
The Genomic and Transcriptomic Analyses of Floccularia luteovirens, a Rare Edible Fungus in the Qinghai–Tibet Plateau, Provide Insights into the Taxonomy Placement and Fruiting Body Formation



Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation of F. luteovirens and Cultivation Method
2.2. Library Preparation and High-Throughput Sequencing
2.3. Transcriptome Reads Mapping
2.4. Analysis of Genome Composition
2.5. Gene Functional Annotation
2.6. Phylogenetic and Comparative Genomic Analyses
2.7. Differential Expression Analysis and Functional Enrichment
2.8. RT-qPCR Validation
2.9. Data Availability
3. Results
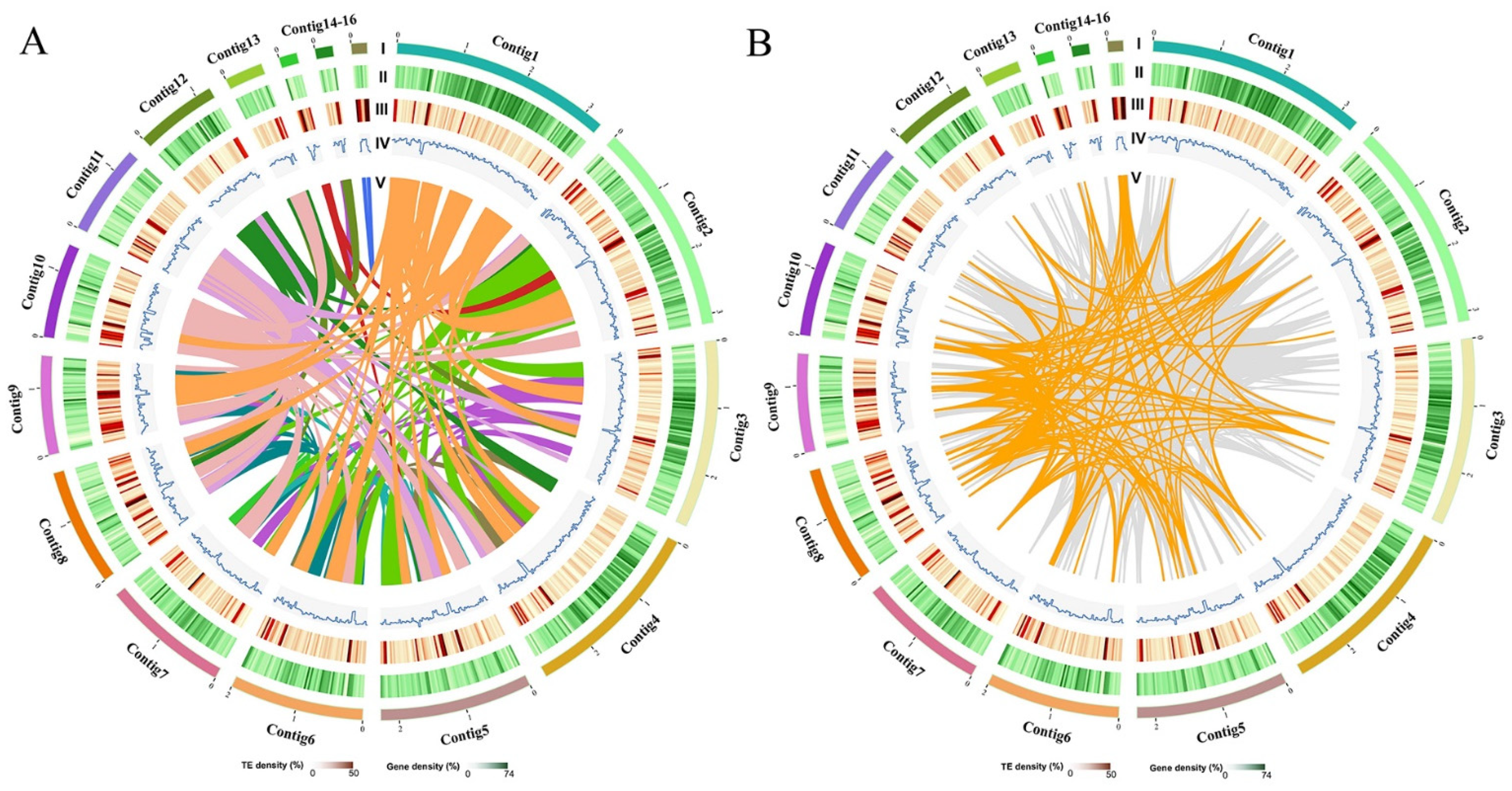
3.1. Strain Isolation and General Genome Features of F. luteovirens
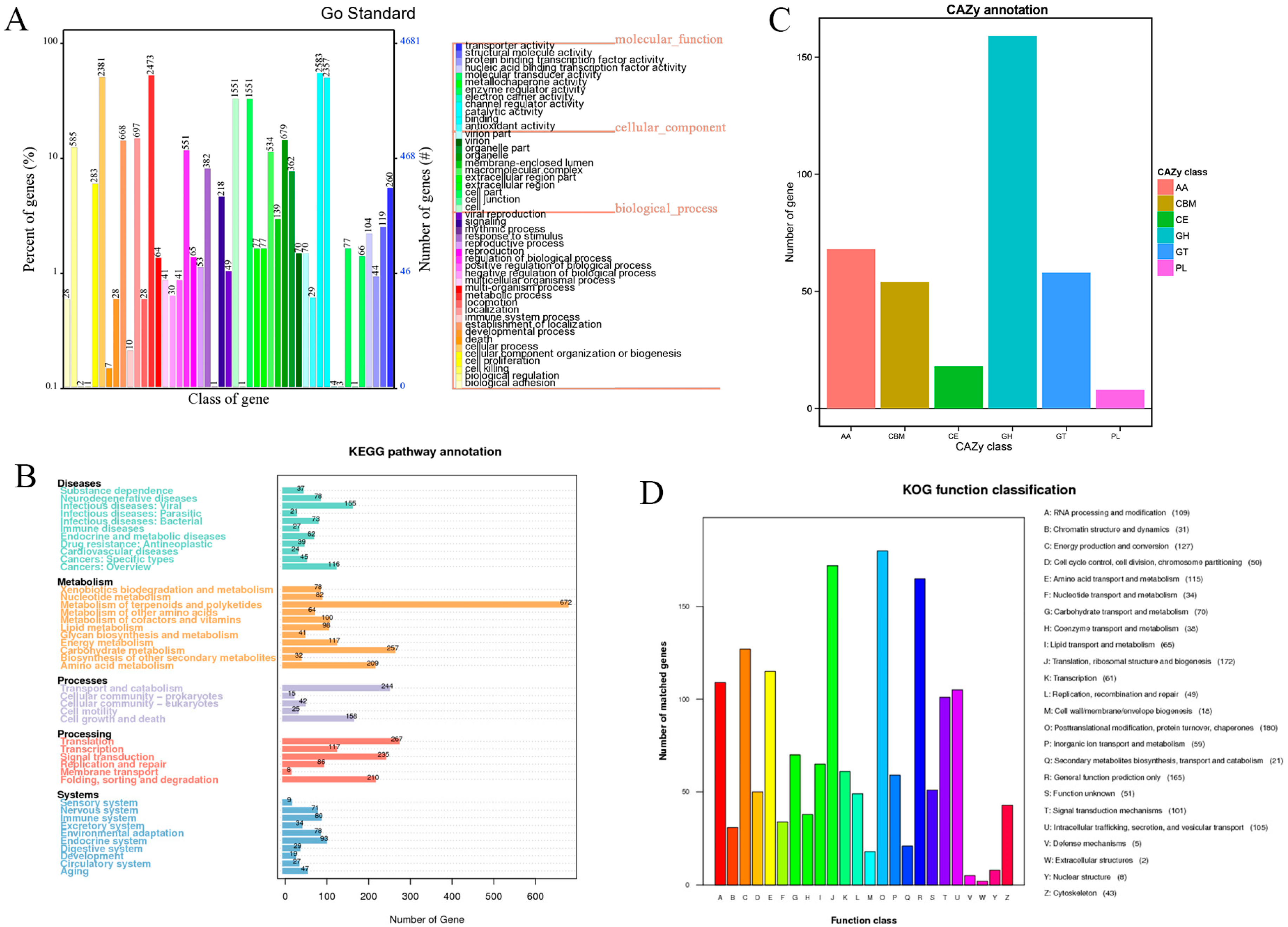
3.2. Gene Function Annotation and Analysis
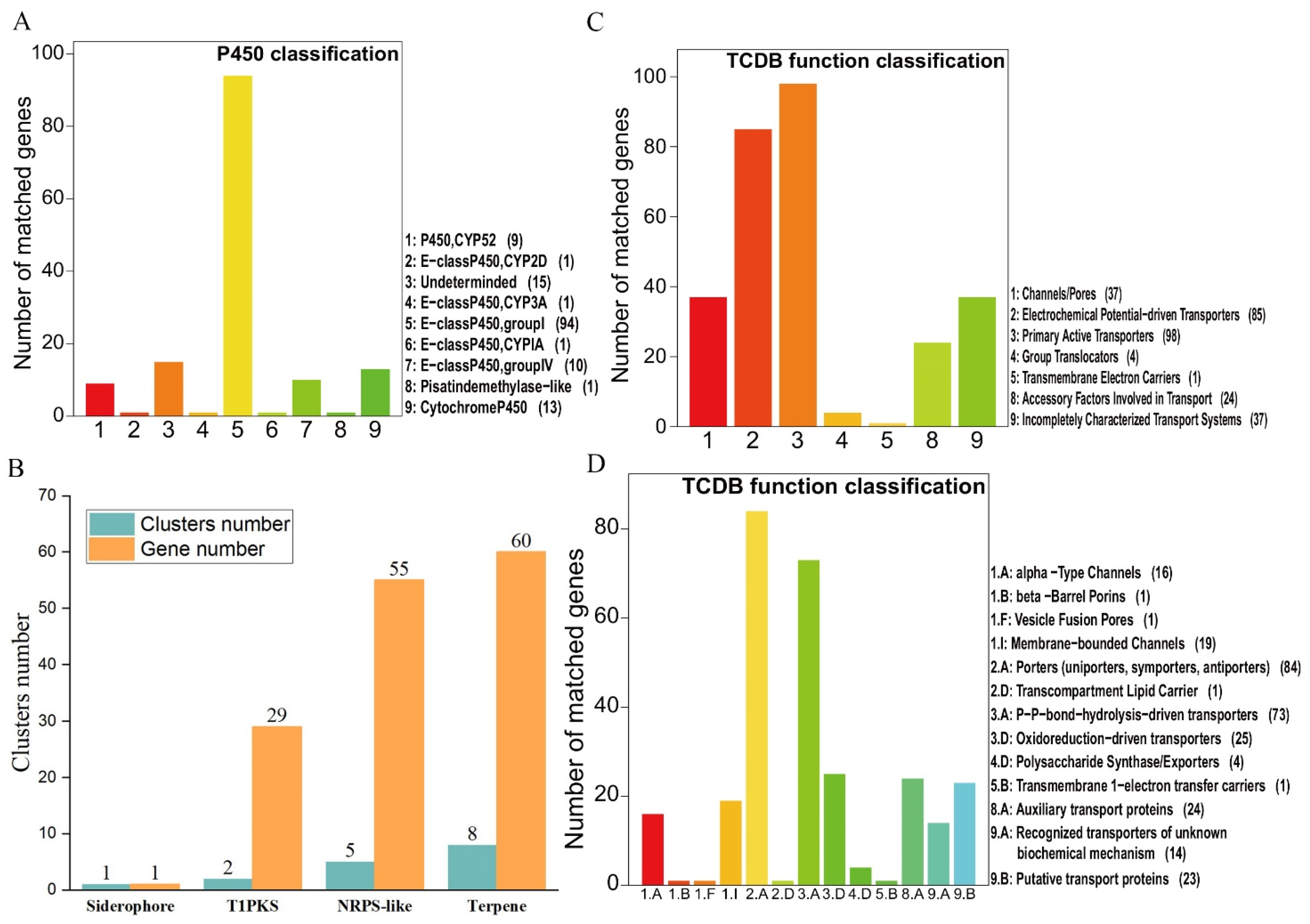
3.3. Cytochrome P450s and Transporters
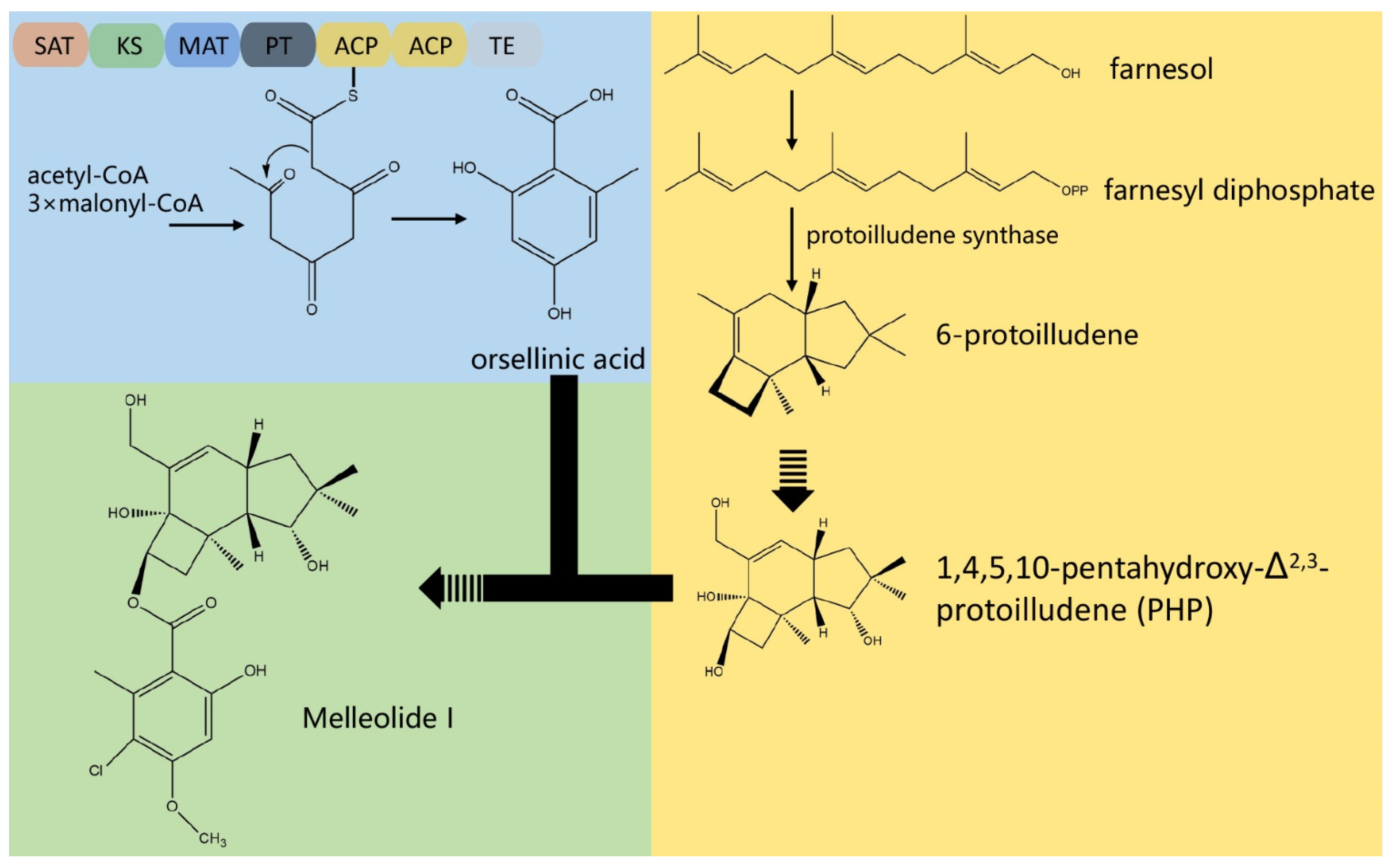
3.4. The Composition of Biosynthesis Gene Clusters (BGCs)
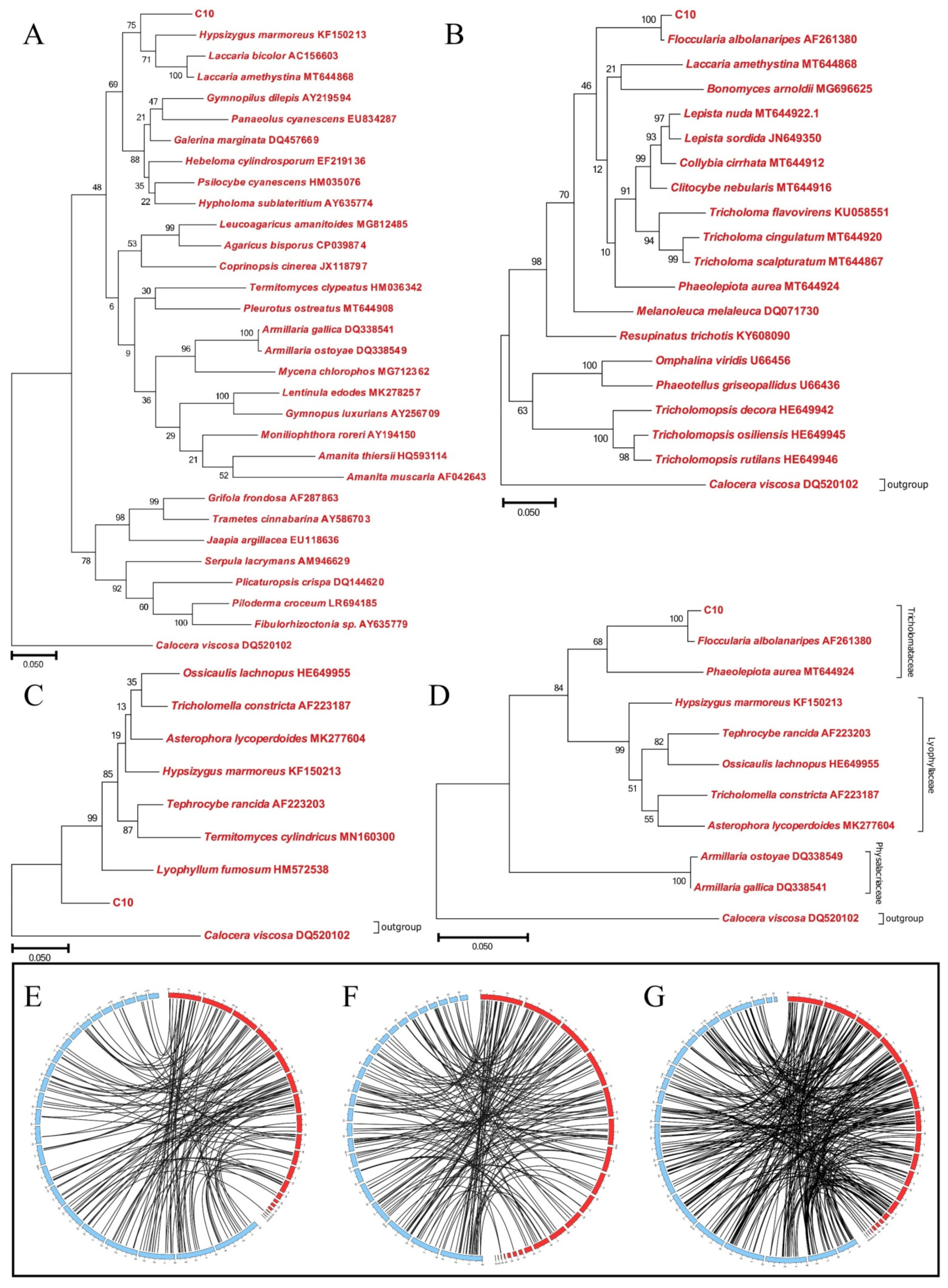
3.5. Phylogenetic Analysis of Strain C10
3.6. Transcriptome Sequencing and Assembly
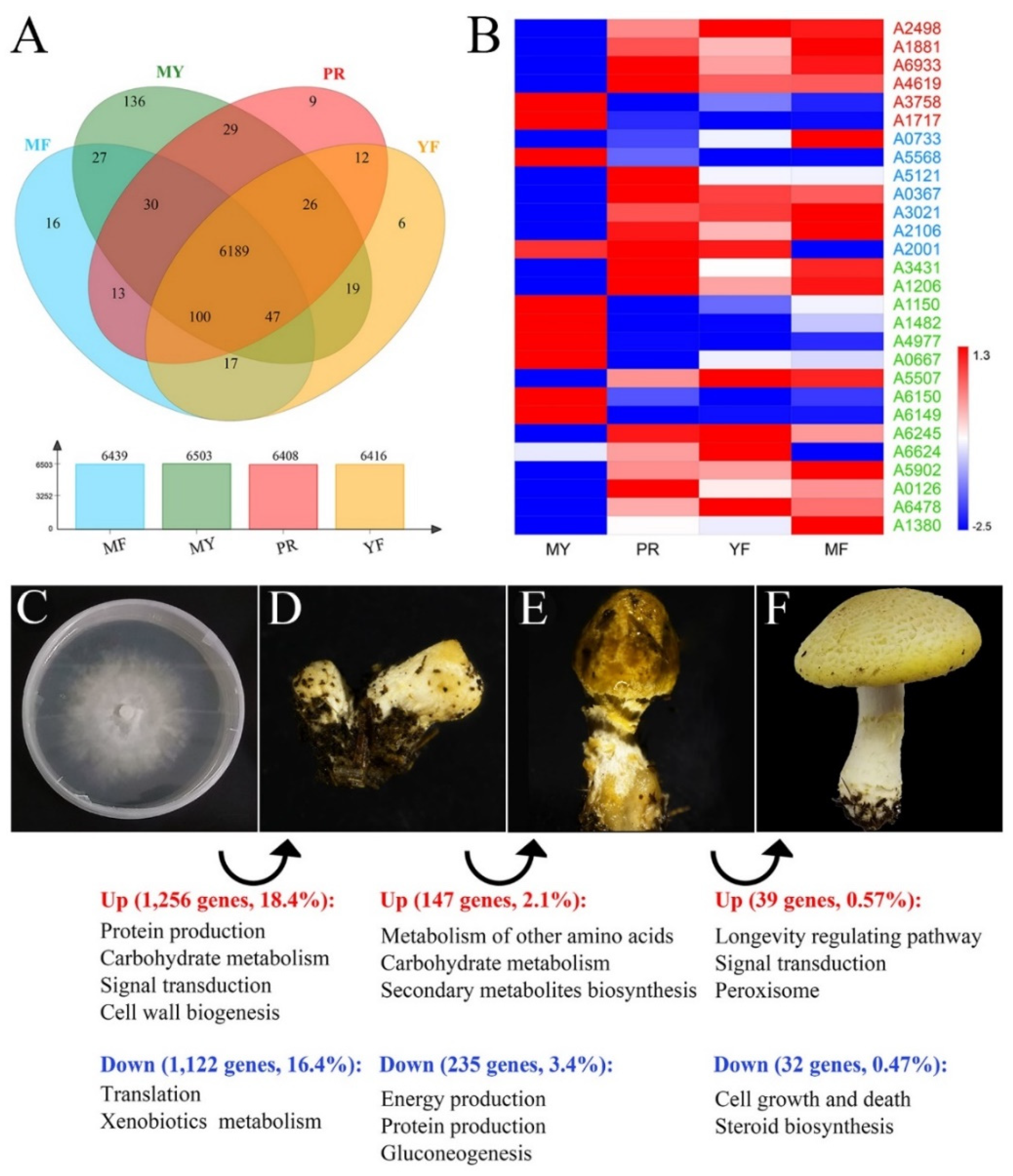
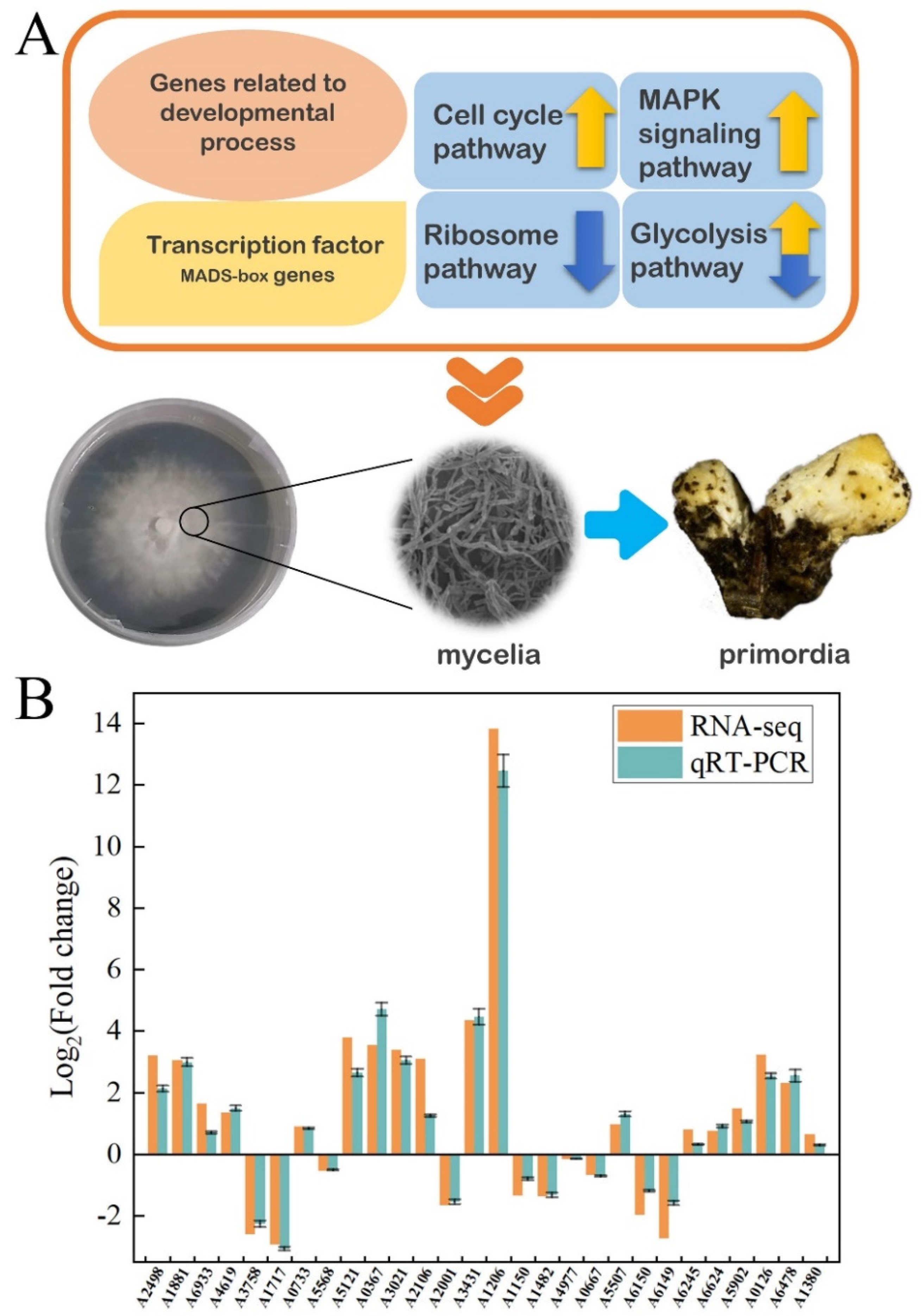
3.7. The Differentially Expressed Genes (DEGs) Regulated Fruiting Body Formation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xing, R.; Gao, Q.B.; Zhang, F.Q.; Fu, P.C.; Wang, J.L.; Yan, H.Y.; Chen, S.L. Genetic variation and phylogenetic relationships of the ectomycorrhizal Floccularia luteovirens on the Qinghai-Tibet Plateau. J. Microbiol. 2017, 55, 600–606. [Google Scholar] [CrossRef]
- Chen, C.; Shao, Y.; Tao, Y.; Wen, H. Optimization of dynamic microwave-assisted extraction of Armillaria polysaccharides using RSM, and their biological activity. LWT-Food Sci. Technol. 2015, 64, 1263–1269. [Google Scholar] [CrossRef]
- Jiao, Y.; Chen, Q.; Zhou, J.; Zhang, H.; Chen, H. Improvement of exo-polysaccharides production and modeling kinetics by Armillaria luteo-virens Sacc. in submerged cultivation. LWT-Food Sci. Technol. 2008, 41, 1694–1700. [Google Scholar] [CrossRef]
