Deficits in Mitochondrial Spare Respiratory Capacity Contribute to the Neuropsychological Changes of Alzheimer’s Disease

 , ,
, ,
Abstract
:1. Background
2. Results
2.1. Patient Demographic Details
2.2. Neuropsychological Profiles
2.3. Neuroimaging Profiles
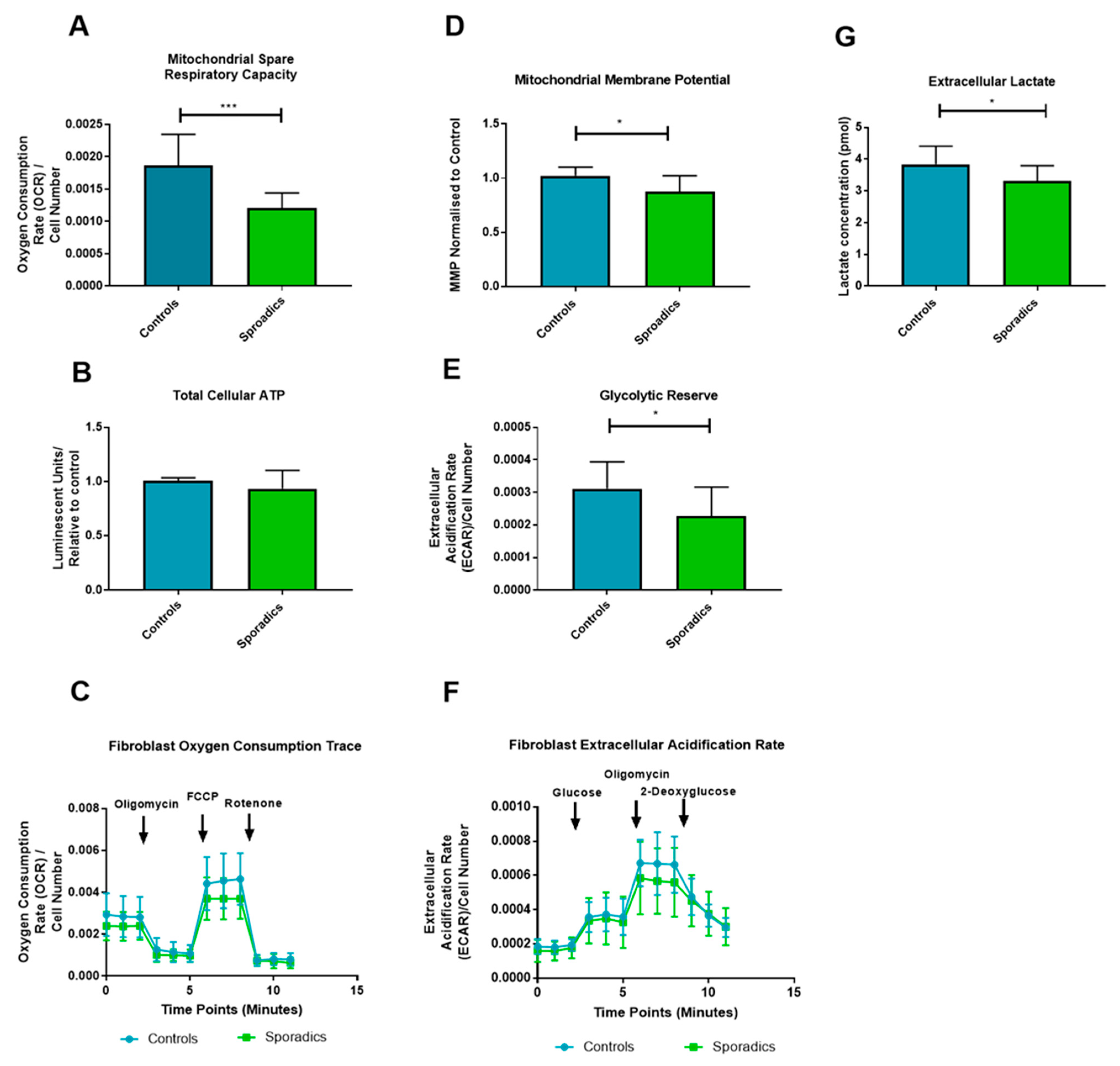
2.4. Fibroblast Metabolic Assessment
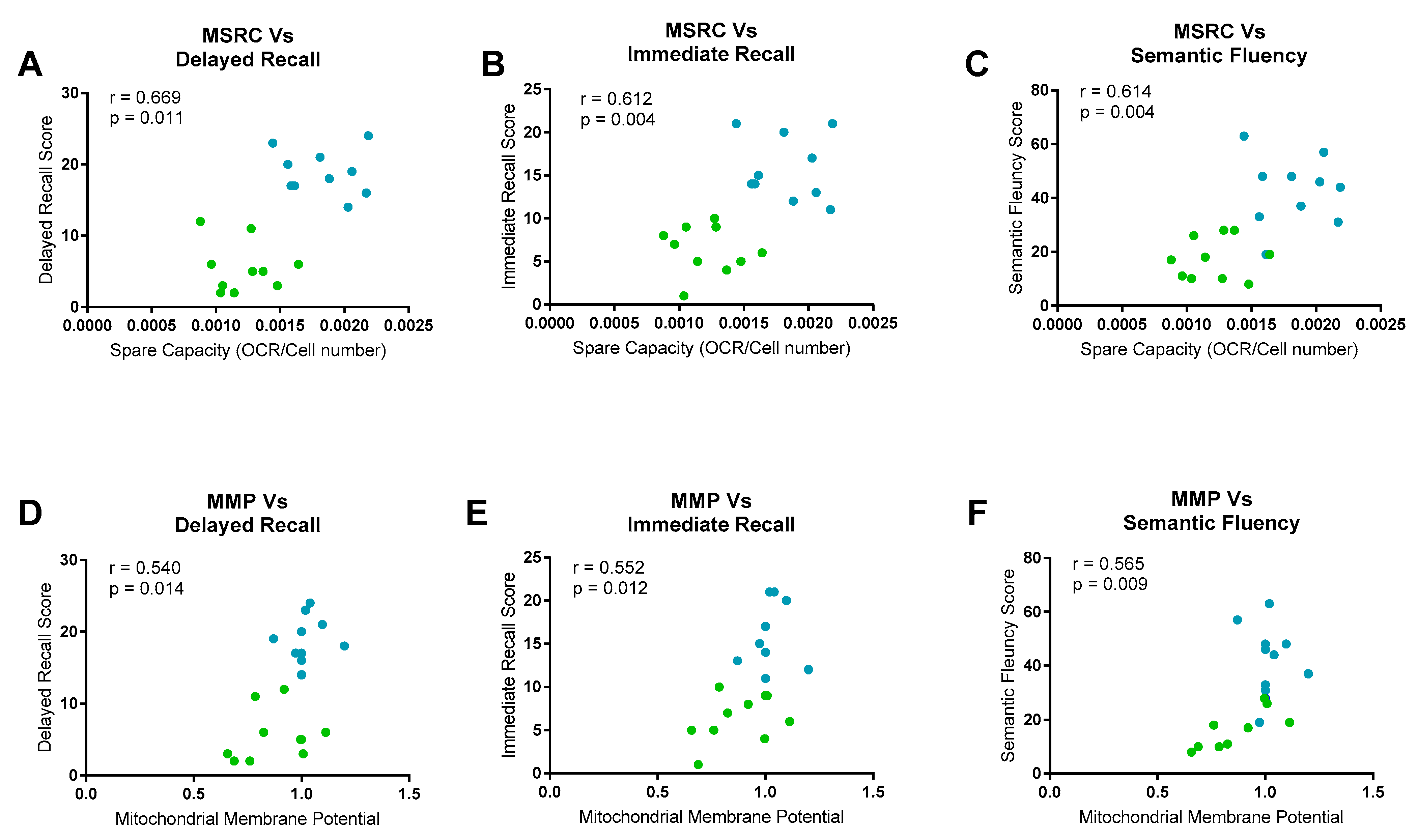
2.5. Neuropsychological/Metabolic Correlations
2.6. Neuroimaging/Metabolic Correlations
3. Discussion
4. Methods
4.1. Patient Details
4.2. Neuropsychological Testing
4.3. MRI Acquisition and Processing
4.4. Tissue Culture
4.5. Intracellular ATP levels
4.6. Mitochondrial Membrane Potential
4.7. Extracellular Lactate Measurement
4.8. Metabolic Flux Assay
4.8.1. Mito Stress Test
4.8.2. Glycolysis Stress Test
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wimo, A.; Guerchet, M.; Ali, G.-C.; Wu, Y.-T.; Prina, A.M.; Winblad, B.; Jönsson, L.; Liu, Z.; Prince, M. The worldwide costs of dementia 2015 and comparisons with 2010. Alzheimer’s Dement. 2017, 13, 1–7. [Google Scholar] [CrossRef]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Brookmeyer, R.; Corrada, M.M.; Curriero, F.C.; Kawas, C. Survival following a diagnosis of Alzheimer disease. Arch. Neurol. 2002, 59, 1764–1767. [Google Scholar] [CrossRef]
- Perl, D.P. Neuropathology of Alzheimer’s Disease. Mt. Sinai J. Med. 2010, 77, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honig, L.S.; Vellas, B.; Woodward, M.; Boada, M.; Bullock, R.; Borrie, M.; Hager, K.; Andreasen, N.; Scarpini, E.; Liu-Seifert, H.; et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [Green Version]
- ALZFORUM. Topline Result for First DIAN-TU Clinical Trial: Negative on Primary. Available online: https://www.alzforum.org/news/research-news/topline-result-first-dian-tu-clinical-trial-negative-primary (accessed on 29 March 2020).
- Savva, G.M.; Wharton, S.B.; Ince, P.G.; Forster, G.; Matthews, F.E.; Brayne, C. Age, neuropathology, and dementia. N. Engl. J. Med. 2009, 360, 2302–2309. [Google Scholar] [CrossRef]
- Le Couteur, D.G.; Hunter, S.; Brayne, C. Solanezumab and the amyloid hypothesis for Alzheimer’s disease. BMJ 2016, 355, i6771. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Haroutunian, V.; Schnaider-Beeri, M.; Schmeidler, J.; Wysocki, M.; Purohit, D.P.; Perl, D.P.; Libow, L.S.; Lesser, G.T.; Maroukian, M.; Grossman, H.T. Role of the neuropathology of Alzheimer disease in dementia in the oldest-old. Arch. Neurol. 