Minimal Residual Disease in Multiple Myeloma: State of the Art and Applications in Clinical Practice

 ,
, 

Abstract
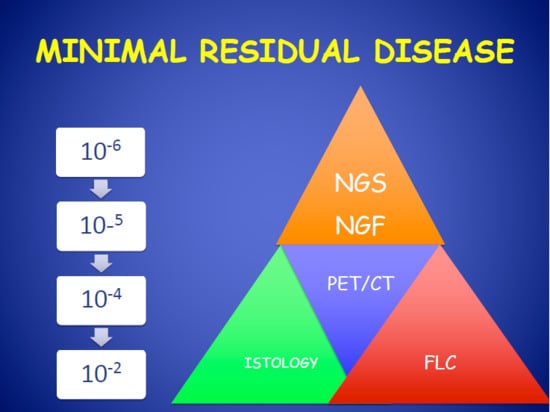
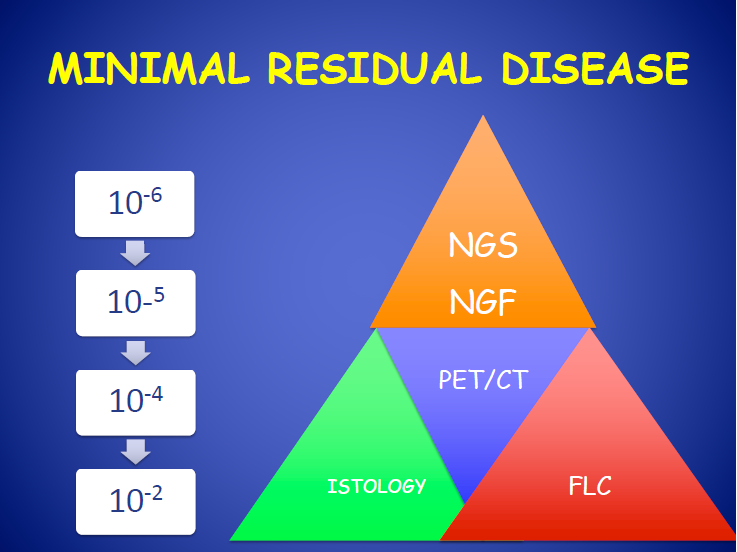
:
1. Introduction
Complete Remission (CR) and Minimal Residual Disease
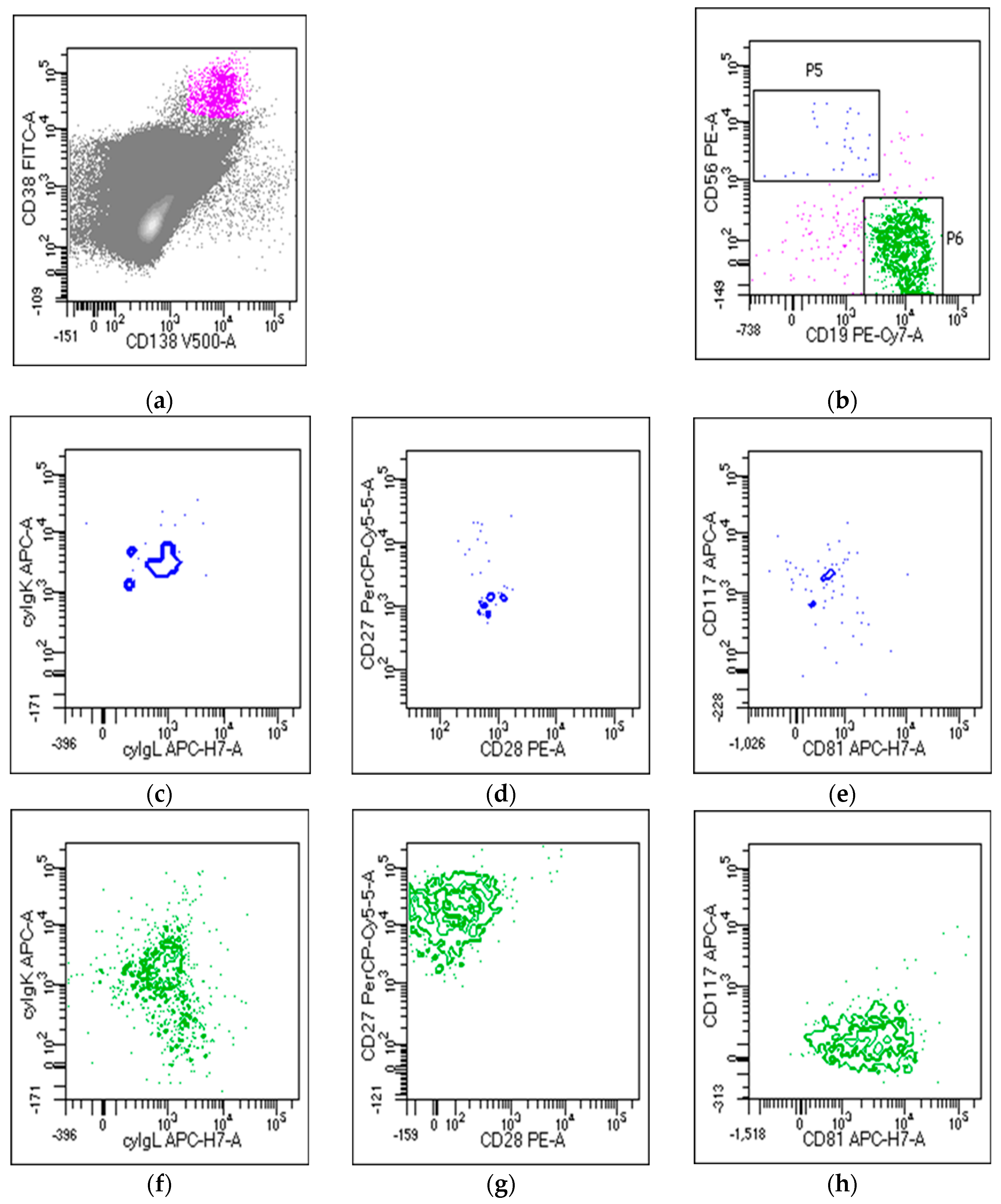
2. Next-Generation Flow Cytometry for Evaluation of Multiple Myeloma Minimal Residual Disease
3. Next-Generation Sequencing (NGS) for Evaluation of Multiple Myeloma MRD
3.1. Quality of Bone Marrow Aspirates May Influence MRD Analysis
3.2. MRD in Peripheral Blood
4. MRD in Clinical Trials
4.1. MRD in High-Risk Cytogenetic MM Patients
4.2. Imaging in MRD Evaluation
5. MRD in Real-World Clinical Practice: More Questions than Answers
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gozzetti, A.; Candi, V.; Papini, G.; Bocchia, M. Therapeutic advancements in multiple myeloma. Front. Oncol. 2014, 4, 241. [Google Scholar] [CrossRef]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. (Eds.) SEER Cancer Statistics Review, 1975–2017; National Cancer Institute: Bethesda, MD, USA, 2020. Available online: https://seer.cancer.gov/csr/1975_2017/ (accessed on 15 July 2020).
- Mohty, M.; Terpos, E.; Mateos, M.V.; Cavo, M.; Lejniece, S.; Beksac, M.; Bekadja, M.A.; Legiec, W.; Dimopoulos, M.; Stankovic, S.; et al. EMMOS Investigators. Multiple Myeloma Treatment in Real-world Clinical Practice: Results of a Prospective, Multinational, Noninterventional Study. Clin. Lymphoma Myeloma Leuk. 2018, 18, e401–e419. [Google Scholar] [CrossRef] [Green Version]
- Brenner, H.; Gondos, A.; Pulte, D. Recent major improvement in long-term survival of younger patients with multiple myeloma. Blood 2008, 111, 2521–2526. [Google Scholar] [CrossRef]
- Cini, M.; Zamagni, E.; Valdré, L.; Palareti, G.; Patriarca, F.; Tacchetti, P.; Legnani, C.; Catalano, L.; Masini, L.; Tosi, P.; et al. Thalidomide–dexamethasone as upfront therapy for patients with newly diagnosed multiple myeloma: Thrombophilic alterations, thrombotic complications, and thromboprophylaxis with low-dose warfarin. Eur. J. Haematol. 2010, 84, 484–492. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, S.V.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Zeldenrust, S.R.; Dingli, D.; Russell, S.J.; Lust, J.A.; et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008, 111, 2516–2520. [Google Scholar] [CrossRef] [Green Version]
- Gozzetti, A.; Cerase, A.; Lotti, F.; Rossi, D.; Palumbo, A.; Petrucci, M.T.; Patriarca, F.; Nozzoli, C.; Cavo, M.; Offidani, M.; et al. GIMEMA (Gruppo Italiano Malattie Ematologiche dell’Adulto) Myeloma Working Party Extramedullary intracranial localizations of multiple myeloma and treatment with novel agents: A retrospective survey of 50 patients. Cancer 2012, 118, 1574–1584. [Google Scholar] [CrossRef]
- Jurczyszyn, A.; Grzasko, N.; Gozzetti, A.; Czepiel, J.; Cerase, A.; Hungria, V.; Crusoe, E.; Silva Dias, A.L.; Vij, R.; Fiala, M.A.; et al. Central nervous system involvement by multiple myeloma: A multi-institutional retrospective study of 172 patients in daily clinical practice. Am. J. Hematol. 2016, 91, 575–580. [Google Scholar] [CrossRef] [Green Version]
- Gozzetti, A.; Cerase, A. Novel agents in CNS myeloma treatment. Cent. Nerv. Syst. Agents Med. Chem. 2014, 14, 23–27. [Google Scholar] [CrossRef]