- Feng, K.; Liu, Q.H.; Ng, T.B.; Liu, H.Z.; Li, J.Q.; Chen, G.; Sheng, H.Y.; Xie, Z.L.; Wang, H.X. Isolation and characterization of a novel lectin from the mushroom Armillaria luteo-virens. Biochem. Biophys. Res. Commun. 2006, 345, 1573–1578. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Tao, Y.; Wang, W.; Mei, L.; Shao, Y.; Wang, Q.; Dang, J. Chemical Constituents of Fruit Body of Armillaria luteo-virens. Chem. Nat. Compd. 2019, 55, 373–375. [Google Scholar] [CrossRef]
- Xu, L.J.; Chen, Q.J.; Wang, H.X.; Zhang, G.Q. Purification and characterization of a ribonuclease from the wild edible mushroom Armillaria luteo-virens. Indian J. Biochem. Biophys. 2013, 50, 196–201. [Google Scholar]
- Xiong, H.Y.; Fei, D.Q.; Zhou, J.S.; Yang, C.J.; Ma, G.L. Steroids and other constituents from the mushroom Armillaria lueo-virens. Chem. Nat. Compd. 2009, 45, 759–761. [Google Scholar] [CrossRef]
- Xu, D.; Fu, M.; Chen, Q.; Liu, J. Effect of submerged culture conditions on exopolysaccharides production by Armillaria luteo-virens Sacc QH and kinetic modeling. Bioprocess. Biosyst. Eng. 2011, 34, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Xing, R.; Gao, Q.; Zhang, F.; Li, Y.; Fu, P.; Zhang, J.; Wang, J.; Khan, G.; Chen, S. Genetic diversity and population structure of Armillaria luteovirens (Physalacriaceae) in Qinghai-Tibet Plateau revealed by SSR markers. Biochem. Syst. Ecol. 2014, 56, 1–7. [Google Scholar] [CrossRef]
- Sun, L.; Cao, M.; Liu, F.; Wang, Y.; Wan, J.; Wang, R.; Zhou, H.; Wang, W.; Xu, J. The volatile organic compounds of Floccularia luteovirens modulate plant growth and metabolism in Arabidopsis thaliana. Plant Soil 2020, 456, 207–221. [Google Scholar] [CrossRef]
- Dang, J.; Chen, C.; Ma, J.; Dawa, Y.; Wang, Q.; Tao, Y.; Wang, Q.; Ji, T. Preparative isolation of highly polar free radical inhibitor from Floccularia luteovirens using hydrophilic interaction chromatography directed by on-line HPLC-DPPH assay. J. Chromatogr. B 2020, 1142, 122043. [Google Scholar] [CrossRef]