2008, 65, 1211–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weintraub, S.; Wicklund, A.H.; Salmon, D.P. The neuropsychological profile of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006171. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. A Cellular Perspective on Brain Energy Metabolism and Functional Imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [Green Version]
- Ewers, M.; Brendel, M.; Rizk-Jackson, A.; Rominger, A.; Bartenstein, P.; Schuff, N.; Weiner, M.W. Alzheimer’s Disease Neuroimaging I Reduced FDG-PET brain metabolism and executive function predict clinical progression in elderly healthy subjects. NeuroImage. Clin. 2013, 4, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Kalpouzos, G.; Eustache, F.; de la Sayette, V.; Viader, F.; Chételat, G.; Desgranges, B. Working memory and FDG–PET dissociate early and late onset Alzheimer disease patients. J. Neurol. 2005, 252, 548–558. [Google Scholar] [CrossRef]
- Ishii, K.; Sasaki, M.; Yamaji, S.; Sakamoto, S.; Kitagaki, H.; Mori, E. Demonstration of decreased posterior cingulate perfusion in mild Alzheimer’s disease by means of H215O positron emission tomography. Eur. J. Nucl. Med. 1997, 24, 670–673. [Google Scholar]
- Morgen, K.; Frolich, L. The metabolism hypothesis of Alzheimer’s disease: From the concept of central insulin resistance and associated consequences to insulin therapy. J. Neural Transm. 2015, 122, 499–504. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670. [Google Scholar] [CrossRef] [Green Version]
- Blass, J.P.; Sheu, R.K.; Gibson, G.E. Inherent abnormalities in energy metabolism in Alzheimer disease. Interaction with cerebrovascular compromise. Ann. N. Y. Acad. Sci. 2000, 903, 204–221. [Google Scholar] [CrossRef]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507.e496. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef]
- Hartl, D.; Schuldt, V.; Forler, S.; Zabel, C.; Klose, J.; Rohe, M. Presymptomatic Alterations in Energy Metabolism and Oxidative Stress in the APP23 Mouse Model of Alzheimer Disease. J. Proteome Res. 2012, 11, 3295–3304. [Google Scholar] [CrossRef]
- Gan, X.Q.; Huang, S.B.; Wu, L.; Wang, Y.F.; Hu, G.; Li, G.Y.; Zhang, H.J.; Yu, H.Y.; Swerdlow, R.H.; Chen, J.X.; et al. Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosconi, L.; Tsui, W.H.; De Santi, S.; Li, J.; Rusinek, H.; Convit, A.; Li, Y.; Boppana, M.; de Leon, M.J. Reduced hippocampal metabolism in MCI and AD: Automated FDG-PET image analysis. Neurology 2005, 64, 1860–1867. [Google Scholar] [CrossRef] [PubMed]