- Castillo, J.J.; Jurczyszyn, A.; Brozova, L.; Crusoe, E.; Czepiel, J.; Davila, J.; Dispenzieri, A.; Eveillard, M.; Fiala, M.A.; Ghobrial, I.M.; et al. IgM myeloma: A multicenter retrospective study of 134 patients. Am. J. Hematol. 2017, 92, 746–751. [Google Scholar] [CrossRef] [PubMed]
- Jurczyszyn, A.; Olszewska-Szopa, M.; Hungria, V.; Crusoe, E.; Pika, T.; Delforge, M.; Leleu, X.; Rasche, L.; Nooka, A.K.; Druzd-Sitek, A.; et al. Cutaneous involvement in multiple myeloma: A multi-institutional retrospective study of 53 patients. Leuk. Lymphoma 2016, 57, 2071–2076. [Google Scholar] [CrossRef] [PubMed]
- Ocio, E.M.; Richardson, P.G.; Rajkumar, S.V.; Palumbo, A.; Mateos, M.V.; Orlowski, R.; Kumar, S.; Usmani, S.; Roodman, D.; Niesvizky, R.; et al. New drugs and novel mechanisms of action in multiple myeloma 2013: A report from the International Myeloma Working group (IMWG). Leukemia 2014, 28, 525–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajan, A.M.; Rajkumar, S.V. Interpretation of cytogenetic results in multiple myeloma for clinical practice. Blood Cancer J. 2015, 5, e365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gozzetti, A.; Le Beau, M.M. Fluorescence in situ hybridization: Uses and limitations. Semin Hematol. 2000, 37, 320–333. [Google Scholar] [CrossRef]
- Landgren, O.; Rajkumar, S.V. New Developments in Diagnosis, Prognosis, and Assessment of Response in Multiple Myeloma. Clin. Cancer Res. 2016, 22, 5428–5433. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Paiva, B.; Anderson, K.C.; Durie, B.; Landgren, O.; Moreau, P.; Munshi, N.; Lonial, S.; Bladé, J.; Mateos, M.V.; et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016, 17, e328–e346. [Google Scholar] [CrossRef]
- Paiva, B.; Gutierrez, N.C.; Rosinol, L.; Vidriales, M.B.; Montalban, M.A.; Martinez-Lopez, J.; Mateos, M.V.; Cibeira, M.T.; Cordon, L.; Oriol, A.; et al. High-risk cytogenetics and persistent minimal residual disease by multiparametric flow cytometry predict unsustained complete response after autologous stem cell transplantation in multiple myeloma. Blood 2012, 119, 687–691. [Google Scholar] [CrossRef]
- Flores-Montero, J.; Sanoja-Flores, L.; Paiva, B.; Puig, N.; García-Sánchez, O.; Böttcher, S.; van der Velden, V.; Pérez-Morán, J.J.; Vidriales, M.B.; García-Sanz, R.; et al. Next Generation Flow for highly sensitive and standardized detection of minimal residual disease in multiple myeloma. Leukemia 2017, 31, 2094–2103. [Google Scholar] [CrossRef] [Green Version]
- Arroz, M.; Came, N.; Lin, P.; Chen, W.; Yuan, C.; Lagoo, A.; Monreal, M.; De Tute, R.; Vergilio, J.A.; Rawstron, A.C.; et al. Consensus guidelines on plasma cell myeloma minimal residual disease analysis and reporting. Cytom. B Clin. Cytom. 2016, 90, 31–39. [Google Scholar] [CrossRef]
- Oberle, A.; Brandt, A.; Alawi, M.; Langebrake, C.; Janjetovic, S.; Wolschke, C.; Schütze, K.; Bannas, P.; Kröger, N.; Koch-Nolte, F.; et al. Long-term CD38 saturation by daratumumab interferes with diagnostic myeloma cell detection. Haematologica 2017, 102, e368–e370. [Google Scholar] [CrossRef] [Green Version]
- Courville, E.L.; Yohe, S.; Shivers, P.; Linden, M.A. VS38 Identifies Myeloma Cells With Dim CD38 Expression and Plasma Cells Following Daratumumab Therapy, Which Interferes With CD38 Detection for 4 to 6 Months. Am. J. Clin. Pathol. 2020, 153, 221–228. [Google Scholar] [CrossRef]