- Xing, R.; Yan, H.Y.; Gao, Q.B.; Zhang, F.Q.; Wang, J.L.; Chen, S.L. Microbial communities inhabiting the fairy ring of Floccularia luteovirens and isolation of potential mycorrhiza helper bacteria. J. Basic Microbiol. 2018, 58, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Yap, H.Y.; Chooi, Y.H.; Firdaus-Raih, M.; Fung, S.Y.; Ng, S.T.; Tan, C.S.; Tan, N.H. The genome of the Tiger Milk mushroom, Lignosus rhinocerotis, provides insights into the genetic basis of its medicinal properties. BMC Genom. 2014, 15, 635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiner, J.; Pisani, L.; Qiao, W.; Singh, R.; Yang, Y.; Shi, L.; Khan, W.A.; Sebra, R.; Cohen, N.; Babu, A.; et al. Cytogenomic identification and long-read single molecule real-time (SMRT) sequencing of a Bardet–Biedl Syndrome 9 (BBS9) deletion. NPJ Genom. Med. 2018, 3, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Bridges, S.; Magbanua, Z.V.; Peterson, D.G. Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 2008, 36, 2284–2294. [Google Scholar] [CrossRef] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Gardner, P.P.; Daub, J.; Tate, J.G.; Nawrocki, E.P.; Kolbe, D.L.; Lindgreen, S.; Wilkinson, A.C.; Finn, R.D.; Griffiths-Jones, S.; Eddy, S.R.; et al. Rfam: Updates to the RNA families database. Nucleic Acids Res. 2009, 37, D136–D140. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jaroszewski, L.; Godzik, A. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 2002, 18, 77–82. [Google Scholar] [CrossRef]
- Moktali, V.; Park, J.; Fedorova-Abrams, N.D.; Park, B.; Choi, J.; Lee, Y.H.; Kang, S. Systematic and searchable classification of cytochrome P450 proteins encoded by fungal and oomycete genomes. BMC Genom. 2012, 13, 525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [Green Version]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nix, D.A.; Eisen, M.B. GATA: A graphic alignment tool for comparative sequence analysis. BMC Bioinform. 2005, 6, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- Gan, X.; Cao, D.; Zhang, Z.; Cheng, S.; Wei, L.; Li, S.; Liu, B. Draft Genome Assembly of Floccularia luteovirens, an Edible and Symbiotic Mushroom on Qinghai-Tibet Plateau. G3-Genes Genomes Genet. 