- Vlassenko, A.G.; Vaishnavi, S.N.; Couture, L.; Sacco, D.; Shannon, B.J.; Mach, R.H.; Morris, J.C.; Raichle, M.E.; Mintun, M.A. Spatial correlation between brain aerobic glycolysis and amyloid-beta (Abeta) deposition. Proc. Natl. Acad. Sci. USA 2010, 107, 17763–17767. [Google Scholar] [CrossRef] [Green Version]
- Vlassenko, A.G.; Gordon, B.A.; Goyal, M.S.; Su, Y.; Blazey, T.M.; Durbin, T.J.; Couture, L.E.; Christensen, J.J.; Jafri, H.; Morris, J.C.; et al. Aerobic glycolysis and tau deposition in preclinical Alzheimer’s disease. Neurobiol. Aging 2018, 67, 95–98. [Google Scholar] [CrossRef]
- Bell, S.M.; Barnes, K.; Clemmens, H.; Al-Rafiah, A.R.; Al-Ofi, E.A.; Leech, V.; Bandmann, O.; Shaw, P.J.; Blackburn, D.J.; Ferraiuolo, L.; et al. Ursodeoxycholic Acid Improves Mitochondrial Function and Redistributes Drp1 in Fibroblasts from Patients with Either Sporadic or Familial Alzheimer’s Disease. J. Mol. Biol. 2018, 430, 3942–3953. [Google Scholar] [CrossRef]
- Pickart, L.; Vasquez-Soltero, J.M.; Margolina, A. The Effect of the Human Peptide GHK on Gene Expression Relevant to Nervous System Function and Cognitive Decline. Brain Sci. 2017, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Desler, C.; Hansen, T.L.; Frederiksen, J.B.; Marcker, M.L.; Singh, K.K.; Juel Rasmussen, L. Is There a Link between Mitochondrial Reserve Respiratory Capacity and Aging? J. Aging Res. 2012, 2012, 192503. [Google Scholar] [CrossRef] [Green Version]
- Martino Adami, P.V.; Quijano, C.; Magnani, N.; Galeano, P.; Evelson, P.; Cassina, A.; Do Carmo, S.; Leal, M.C.; Castaño, E.M.; Cuello, A.C.; et al. Synaptosomal bioenergetic defects are associated with cognitive impairment in a transgenic rat model of early Alzheimer’s disease. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2017, 37, 69–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capitani, E.; Rosci, C.; Saetti, M.C.; Laiacona, M. Mirror asymmetry of Category and Letter fluency in traumatic brain injury and Alzheimer’s patients. Neuropsychologia 2009, 47, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Doody, R.S.; Massman, P.; Dunn, J.K. A method for estimating progression rates in Alzheimer disease. Arch. Neurol. 