- Lionetti, M.; Neri, A. Utilizing next-generation sequencing in the management of multiple myeloma. Expert Rev. Mol. Diagn. 2017, 17, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Orfao, A.; Chim, C.S. Molecular Detection of Minimal Residual Disease in Multiple Myeloma. Br. J. Haematol. 2018, 181, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Q.; Bai, Y.; Orfao, A.; Chim, C.S. Standardized Minimal Residual Disease Detection by Next-Generation Sequencing in Multiple Myeloma. Front. Oncol. 2019, 9, 449. [Google Scholar] [CrossRef] [PubMed]
- Della Starza, I.; Nunes, V.; Cavalli, M.; De Novi, L.A.; Ilari, C.; Apicella, V.; Vitale, A.; Testa, A.M.; Del Giudice, I.; Chiaretti, S.; et al. Comparative Analysis Between RQ-PCR and digital-droplet-PCR of immunoglobulin/T-cell Receptor Gene Rearrangements to Monitor Minimal Residual Disease in Acute Lymphoblastic Leukaemia. Br. J. Haematol. 2016, 174, 541–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korde, N.; Mailankody, S.; Roschewski, M.; Faham, M.; Kotwaliwale, C.; Moorhead, M.; Kwok, M.L.; Manasanch, E.E.; Bhutani, M.; Tageja, N.; et al. Minimal Residual Disease (MRD) Testing in Newly Diagnosed Multiple myeloma (MM) Patients: A Prospective Head-to-Head Assessment of Cell-Based, Molecular, and Molecular-Imaging Modalities. Blood 2014, 124, 2105. [Google Scholar] [CrossRef]
- Perrot, A.; Lauwers-Cances, V.; Corre, J.; Robillard, N.; Hulin, C.; Chretien, M.L.; Dejoie, T.; Maheo, S.; Stoppa, A.M.; Pegourie, B.; et al. Minimal residual disease negativity using deep sequencing is a major prognostic factor in multiple myeloma. Blood 2018, 132, 2456–2464. [Google Scholar] [CrossRef] [Green Version]
- Avet-Loiseau, H.; Bene, M.C.; Wuilleme, S.; Corre, J.; Attal, M.; Arnulf, B.; Garderet, L.; Macro, M.; Stoppa, A.M.; Delforge, M.; et al. Concordance of Post-consolidation Minimal Residual Disease Rates by Multiparametric Flow Cytometry and Next-generation Sequencing in CASSIOPEIA. Clin. Lymphoma Myeloma Leuk. 2019, 19, E3–E4. [Google Scholar] [CrossRef]
- Martinez-Lopez, J.; Lahuerta, J.J.; Pepin, F.; Gonzalez, M.; Barrio, S.; Alaya, R.; Puig, N.; Montalban, M.A.; Paiva, B.; Weng, L.; et al. Prognostic value of deep sequencing method for minimal residual disease detection in multiple myeloma. Blood 2014, 123, 3073–3079. [Google Scholar] [CrossRef]
- Oliva, S.; Gambella, M.; Gilestro, M.; Muccio, V.E.; Gay, F.; Drandi, D.; Ferrero, S.; Passera, R.; Pautasso, C.; Bernardini, A.; et al. Minimal residual disease after transplantation or lenalidomide-based consolidation in myeloma patients: A prospective analysis. Oncotarget 2017, 8, 5924–5935. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Orfao, A.; Beksac, M.; Bezdickova, L.; Brooimans, R.A.; Bumbea, H.; Dalva, K.; Fuhler, G.; Gratama, J.; Hose, D.; et al. European Myeloma Network. Report of the European Myeloma Network on Multiparametric Flow Cytometry in Multiple Myeloma and Related Disorders. Haematologica 2008, 93, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Brooimans, R.A.; Kraan, J.; Van Putten, W.; Cornelissen, J.J.; Löwenberg, B.; Gratama, J.W. Flow cytometric differential of leukocytes populations in normal bone marrow: Influence of peripheral blood contamination. Cytom. B Clin. Cytom. 