2020, 10, 1167–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Zeng, X.; Yang, Y.L.; Xing, Y.M.; Zhang, Q.; Li, J.M.; Ma, K.; Liu, H.W.; Guo, S.X. Genomic and transcriptomic analyses reveal differential regulation of diverse terpenoid and polyketides secondary metabolites in Hericium erinaceus. Sci. Rep. 2017, 7, 10151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Xu, J.; Liu, C.; Zhu, Y.; Nelson, D.R.; Zhou, S.; Li, C.; Wang, L.; Guo, X.; Sun, Y.; et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012, 3, 913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.Y.; Fan, W.L.; Wang, W.F.; Chen, T.; Tang, Y.C.; Chu, F.H.; Chang, T.T.; Wang, S.Y.; Li, M.Y.; Chen, Y.H.; et al. Genomic and transcriptomic analyses of the medicinal fungus Antrodia cinnamomea for its metabolite biosynthesis and sexual development. Proc. Natl. Acad. Sci. USA 2014, 111, E4743–E4752. [Google Scholar] [CrossRef] [Green Version]
- Črešnar, B.; Petrič, Š. Cytochrome P450 enzymes in the fungal kingdom. BBA-Proteins Proteom. 2011, 1814, 29–35. [Google Scholar] [CrossRef]
- Nelson, D.R. The cytochrome p450 homepage. Hum. Genom. 2009, 4, 59. [Google Scholar] [CrossRef] [Green Version]
- Platta, H.W.; Erdmann, R. The peroxisomal protein import machinery. FEBS Lett. 2007, 581, 2811–2819. [Google Scholar] [CrossRef]
- Holmes, T.C.; May, A.E.; Zaleta-Rivera, K.; Ruby, J.G.; Skewes-Cox, P.; Fischbach, M.A.; DeRisi, J.L.; Iwatsuki, M.; Omura, S.; Khosla, C. Molecular insights into the biosynthesis of guadinomine: A type III secretion system inhibitor. J. Am. Chem. Soc. 2012, 134, 17797–17806. [Google Scholar] [CrossRef] [Green Version]
- Konig, S.; Romp, E.; Krauth, V.; Ruhl, M.; Dorfer, M.; Liening, S.; Hofmann, B.; Hafner, A.K.; Steinhilber, D.; Karas, M.; et al. Melleolides from Honey Mushroom Inhibit 5-Lipoxygenase via Cys159. Cell Chem. Biol. 2019, 26, 60–70.e64. [Google Scholar] [CrossRef]
- Bohnert, M.; Scherer, O.; Wiechmann, K.; Konig, S.; Dahse, H.M.; Hoffmeister, D.; Werz, O. Melleolides induce rapid cell death in human primary monocytes and cancer cells. Bioorg. Med. Chem. 2014, 22, 3856–3861. [Google Scholar] [CrossRef]