2001, 58, 449–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scahill, R.I.; Schott, J.M.; Stevens, J.M.; Rossor, M.N.; Fox, N.C. Mapping the evolution of regional atrophy in Alzheimer’s disease: Unbiased analysis of fluid-registered serial MRI. Proc. Natl. Acad. Sci. USA 2002, 99, 4703–4707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boller, B.; Mellah, S.; Ducharme-Laliberte, G.; Belleville, S. Relationships between years of education, regional grey matter volumes, and working memory-related brain activity in healthy older adults. Brain Imaging Behav. 2017, 11, 304–317. [Google Scholar] [CrossRef]
- Rzezak, P.; Squarzoni, P.; Duran, F.L.; de Toledo Ferraz Alves, T.; Tamashiro-Duran, J.; Bottino, C.M.; Ribeiz, S.; Lotufo, P.A.; Menezes, P.R.; Scazufca, M.; et al. Relationship between Brain Age-Related Reduction in Gray Matter and Educational Attainment. PLoS ONE 2015, 10, e0140945. [Google Scholar] [CrossRef]
- Harada, C.N.; Natelson Love, M.C.; Triebel, K.L. Normal cognitive aging. Clin. Geriatr. Med. 2013, 29, 737–752. [Google Scholar] [CrossRef] [Green Version]
- Stern, Y. Cognitive reserve: Implications for assessment and intervention. Folia Phoniatr. Logop. Off. Organ. Int. Assoc. Logop. Phoniatr. IALP 2013, 65, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Schniertshauer, D.; Gebhard, D.; Bergemann, J. Age-Dependent Loss of Mitochondrial Function in Epithelial Tissue Can Be Reversed by Coenzyme Q10. J. Aging Res. 2018, 2018, 6354680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriskanthadevan, S.; Jeyaraju, D.V.; Chung, T.E.; Prabha, S.; Xu, W.; Skrtic, M.; Jhas, B.; Hurren, R.; Gronda, M.; Wang, X.; et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood 2015, 125, 2120–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadava, N.; Nicholls, D.G. Spare Respiratory Capacity Rather Than Oxidative Stress Regulates Glutamate Excitotoxicity after Partial Respiratory Inhibition of Mitochondrial Complex I with Rotenone. J. Neurosci. 2007, 27, 7310–7317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortiboys, H.; Johansen, K.K.; Aasly, J.O.; Bandmann, O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 2010, 75, 2017–2020. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Pasinelli, P.; Trotti, D. Role of mitochondria in mutant SOD1 linked amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2014, 1842, 1295–1301. [Google Scholar] [CrossRef] [Green Version]
