggertsson, H.P.; Moore, K.H.S.; Hauswedell, H.; Eiriksson, O.; Ulfarsson, M.O.; Palsson, G.; Hardarson, M.T.; Oddsson, A.; Jensson, B.O.; et al. The sequences of 150,119 genomes in the UK biobank. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hu, Y.; Lu, Q.; Powles, R.; Yao, X.; Yang, C.; Fang, F.; Xu, X.; Zhao, H. Leveraging functional annotations in genetic risk prediction for human complex diseases. PLoS Comput. Biol. 2017, 13, e1005589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, Y.; Anne Feng, Y.-C.; Chen, C.-Y.; Lam, M.; Sawa, A.; Martin, A.R.; Qin, S.; Huang, H.; Ge, T. Improving polygenic prediction in ancestrally diverse populations. medRxiv 2021. [Google Scholar] [CrossRef]
- Privé, F.; Arbel, J.; Vilhjálmsson, B.J. LDpred2: Better, faster, stronger. Bioinformatics 2020, 36, 5424–5431. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Yi, Y.; Song, J.; Wu, Y.; Zhong, X.; Lin, Y.; Hohman, T.J.; Fletcher, J.; Lu, Q. PUMAS: Fine-tuning polygenic risk scores with GWAS summary statistics. Genome Biol. 2021, 22, 257. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef]
- Mills, M.C.; Rahal, C. The GWAS diversity monitor tracks diversity by disease in real time. Nat. Genet. 2020, 52, 242–243. [Google Scholar] [CrossRef]
- Martin, A.R.; Kanai, M.; Kamatani, Y.; Okada, Y.; Neale, B.M.; Daly, M.J. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 2019, 51, 584–591. [Google Scholar] [CrossRef]
- Mostafavi, H.; Harpak, A.; Agarwal, I.; Conley, D.; Pritchard, J.K.; Przeworski, M. Variable prediction accuracy of polygenic scores within an ancestry group. eLife 2020, 9, e48376. [Google Scholar] [CrossRef]
- Friedmann, T. A brief history of gene therapy. Nat. Genet. 1992, 2, 93–98. [Google Scholar] [CrossRef]
- Rogers, S.; Lowenthal, A.; Terheggen, H.G.; Columbo, J.P. Induction of arginase activity with the Shope papilloma virus in tissue culture cells from an argininemic patient. J. Exp. Med. 1973, 137, 1091–1096. [Google Scholar] [CrossRef]
- Tabernero, J.; Shapiro, G.I.; Lorusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013, 3, 406–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Spreafico, R.; Soriaga, L.B.; Grosse, J.; Virgin, H.W.; Telenti, A. Advances in Genomics for Drug Development. Genes 2020, 11, 942. [Google Scholar] [CrossRef] [PubMed]
- Aschenbrenner, D.S. Two New Drugs for Sickle Cell Disease. Am. J. Nurs. 2020, 120, 24. [Google Scholar] [CrossRef]
- Ataga, K.I.; Kutlar, A.; Kanter, J.; Liles, D.; Cancado, R.; Friedrisch, J.; Guthrie, T.H.; Knight-Madden, J.; Alvarez, O.A.; Gordeuk, V.R.; et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 429–439. [Google Scholar] [CrossRef]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Al Kindi, S.; Brown, R.C.; et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N. Engl. J. Med. 2019, 381, 509–519. [Google Scholar] [CrossRef]
- Sebastiani, P.; Solovieff, N.; Hartley, S.W.; Milton, J.N.; Riva, A.; Dworkis, D.A.; Melista, E.; Klings, E.; Garrett, M.E.; Telen, M.J.; et al. Genetic modifiers of the severity of sickle cell anemia identified through a genome-wide association study. Am. J. Hematol. 2010, 85, 29–35. [Google Scholar] [CrossRef] [Green Version]
- Esrick, E.B.; Lehmann, L.E.; Biffi, A.; Achebe, M.; Brendel, C.; Ciuculescu, M.F.; Daley, H.; MacKinnon, B.; Morris, E.; Federico, A.; et al. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. N. Engl. J. Med. 2021, 384, 205–215. [Google Scholar] [CrossRef]