- Momose, I.; Sekizawa, R.; Hosokawa, N.; Iinuma, H.; Matsui, S.; Nakamura, H.; Naganawa, H.; Hamada, M.; Takeuchi, T. Melleolides K, L and M, new melleolides from Armillariella mellea. J. Antibiot. 2000, 53, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhelifonova, V.P.; Antipova, T.V.; Litvinova, E.A.; Baskunov, B.P.; Litovka, Y.A.; Pavlov, I.N.; Kozlovsky, A.G. Biosynthesis of Protoilludene Sesquiterpene Aryl Esters by Siberian Strains of the Genus Armillaria Fungi. Appl. Biochem. Microbiol. 2019, 55, 277–283. [Google Scholar] [CrossRef]
- Wick, J.; Heine, D.; Lackner, G.; Misiek, M.; Tauber, J.; Jagusch, H.; Hertweck, C.; Hoffmeister, D. A fivefold parallelized biosynthetic process secures chlorination of Armillaria mellea (honey mushroom) toxins. Appl. Environ. Microbiol. 2016, 82, 1196–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lackner, G.; Bohnert, M.; Wick, J.; Hoffmeister, D. Assembly of melleolide antibiotics involves a polyketide synthase with cross-coupling activity. Chem. Biol. 2013, 20, 1101–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dörfer, M.; Heine, D.; König, S.; Gore, S.; Werz, O.; Hertweck, C.; Gressler, M.; Hoffmeister, D. Melleolides impact fungal translation: Via elongation factor 2. Org. Biomol. Chem. 2019, 17, 4906–4916. [Google Scholar] [CrossRef]
- Bohnert, M.; Nützmann, H.W.; Schroeckh, V.; Horn, F.; Dahse, H.M.; Brakhage, A.A.; Hoffmeister, D. Cytotoxic and antifungal activities of melleolide antibiotics follow dissimilar structure-activity relationships. Phytochemistry 2014, 105, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, L.; Chen, B.; Mukhtar, I.; Xie, B.; Li, Z.; Meng, L. Characterization and expression pattern of homeobox transcription factors in fruiting body development of straw mushroom Volvariella volvacea. Fungal Biol. 2019, 123, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Vonk, P.J.; Ohm, R.A. The role of homeodomain transcription factors in fungal development. Fungal Biol. Rev. 2018, 32, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Dai, Y.; Yang, C.; Wei, P.; Song, B.; Yang, Y.; Sun, L.; Zhang, Z.