- Carling, P.J.; Mortiboys, H.; Green, C.; Mihaylov, S.; Sandor, C.; Schwartzentruber, A.; Taylor, R.; Wei, W.; Hastings, C.; Wong, S.; et al. Deep phenotyping of peripheral tissue facilitates mechanistic disease stratification in sporadic Parkinson’s disease. Prog. Neurobiol. 2020, 187, 101772. [Google Scholar] [CrossRef]
- Milanese, C.; Payán-Gómez, C.; Galvani, M.; Molano González, N.; Tresini, M.; Nait Abdellah, S.; van Roon-Mom, W.M.; Figini, S.; Marinus, J.; van Hilten, J.J.; et al. Peripheral mitochondrial function correlates with clinical severity in idiopathic Parkinson’s disease. Mov. Disord. 2019, 34, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, E.; Armour, S.M.; Harris, M.H.; Thompson, C.B. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003, 10, 709–717. [Google Scholar] [CrossRef]
- Grogan, A.; Green, D.W.; Ali, N.; Crinion, J.T.; Price, C.J. Structural correlates of semantic and phonemic fluency ability in first and second languages. Cereb. Cortex 2009, 19, 2690–2698. [Google Scholar] [CrossRef] [Green Version]
- Venneri, A.; Jahn-Carta, C.; De Marco, M.; Quaranta, D.; Marra, C. Diagnostic and prognostic role of semantic processing in preclinical Alzheimer’s disease. Biomark. Med. 2018, 12, 637–651. [Google Scholar] [CrossRef] [Green Version]
- Baldo, J.V.; Schwartz, S.; Wilkins, D.; Dronkers, N.F. Role of frontal versus temporal cortex in verbal fluency as revealed by voxel-based lesion symptom mapping. J. Int. Neuropsychol. Soc. 2006, 12, 896–900. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.S.; Reiman, E.M.; Valla, J.; Dunckley, T.; Beach, T.G.; Grover, A.; Niedzielko, T.L.; Schneider, L.E.; Mastroeni, D.; Caselli, R.; et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 4441–4446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosconi, L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 486–510. [Google Scholar] [CrossRef] [PubMed]
- Ramanoël, S.; Hoyau, E.; Kauffmann, L.; Renard, F.; Pichat, C.; Boudiaf, N.; Krainik, A.; Jaillard, A.; Baciu, M. Gray Matter Volume and Cognitive Performance During Normal Aging. A Voxel-Based Morphometry Study. Front. Aging Neurosci. 2018, 10, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, S.; Hejl, A.M.; Dinh, T.S.T.; Keijzers, G.; Hansen, Å.M.; Desler, C.; Moreno-Villanueva, M.; Bürkle, A.; Rasmussen, L.J.; Waldemar, G.; et al. Defective mitochondrial respiration, altered dNTP pools and reduced AP endonuclease 1 activity in peripheral blood mononuclear cells of Alzheimer’s disease patients. Aging 2015, 7, 793. [Google Scholar] [CrossRef] [Green Version]