- Cavazzana, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef]
- Stoica, L.; Sena-Esteves, M. Adeno Associated Viral Vector Delivered RNAi for Gene Therapy of SOD1 Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2016, 9, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, S.B.; Ranum, P.T.; Moteki, H.; Pan, B.; Goodwin, A.T.; Goodman, S.S.; Abbas, P.J.; Holt, J.R.; Smith, R.J.; Shibata, S.B.; et al. RNA Interference Prevents Autosomal-Dominant Hearing Loss. Am. J. Hum. Genet. 2016, 98, 1101–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathwani, A.C.; Reiss, U.M.; Tuddenham, E.G.; Rosales, C.; Chowdary, P.; McIntosh, J.; Della Peruta, M.; Lheriteau, E.; Patel, N.; Raj, D.; et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014, 371, 1994–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batty, P.; Lillicrap, D. Hemophilia Gene Therapy: Approaching the First Licensed Product. Hemasphere 2021, 5, e540. [Google Scholar] [CrossRef] [PubMed]
- Hauswirth, W.; Aleman, T.S.; Kaushal, S.; Cideciyan, A.V.; Schwartz, S.B.; Wang, L.; Conlon, T.J.; Boye, S.L.; Flotte, T.R.; Byrne, B.J.; et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: Short-term results of a phase I trial. Hum. Gene Ther. 2008, 19, 979–990. [Google Scholar] [CrossRef] [Green Version]
- Maguire, A.M.; High, K.A.; Auricchio, A.; Wright, J.F.; Pierce, E.A.; Testa, F.; Mingozzi, F.; Bennicelli, J.L.; Ying, G.-S.; Rossi, S.; et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: A phase 1 dose-escalation trial. Lancet 2009, 374, 1597–1605. [Google Scholar] [CrossRef] [Green Version]
- Bainbridge, J.W.; Mehat, M.S.; Sundaram, V.; Robbie, S.J.; Barker, S.E.; Ripamonti, C.; Georgiadis, A.; Mowat, F.; Beattie, S.G.; Gardner, P.; et al. Long-term effect of gene therapy on Leber’s congenital amaurosis. N. Engl. J. Med. 2015, 372, 1887–1897. [Google Scholar] [CrossRef] [Green Version]
- Wright, A.F. Long-term effects of retinal gene therapy in childhood blindness. N. Engl. J. Med. 2015, 372, 1954–1955. [Google Scholar] [CrossRef]
- Bennett, J.; Wellman, J.; Marshall, K.A.; McCague, S.; Ashtari, M.; DiStefano-Pappas, J.; Elci, O.U.; Chung, D.C.; Sun, J.; Wright, J.F.; et al. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: A follow-on phase 1 trial. Lancet 2016, 388, 661–672. [Google Scholar] [CrossRef] [Green Version]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Griesenbach, U.; Pytel, K.M.; Alton, E.W. Cystic Fibrosis Gene Therapy in the UK and Elsewhere. Hum. Gene Ther. 2015, 26, 266–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- U.S. Food and Drug Administration. Approved Cellular and Gene Therapy Products. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products (accessed on 26 October 2021).
- U.S. Food and Drug Administration. ABECMA (Idecabtagene Vicleucel). Available online: https://www.fda.gov/vaccines-blood-biologics/abecma-idecabtagene-vicleucel (accessed on 21 April 2021).
- U.S. Food and Drug Administration. BREYANZI (Lisocabtagene Maraleucel). Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/breyanzi-lisocabtagene-maraleucel (accessed on 4 March 2021).
- U.S. Food and Drug Administration. IMLYGIC. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/imlygic (accessed on 9 December 2021).