-W.; Li, Y. Comparative Transcriptome Analysis Identified Candidate Genes Related to Bailinggu Mushroom Formation and Genetic Markers for Genetic Analyses and Breeding. Sci. Rep. 2017, 7, 9266. [Google Scholar] [CrossRef] [Green Version]
- Kamada, T.; Sano, H.; Nakazawa, T.; Nakahori, K. Regulation of fruiting body photomorphogenesis in Coprinopsis cinerea. Fungal Genet. Biol. 2010, 47, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Nakamura, M.; Babasaki, K. Molecular cloning of developmentally specific genes by representational difference analysis during the fruiting body formation in the basidiomycete Lentinula edodes. Fungal Genet. Biol. 2005, 42, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.K.; Au, C.H.; Wilke, S.K.; Stajich, J.E.; Zolan, M.E.; Pukkila, P.J.; Kwan, H.S. 5′-Serial Analysis of Gene Expression studies reveal a transcriptomic switch during fruiting body development in Coprinopsis cinerea. BMC Genom. 2013, 14, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, Y.; Irie, T.; Sato, T. Isolation and characterization of a fruiting body-specific exo-beta-1,3-glucanase-encoding gene, exg1, from Lentinula edodes. Curr. Genet. 2005, 47, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Mu, D.; Li, C.; Zhang, X.; Li, X.; Shi, L.; Ren, A.; Zhao, M. Functions of the nicotinamide adenine dinucleotide phosphate oxidase family in G anoderma lucidum: An essential role in ganoderic acid biosynthesis regulation, hyphal branching, fruiting body development, and oxidative--stress resistance. Environ. Microbiol. 2014, 16, 1709–1728. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Ando, Y.; Hata, T.; Nakahori, K. A mutation in the Cc.arp9 gene encoding a putative actin-related protein causes defects in fruiting initiation and asexual development in the agaricomycete Coprinopsis cinerea. Curr. Genet. 2016, 62, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Masloff, S.; Jacobsen, S.; Poggeler, S.; Kuck, U. Functional analysis of the C6 zinc finger gene pro1 involved in fungal sexual development. Fungal Genet. Biol. 2002, 36, 107–116. [Google Scholar] [CrossRef]