2009, 76, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Bhaskar, A.; Kumar, L.; Sharma, A.; Jain, P. Flow Cytometric Immunophenotyping and Minimal Residual Disease Analysis in Multiple Myeloma. Am. J. Clin. Pathol. 2009, 132, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.A.; Guillén-Grima, F.; Moreno, C.; Panizo, C.; Pérez-Robles, C.; Mata, J.J.; Moreno, L.; Arana, P.; Chocarro, S.; Merino, J. A simple flow-cytometry method to evaluate peripheral blood contamination of bone marrow aspirates. J. Immunol. Methods 2017, 442, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Loken, M.R.; Chu, S.C.; Fritschle, W.; Kalnoski, M.; Wells, D.A. Normalization of bone marrow aspirates for hemodilution in flow cytometric analyses. Cytom. B Clin. Cytom. 2008, 76, 27–36. [Google Scholar] [CrossRef]
- Paiva, B.; Puig, N.; Cedena, M.T.; De Jong, B.G.; Ruiz, Y.; Rapado, I.; Martinez-Lopez, J.; Cordon, L.; D’Alignani, D.; Delgado, J.A.; et al. Differentiation stage of myeloma plasma cells: Biological and clinical significance. Leukemia 2017, 31, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanoja-Flores, L.; Flores-Montero, J.; Puig, N.; Contreras-Sanfeliciano, T.; Pontes, R.; Corral-Mateos, A.; García-Sánchez, O.; Díez-Campelo, M.; de Pessoa Magalhães, R.J.; García-Martín, L.; et al. Blood monitoring of circulating tumor plasma cells by next generation flow in multiple myeloma after therapy. Blood 2019, 134, 2218–2222. [Google Scholar] [CrossRef] [Green Version]
- Oberle, A.; Brandt, A.; Voigtlaender, M.; Thiele, B.; Radloff, J.; Schulenkorf, A.; Alawi, M.; Akyüz, N.; März, M.; Ford, C.T.; et al. Monitoring multiple myeloma by next-generation sequencing of V(D)J rearrangements from circulating myeloma cells and cell-free myeloma DNA. Haematologica 2017, 102, 1105–1111. [Google Scholar] [CrossRef]
- Biancon, G.; Gimondi, S.; Vendramin, A.; Carniti, C.; Corradini, P. Noninvasive molecular monitoring in multiple myeloma patients using cell-free tumor DNA: A pilot study. J. Mol. Diagn. 2018, 20, 859–870. [Google Scholar] [CrossRef] [Green Version]
- Mazzotti, C.; Buisson, L.; Maheo, S.; Perrot, A.; Chretien, M.L.; Leleu, X.; Hulin, C.; Manier, S.; Hébraud, B.; Roussel, M.; et al. Myeloma MRD by deep sequencing from circulating tumor DNA does not correlate with results obtained in the bone marrow. Blood Adv. 2018, 2, 2811–2813. [Google Scholar] [CrossRef] [Green Version]
- Landgren, O.; Owen, R.G. Better therapy requires better response evaluation: Paving the way for minimal residual disease testing for every myeloma patient. Cytom. B Clin. Cytom. 2016, 90, 14–20. [Google Scholar] [CrossRef]
- Durie, B.G.; Hoering, A.; Abidi, M.H.; Rajkumar, S.V.; Epstein, J.; Kahanic, S.P.; Thakuri, M.; Reu, F.; Reynolds, C.M.; Sexton, R.; et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem–cell transplant (SWOG S0777): A randomised, open–label, phase 3 trial. Lancet 2017, 389, 519–527. [Google Scholar] [CrossRef] [Green Version]