- Fisar, Z.; Hroudová, J.; Hansíková, H.; Lelková, P.; Wenchich, L.; Jirák, R.; Zeman, J.; Martásek, P.; Raboch, J. Mitochondrial Respiration in the Platelets of Patients with Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 930–941. [Google Scholar] [CrossRef]
- Martin-Maestro, P.; Gargini, R.; Garcia, E.; Perry, G.; Avila, J. Slower Dynamics and Aged Mitochondria in Sporadic Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2017, 2017, 9302761. [Google Scholar] [CrossRef] [Green Version]
- Sonntag, K.C.; Ryu, W.I.; Amirault, K.M.; Healy, R.A.; Siegel, A.J.; McPhie, D.L.; Forester, B.; Cohen, B.M. Late-onset Alzheimer’s disease is associated with inherent changes in bioenergetics profiles. Sci. Rep. 2017, 7, 14038. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Mertens, J.; Paquola, A.C.; Ku, M.; Hatch, E.; Böhnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef] [Green Version]
- Wakefield, S.J.; McGeown, W.J.; Shanks, M.F.; Venneri, A. Differentiating normal from pathological brain ageing using standard neuropsychological tests. Curr. Alzheimer Res. 2014, 11, 765–772. [Google Scholar] [CrossRef] [Green Version]
- De Marco, M.; Duzzi, D.; Meneghello, F.; Venneri, A. Cognitive Efficiency in Alzheimer’s Disease is Associated with Increased Occipital Connectivity. J. Alzheimers Dis. 2017, 57, 541–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, M.J.; Leung, K.; Modat, M.; Keihaninejad, S.; Cash, D.; Barnes, J.; Fox, N.C.; Ourselin, S. STEPS: Similarity and Truth Estimation for Propagated Segmentations and its application to hippocampal segmentation and brain parcelation. Med. Image Anal. 2013, 17, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, A.; Vijayakumar, A. Comparison of hippocampal volume in dementia subtypes. ISRN Radiol. 2013, 2013, 174524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortiboys, H.; Thomas, K.J.; Koopman, W.J.; Klaffke, S.; Abou-Sleiman, P.; Olpin, S.; Wood, N.W.; Willems, P.H.; Smeitink, J.A.; Cookson, M.R. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann. Neurol. 2008, 64, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Agilent Technologies, Agilent Seahorse XF Glycolysis Stress Test Kit. Available online: https://www.agilent.com/cs/library/usermanuals/public/XF_Glycolysis_Stress_Test_Kit_User_Guide.pdf (accessed on 29 March 2020).