- Wagemaker, M.J.; Eastwood, D.C.; Welagen, J.; van der Drift, C.; Jetten, M.S.; Burton, K.; Van Griensven, L.J.; den Camp, H.J.O. The role of ornithine aminotransferase in fruiting body formation of the mushroom Agaricus bisporus. Mycol. Res. 2007, 111, 909–918. [Google Scholar] [CrossRef]
- Zhang, J.; Ren, A.; Chen, H.; Zhao, M.; Shi, L.; Chen, M.; Wang, H.; Feng, Z. Transcriptome analysis and its application in identifying genes associated with fruiting body development in basidiomycete Hypsizygus marmoreus. PLoS ONE 2015, 10, e0123025. [Google Scholar] [CrossRef] [Green Version]
- Palmer, G.E.; Horton, J.S. Mushrooms by magic: Making connections between signal transduction and fruiting body development in the basidiomycete fungus Schizophyllum commune. FEMS Microbiol. Lett. 2006, 262, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.-C.; Yang, R.-M.; Chang, T.-T.; Chou, J.-C. Fructification of Antrodia cinnamomea Was Strain Dependent in Malt Extract Media and Involved Specific Gene Expression. J. Agric. Food Chem. 2010, 58, 257–261. [Google Scholar] [CrossRef]
- Hsu, K.-H.; Lee, Y.-R.; Lin, Y.-L.; Chu, F.-H. Cytochrome P450 Genes in Medicinal Mushroom Antrodia cinnamomea T.T. Chang et W.N. Chou (Higher Basidiomycetes) are Strongly Expressed during Fruiting Body Formation. Int. J. Med. Mushrooms 2011, 13, 513–523. [Google Scholar] [CrossRef]
- Szeto, C.Y.; Leung, G.S.; Kwan, H.S. Le. MAPK and its interacting partner, Le.DRMIP, in fruiting body development in Lentinula edodes. Gene 2007, 393, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fu, M.L.; Chen, Q.H. Biotransformation optimization of betulin into betulinic acid production catalysed by cultured Armillaria luteo-virens Sacc ZJUQH100-6 cells. J. Appl. Microbiol. 