- U.S. Food and Drug Administration. KYMRIAH (Tisagenlecleucel). Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/kymriah-tisagenlecleucel (accessed on 14 June 2021).
- U.S. Food and Drug Administration. LUXTURNA. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/luxturna (accessed on 26 July 2018).
- U.S. Food and Drug Administration. PROVENGE (sipuleucel-T). Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/provenge-sipuleucel-t (accessed on 28 May 2019).
- U.S. Food and Drug Administration. TECARTUS (Brexucabtagene Autoleucel). Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/tecartus-brexucabtagene-autoleucel (accessed on 17 November 2021).
- U.S. Food and Drug Administration. YESCARTA (Axicabtagene Ciloleucel). Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/yescarta-axicabtagene-ciloleucel (accessed on 11 May 2021).
- U.S. Food and Drug Administration. ZOLGENSMA. Available online: https://www.fda.gov/vaccines-blood-biologics/zolgensma (accessed on 26 October 2021).
- Roden, D.M.; Wilke, R.A.; Kroemer, H.K.; Stein, C.M. Pharmacogenomics: The genetics of variable drug responses. Circulation 2011, 123, 1661–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schärfe, C.P.I.; Tremmel, R.; Schwab, M.; Kohlbacher, O.; Marks, D.S. Genetic variation in human drug-related genes. Genome Med. 2017, 9, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aneesh, T.P.; Sekhar, M.S.; Jose, A.; Chandran, L.; Zachariaha, S.M. Pharmacogenomics: The right drug to the right person. J. Clin. Med. Res. 2009, 1, 191–194. [Google Scholar] [CrossRef] [Green Version]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Cobain, E.F.; Wu, Y.-M.; Vats, P.; Chugh, R.; Worden, F.; Smith, D.C.; Schuetze, S.M.; Zalupski, M.M.; Sahai, V.; Alva, A.; et al. Assessment of Clinical Benefit of Integrative Genomic Profiling in Advanced Solid Tumors. JAMA Oncol. 2021, 7, 525–533. [Google Scholar] [CrossRef]
- Relling, M.V.; Evans, W.E. Pharmacogenomics in the clinic. Nature 2015, 526, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Köhler, S.; Doelken, S.C.; Mungall, C.J.; Bauer, S.; Firth, H.V.; Bailleul-Forestier, I.; Black, G.C.M.; Brown, D.L.; Brudno, M.; Campbell, J.; et al. The Human Phenotype Ontology project: Linking molecular biology and disease through phenotype data. Nucleic Acids Res. 2014, 42, D966–D974. [Google Scholar] [CrossRef] [Green Version]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef]
- Hwang, T.; Atluri, G.; Xie, M.; Dey, S.; Hong, C.; Kumar, V.; Kuang, R. Co-clustering phenome-genome for phenotype classification and disease gene discovery. Nucleic Acids Res. 2012, 40, e146. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Rico, M.; Alvarado, J.M. A Machine Learning Approach for Studying the Comorbidities of Complex Diagnoses. Behav. Sci. 2019, 9, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narita, A.; Nagai, M.; Mizuno, S.; Ogishima, S.; Tamiya, G.; Ueki, M.; Sakurai, R.; Makino, S.; Obara, T.; Ishikuro, M.; et al. Clustering by phenotype and genome-wide association study in autism. Transl. Psychiatry 2020, 10, 290. [Google Scholar] [CrossRef] [PubMed]