- Attal, M.; Lauwers-Cances, V.; Marit, G.; Caillot, D.; Moreau, P.; Facon, T.; Stoppa, A.M.; Hulin, C.; Benboubker, L.; Garderet, L.; et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N. Engl. J. Med. 2012, 366, 1782–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, P.L.; Owzar, K.; Hofmeister, C.C.; Hurd, D.D.; Hassoun, H.; Richardson, P.G.; Giralt, S.; Stadtmauer, E.A.; Weisdorf, D.J.; Vij, R.; et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N. Engl. J. Med. 2012, 366, 1770–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avet-Loiseau, H.; Ludwig, H.; Landgren, O.; Paiva, B.; Morris, C.; Yang, H.; Zhou, K.; Ro, S.; Mateos, M.V. Minimal Residual Disease Status as a Surrogate Endpoint for Progression-free Survival in Newly Diagnosed Multiple Myeloma Studies: A Meta-analysis. Clin. Lymphoma Myeloma Leuk. 2020, 20, e30–e37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandolfi, S.; Prada, C.P.; Richardson, P.G. How I treat the young patient with multiple myeloma. Blood 2018, 132, 1114–1124. [Google Scholar] [CrossRef]
- Touzeau, C.; Moreau, P.; Dumontet, C. Monoclonal antibody therapy in multiple myeloma. Leukemia 2017, 31, 1039–1047. [Google Scholar] [CrossRef]
- Paiva, B.; Vidriales, M.B.; Cervero, J.; Mateo, G.; Pérez, J.J.; Montalbán, M.A.; Sureda, A.; Montejano, L.; Gutiérrez, N.C.; de García Coca, A.; et al. Multiparameter flow cytometric remission is the most relevant prognostic factor for multiple myeloma patients who undergo autologous stem cell transplantation. Blood 2008, 112, 4017–4023. [Google Scholar] [CrossRef] [Green Version]
- Korde, N.; Roschewski, M.; Zingone, A.; Kwok, M.; Manasanch, E.E.; Bhutani, M.; Tageja, N.; Kazandjian, D.; Mailankody, S.; Wu, P.; et al. Treatment with carfilzomib–lenalidomide–dexamethasone with lenalidomide extension in patients with smoldering or newly diagnosed multiple myeloma. JAMA Oncol. 2015, 1, 746–754. [Google Scholar] [CrossRef] [Green Version]
- Lonial, S.; Anderson, K.C. Association of response endpoints with survival outcomes in multiple myeloma. Leukemia 2014, 28, 258–268. [Google Scholar] [CrossRef]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. ELOQUENT-2 Investigators. Elotuzumab therapy for relapsed or refractory multiple myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. CASTOR Investigators. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; Masszi, T.; Špička, I.; Oriol, A.; Hájek, R.; Rosiñol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2015, 372, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.; Anderson, K.C. Minimal residual disease in multiple myeloma. J. Clin. Oncol. 2013, 31, 2523–2526. [Google Scholar] [CrossRef] [Green Version]
- Mailankody, S.; Korde, N.; Lesokhin, A.M.; Lendvai, N.; Hassoun, H.; Stetler-Stevenson, M.; Landgren, O. Minimal residual disease in multiple myeloma: Bringing the bench to the bedside. Nat. Rev. Clin. Oncol. 2015, 12, 286–295. [Google Scholar] [CrossRef]
- Rawstron, A.C.; de Tute, R.M.; Haughton, J.; Owen, R.G. Measuring disease levels in myeloma using flow cytometry in combination with other laboratory techniques: Lessons from the past 20 years at the Leeds Haematological Malignancy Diagnostic Service. Cytom. B Clin. Cytom. 2016, 90, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Rawstron, A.C.; Child, J.A.; de Tute, R.M.; Davies, F.E.; Gregory, W.M.; Bell, S.E.; Szubert, A.J.; Navarro-Coy, N.; Drayson, M.T.; Feyler, S.; et al. Minimal residual disease assessed by multiparameter flow cytometry in multiple myeloma: Impact on outcome in the Medical Research Council Myeloma IX Study. J. Clin. Oncol. 