{kind=link}
{kind=link}
{kind=link}
| Patient Number | Age (Years) | Sex | MMSE | Length of Education (Years) | AD Treatment (in Disease Cohort, at Time of Biopsy) |
|---|---|---|---|---|---|
| 1 | 63 | Male | 20 | 16 | None |
| 2 | 57 | Male | 18 | 15 | Donepezil |
| 3 | 53 | Male | 14 | 11 | Donepezil |
| 4 * | 60 | Male | 18 | 9 | None |
| 5 * | 59 | Female | 23 | 11 | Galantamine |
| 6 * | 63 | Female | 26 | 10 | Donepezil |
| 7 * | 60 | Male | 18 | 11 | Memantine |
| 8 | 60 | Male | 18 | 11 | None |
| 9 | 79 | Female | 28 | 15 | None |
| 10 | 61 | Female | 25 | 11 | Donepezil |
| Group Mean (Standard Dev) | 61.33 (7.19) | 20.8 (4.4) | 12.0 (2.4) | ||
| Controls | |||||
| 1 | 66 | Male | 29 | 11 | NA |
| 2 * | 54 | Male | 27 | 17 | NA |
| 3 * | 53 | Male | 29 | 16 | NA |
| 4 * | 56 | Male | 24 | 11 | NA |
| 5 | 61 | Female | 30 | NA | NA |
| 6 | 54 | Female | 29 | 17 | NA |
| 7 * | 100 | Female | 24 # | 14 | NA |
| 8 | 75 | Female | 28 | 18 | NA |
| 9 | 73 | Female | 26 | 12 | NA |
| 10 | 75 | Male | 27 | 10 | NA |
| Group Mean (Standard Dev) | 65.77 (14.7) | 27.6 (1.8) | 14 (3.1) |
| Brain Volume | F-Test | p-Value |
|---|---|---|
| Left Hippocampal Volume | 9.420 | 0.001 |
| Left Parietal Volume | 7.882 | 0.002 |
| Right Parietal Volume | 10.051 | <0.0001 |
| Anterior Medial Prefrontal Cortex | 0.056 | 0.017 |
| Posterior Cingulate Cortex | 0.752 | 0.575 |
| Psychological Test | Fibroblast Marker | R Value | p-Value |
|---|---|---|---|
| Immediate Episodic Recall | |||
| MSRC | 0.605 | 0.013 | |
| MMP | 0.196 | 0.466 | |
| Delayed Episodic Recall | |||
| MSRC | 0.695 | 0.003 | |
| MMP | 0.204 | 0.448 | |
| Semantic Memory | |||
| MSRC | 0.610 | 0.012 | |
| MMP | 0.345 | 0.191 |
| Grey Matter Volume | Fibroblast Marker | R Value | p-Value |
|---|---|---|---|
| Left Hippocampal Volume | |||
| MSRC | 0.371 | 0.157 | |
| MMP | 0.041 | 0.881 | |
| Left Parietal Volume | |||
| MSRC | 0.341 | 0.196 | |
| MMP | −0.047 | 0.862 | |
| Right Parietal Volume | |||
| MSRC | 0.418 | 0.107 | |
| MMP | −0.043 | 0.875 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bell, S.M.; De Marco, M.; Barnes, K.; Shaw, P.J.; Ferraiuolo, L.; Blackburn, D.J.; Mortiboys, H.; Venneri, A. Deficits in Mitochondrial Spare Respiratory Capacity Contribute to the Neuropsychological Changes of Alzheimer’s Disease. J. Pers. Med. 2020, 10, 32. https://doi.org/10.3390/jpm10020032
Bell SM, De Marco M, Barnes K, Shaw PJ, Ferraiuolo L, Blackburn DJ, Mortiboys H, Venneri A. Deficits in Mitochondrial Spare Respiratory Capacity Contribute to the Neuropsychological Changes of Alzheimer’s Disease. Journal of Personalized Medicine. 2020; 10(2):32. https://doi.org/10.3390/jpm10020032
Chicago/Turabian StyleBell, Simon M., Matteo De Marco, Katy Barnes, Pamela J. Shaw, Laura Ferraiuolo, Daniel J. Blackburn, Heather Mortiboys, and Annalena Venneri. 2020. "Deficits in Mitochondrial Spare Respiratory Capacity Contribute to the Neuropsychological Changes of Alzheimer’s Disease" Journal of Personalized Medicine 10, no. 2: 32. https://doi.org/10.3390/jpm10020032