- Westbury, S.K.; on behalf of the BRIDGE-BPD Consortium; Turro, E.; Greene, D.; Lentaigne, C.; Kelly, A.M.; Bariana, T.K.; Simeoni, I.; Pillois, X.; Attwood, A.; et al. Human phenotype ontology annotation and cluster analysis to unravel genetic defects in 707 cases with unexplained bleeding and platelet disorders. Genome Med. 2015, 7, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashley, E.A. Towards precision medicine. Nat. Rev. Genet. 2016, 17, 507–522. [Google Scholar] [CrossRef]

| Name | Manufacturer | Target Disease | Gene of Interest | FDA Approval Date |
|---|---|---|---|---|
| Abecma (idecabtagene vicleucel) | Celgene Corporation (Bristol-Myers Squibb Company) | Relapsed or refractory multiple myeloma | BCMA (B-cell maturation antigen) | March 2021 [133] |
| Breyanzi (lisocabtagene maraleucel) | Juno Therapeutics (Bristol-Myers Squibb Company) | Relapsed or refractory large B-cell lymphoma | CD137 (4-1BB TNF- receptor) and CD3-zeta | February 2021 [134] |
| Imlygic (talimogene laherparepvec) | BioVex (Subsidiary of Amgen) | Melanoma (unresectable cutaneous, subcutaneous, and nodal lesions) | GM-CSF (immune stimulatory protein) | October 2015 [135] |
| Kymriah (tisagenlecleucel) | Novartis Pharmaceuticals Corporation | Pediatric B-cell precursor acute lymphoblastic leukemia (ALL) | CD137 (4-1BB TNF- receptor) and CD3-zeta | August 2017 [136] |
| Relapsed or refractory large B-cell lymphoma in adult | CD137 (4-1BB TNF- receptor) and CD3-zeta | May 2018 [136] | ||
| Luxturna (voretigene neparvovec-rzyl) | Spark Therapeutics | Retinal dystrophy (biallelic RPE65 mutation- associated) | RPE65 (human retinal pigment epithelial 65 kDa protein) | December 2017 [137] |
| Provenge (sipuleucel-t) | Dendreon Corporation | Asymptomatic or minimally symptomatic metastatic castration-resistant prostate cancer (mCRPC) | ACP3 (prostate acid phosphatase) | April 2010 [138] |
| Tecartus (brexucabtagene autoleucel) | Kite Pharma | Relapsed or refractory mantle cell lymphoma (MCL) in adult | CD28 and CD3-zeta | July 2020 [139] |
| Relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL) in adult | CD28 and CD3-zeta | October 2021 [139] | ||
| Yescarta (axicabtagene ciloleucel) | Kite Pharma | Relapsed or refractory large B-cell lymphoma | CD28 and CD3-zeta | October 2017 [140] |
| Relapsed or refractory follicular lymphoma | CD28 and CD3-zeta | March 2021 [140] | ||
| Zolgensma (onasemnogene abeparvovec-xioi) | Novartis Gene Therapies (Formerly AveXis) | Spinal muscular atrophy (Type I) | SMN1 (human survival motor neuron 1 protein) | May 2019 [141] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.-C.; Wu, Y.; Choi, J.; Allington, G.; Zhao, S.; Khanfar, M.; Yang, K.; Fu, P.-Y.; Wrubel, M.; Yu, X.; et al. Computational Genomics in the Era of Precision Medicine: Applications to Variant Analysis and Gene Therapy. J. Pers. Med. 2022, 12, 175. https://doi.org/10.3390/jpm12020175
Wang Y-C, Wu Y, Choi J, Allington G, Zhao S, Khanfar M, Yang K, Fu P-Y, Wrubel M, Yu X, et al. Computational Genomics in the Era of Precision Medicine: Applications to Variant Analysis and Gene Therapy. Journal of Personalized Medicine. 2022; 12(2):175. https://doi.org/10.3390/jpm12020175
Chicago/Turabian StyleWang, Yung-Chun, Yuchang Wu, Julie Choi, Garrett Allington, Shujuan Zhao, Mariam Khanfar, Kuangying Yang, Po-Ying Fu, Max Wrubel, Xiaobing Yu, and et al. 2022. "Computational Genomics in the Era of Precision Medicine: Applications to Variant Analysis and Gene Therapy" Journal of Personalized Medicine 12, no. 2: 175. https://doi.org/10.3390/jpm12020175