2013, 31, 2540–2547. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Harousseau, J.L.; Durie, B.; Anderson, K.C.; Dimopoulos, M.; Kyle, R.; Blade, J.; Richardson, P.; Orlowski, R.; Siegel, D.; et al. Consensus recommendations for the uniform reporting of clinical trials: Report of the International Myeloma Workshop Consensus Panel. Blood 2011, 117, 4691–4695. [Google Scholar] [CrossRef] [Green Version]
- Attal, M.; Lauwers-Cances, V.; Hulin, C.; Leleu, X.; Caillot, D.; Escoffre, M.; Arnulf, B.; Macro, M.; Belhadj, K.; Garderet, L.; et al. IFM 2009 Study. Lenalidomide, Bortezomib, and Dexamethasone with Transplantation for Myeloma. N. Engl. J. Med. 2017, 376, 1311–1320. [Google Scholar] [CrossRef]
- Mateos, M.V.; Oriol, A.; López, J.M.; Teruel, A.I.; López de la Guía, A.; López, J.; Bengoechea, E.; Pérez, M.; Martínez, R.; Palomera, L.; et al. GEM2005 Trial Update Comparing VMP/VTP as Induction in Elderly Multiple Myeloma Patients: Do We Still Need Alkylators? Blood 2014, 124, 1887–1893. [Google Scholar] [CrossRef]
- Martínez-Sánchez, P.; Montejano, L.; Sarasquete, M.E.; García-Sanz, R.; Fernández-Redondo, E.; Ayala, R.; Montalbán, M.A.; Martínez, R.; García Laraña, J.; Alegre, A.; et al. Evaluation of minimal residual disease in multiple myeloma patients by fluorescent-polymerase chain reaction: The prognostic impact of achieving molecular response. Br. J. Haematol. 2008, 142, 766–774. [Google Scholar] [CrossRef]
- Martinez-Lopez, J.; Fernández-Redondo, E.; García-Sánz, R.; Montalbán, M.A.; Martínez-Sánchez, P.; Pavia, B.; Mateos, M.V.; Rosiñol, L.; Martín, M.; Ayala, R.; et al. Clinical applicability and prognostic significance of molecular response assessed by fluorescent-PCR of immunoglobulin genes in multiple myeloma. Results from a GEM/PETHEMA study. Br. J. Haematol. 2013, 163, 581–589. [Google Scholar] [CrossRef]
- Spencer, A.; Lentzsch, S.; Weisel, K.; Avet-Loiseau, H.; Mark, T.M.; Spicka, I.; Masszi, T.; Lauri, B.; Levin, M.D.; Bosi, A. Daratumumab plus bortezomib and dexamethasone versus bortezomib and dexamethasone in relapsed or refractory multiple myeloma: Updated analysis of CASTOR. Haematologica 2018, 103, 2079–2087. [Google Scholar] [CrossRef] [Green Version]
- Dimopoulos, M.A.; San-Miguel, J.; Belch, A.; White, D.; Benboubker, L.; Cook, G.; Leiba, M.; Morton, J.; Ho, P.J.; Kim, K.; et al. Daratumumab plus lenalidomide and dexamethasone versus lenalidomide and dexamethasone in relapsed or refractory multiple myeloma: Updated analysis of POLLUX. Haematologica 2018, 103, 2088–2096. [Google Scholar] [CrossRef] [Green Version]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Béné, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): A randomised, open-label, phase 3 study. Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef]
- Mills, J.R.; Barnidge, D.R.; Dispenzieri, A.; Murray, D.L. High sensitivity blood-based M-protein detection in sCR patients with multiple myeloma. Blood Cancer J. 2017, 7, e590. [Google Scholar] [CrossRef] [PubMed]
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N. Engl. J. Med. 2019, 380, 2104–2115. [Google Scholar] [CrossRef] [PubMed]
- Kazandjian, D.; Korde, N.; Mailankody, S.; Hill, E.; Figg, W.D.; Roschewski, M.; Landgren, O. Remission and Progression-Free Survival in Patients with Newly Diagnosed Multiple Myeloma Treated With Carfilzomib, Lenalidomide, and Dexamethasone: Five-Year Follow-up of a Phase 2 Clinical Trial. JAMA Oncol. 2018, 4, 1781–1783. [Google Scholar] [CrossRef]
- Landgren, O.; Giralt, S. MRD-driven treatment paradigm for newly diagnosed transplant eligible multiple myeloma patients. Bone Marrow Transplant. 2016, 51, 913–914. [Google Scholar] [CrossRef]
- Luoma, S.; Anttila, P.; Säily, M.; Lundan, T.; Heskanen, J.; Siitonen, T.; Kakko, S.; Putkonen, M.; Ollikainen, H.; Terävä, V.; et al. RVD induction and autologous stem cell transplantation followed by lenalidomide maintenance in newly diagnosed multiple myeloma: A phase 2 study of the Finnish Myeloma Group. Ann. Hematol. 2019, 98, 2781–2792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roussel, M.; Lauwers-Cances, V.; Robillard, N.; Hulin, C.; Leleu, X.; Benboubker, L.; Marit, G.; Moreau, P.; Pegourie, B.; Caillot, D.; et al. Front-Line Transplantation Program With Lenalidomide, Bortezomib, and Dexamethasone Combination As Induction and Consolidation Followed by Lenalidomide Maintenance in Patients With Multiple Myeloma: A Phase II Study by the Intergroupe Francophone du Myelome. J. Clin. Oncol. 2014, 32, 2712–2717. [Google Scholar] [CrossRef]
- Cavo, M.; Gay, F.; Beksac, M.; Pantani, L.; Petrucci, M.T.; Dimopoulos, M.A.; Dozza, L.; van der Holt, B.; Zweegman, S.; Oliva, S.; et al. Autologous haematopoietic stem-cell transplantation versus bortezomib–melphalan–prednisone, with or without bortezomib–lenalidomide–dexamethasone consolidation therapy, and lenalidomide maintenance for newly diagnosed multiple myeloma (EMN02/HO95): A multicentre, randomised, open-label, phase 3 study. Lancet Haematol. 2020, 7, e456–e468. [Google Scholar] [CrossRef] [PubMed]
- Gozzetti, A.; Raspadori, D.; Bacchiarri, F.; Pacelli, P.; Di Martino, F.; Liberati, A.M.; Sicuranza, A.; Lombardo, A.; Caffarelli, C.; Antonioli, E.; et al. DART4MM: Daratumumab as consolidation therapy in patients who already achieved optimal response/MRD positivity by next generation flow (NGF): Preliminary results of a phase 2 multicenter study. Clin. Lymphoma Myeloma Leuk. 2019, 19, e161–e162. [Google Scholar] [CrossRef]
- Munshi, N.C.; Avet-Loiseau, H.; Rawstron, A.C.; Owen, R.G.; Child, J.A.; Thakurta, A.; Sherrington, P.; Samur, M.K.; Georgieva, A.; Anderson, K.C.; et al. Association of minimal residual disease with superior survival outcomes in patients with multiple myeloma: A metaanalysis. JAMA Oncol. 2017, 3, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.Z.; Mitchell, A.; Waheed, S.; Crowley, J.; Hoering, A.; Petty, N.; Brown, T.; Bartel, T.; Anaissie, E.; van Rhee, F.; et al. Prognostic implications of serial 18-fluoro-deoxyglucose emission tomography in multiple myeloma treated with total therapy 3. Blood 2013, 121, 1819–1823. [Google Scholar] [CrossRef] [Green Version]
- Bartel, T.B.; Haessler, J.; Brown, T.L.; Shaughnessy Jr, J.D.; van Rhee, F.; Anaissie, E.; Alpe, T.; Angtuaco, E.; Walker, R.; Epstein, J.; et al. F18-fluorodeoxyglucose positron emission tomography in the context of other imaging techniques and prognostic factors in multiple myeloma. Blood 2009, 114, 2068–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Li, F.; Zhou, X.; Mei, J.; Song, P.; An, Z.; Zhao, Q.; Guo, X.; Wang, X.; Zhai, Y. Achieving minimal residual disease-negative by multiparameter flow cytometry may ameliorate a poor prognosis in MM patients with high-risk cytogenetics: A retrospective single-center analysis. Ann. Hematol. 2019, 98, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Jamet, B.; Zamagni, E.; Nanni, C.; Bailly, C.; Carlier, T.; Touzeau, C.; Michaud, A.V.; Moreau, P.; Bodet-Milin, C.; Kraeber-Bodere, F. Functional Imaging for Therapeutic Assessment and Minimal Residual Disease Detection in Multiple Myeloma. Int. J. Mol. Sci. 2020, 21, 5406. [Google Scholar] [CrossRef]
- Zamagni, E.; Patriarca, F.; Nanni, C.; Zannetti, B.; Englaro, E.; Pezzi, A.; Tacchetti, P.; Buttignol, S.; Perrone, G.; Brioli, A.; et al. Prognostic relevance of 18-F FDG PET/CT in newly diagnosed multiple myeloma patients treated with up-front autologous transplantation. Blood 2011, 118, 5989–5995. [Google Scholar] [CrossRef] [Green Version]
- Moreau, P.; Attal, M.; Caillot, D.; Macro, M.; Karlin, L.; Garderet, L.; Facon, T.; Benboubker, L.; Escoffre-Barbe, M.; Stoppa, A.M.; et al. Prospective evaluation of MRI and PET-CT at diagnosis and before maintenance therapy in symptomatic patients with multiple myeloma included in the IFM/DFCI 2009 trial. J. Clin. Oncol. 2017, 35, 2911–2918. [Google Scholar] [CrossRef]
- Terpos, E.; Kostopoulos, I.V.; Kastritis, E.; Ntanasis-Stathopoulos, I.; Migkou, M.; Rousakis, P.; Argyriou, A.T.; Kanellias, N.; Fotiou, D.; Eleutherakis-Papaiakovou, E.; et al. Impact of Minimal Residual Disease Detection by Next-Generation Flow Cytometry in Multiple Myeloma Patients with Sustained Complete Remission after Frontline Therapy. HemaSphere 2019, 3, e300. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Author | NGF | NGS | |||
|---|---|---|---|---|---|
| 1 × 10−4 | 1 × 10−5 | 1 × 10−6 | 1 × 10−5 | 1 × 10−6 | |
| Paiva et al. [17] | * | * | * | ||
| Flores-Montero et al. [18] | * | * | |||
| Oliva et al. [30] | * | * | |||
| Rawstron et al. [21] | * | ||||
| Bai et al. [23] | * | * | |||
| Yao et al. [24] | * | * | * | ||
| Perrot et al. [27] | * | ||||
| Avet-Loiseau et al. [28] | * | ||||
| Martinez-Lopez et al. [29] | * | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gozzetti, A.; Raspadori, D.; Bacchiarri, F.; Sicuranza, A.; Pacelli, P.; Ferrigno, I.; Tocci, D.; Bocchia, M. Minimal Residual Disease in Multiple Myeloma: State of the Art and Applications in Clinical Practice. J. Pers. Med. 2020, 10, 120. https://doi.org/10.3390/jpm10030120
Gozzetti A, Raspadori D, Bacchiarri F, Sicuranza A, Pacelli P, Ferrigno I, Tocci D, Bocchia M. Minimal Residual Disease in Multiple Myeloma: State of the Art and Applications in Clinical Practice. Journal of Personalized Medicine. 2020; 10(3):120. https://doi.org/10.3390/jpm10030120
Chicago/Turabian StyleGozzetti, Alessandro, Donatella Raspadori, Francesca Bacchiarri, Anna Sicuranza, Paola Pacelli, Ilaria Ferrigno, Dania Tocci, and Monica Bocchia. 2020. "Minimal Residual Disease in Multiple Myeloma: State of the Art and Applications in Clinical Practice" Journal of Personalized Medicine 10, no. 3: 120. https://doi.org/10.3390/jpm10030120