2011, 110, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Shim, D.; Park, S.G.; Kim, K.; Bae, W.; Lee, G.W.; Ha, B.S.; Ro, H.S.; Kim, M.; Ryoo, R.; Rhee, S.K.; et al. Whole genome de novo sequencing and genome annotation of the world popular cultivated edible mushroom, Lentinula edodes. J. Biotechnol. 2016, 223, 24–25. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, S.; Ma, X.; Chen, W.; Zhang, J.; Duan, S.; Gao, Y.; Kui, L.; Huang, W.; Wu, P.; et al. The Genome Sequences of 90 Mushrooms. Sci. Rep. 2018, 8, 9982. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Baek, J.H.; Lee, S.; Kim, C.; Rhee, H.; Kim, H.; Seo, J.S.; Park, H.R.; Yoon, D.E.; Nam, J.Y.; et al. Whole genome and global gene expression analyses of the model mushroom Flammulina velutipes reveal a high capacity for lignocellulose degradation. PLoS ONE 2014, 9, e93560. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, Y.; Yu, Z.; Wang, P.; Tang, X.; He, X.; Mi, F.; Liu, C.; Yang, D.; Xu, J. Genome Sequence of Phlebopus portentosus Strain PP33, a Cultivated Bolete. Genome Announc. 2015, 3, e00326-15. [Google Scholar] [CrossRef] [Green Version]
- Zheng, P.; Xia, Y.; Xiao, G.; Xiong, C.; Hu, X.; Zhang, S.; Zheng, H.; Huang, Y.; Zhou, Y.; Wang, S.; et al. Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biol. 2011, 12, R116. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Gong, Y.; Cai, Y.; Liu, W.; Zhou, Y.; Xiao, Y.; Xu, Z.; Liu, Y.; Lei, X.; Wang, G.; et al. Genome Sequence of the Edible Cultivated Mushroom Lentinula edodes (Shiitake) Reveals Insights into Lignocellulose Degradation. PLoS ONE 2016, 11, e0160336. [Google Scholar] [CrossRef] [PubMed]
- Dörfer, M.; Gressler, M.; Hoffmeister, D. Diversity and bioactivity of Armillaria sesquiterpene aryl ester natural products. Mycol. Prog. 2019, 18, 1027–1037. [Google Scholar] [CrossRef]
- Donnelly, D.M.X.; Hutchinson, R.M.; Coveney, D.; Yonemitsu, M. Sesquiterpene aryl esters from Armillaria mellea. Phytochemistry 1990, 29, 2569–2572. [Google Scholar] [CrossRef]
- Arnone, A.; Cardillo, R.; Nasini, G.; Meille, S.V. Secondary mould metabolites. Part 19. Structure elucidation and absolute configuration of melledonals B and C, novel antibacterial sesquiterpenoids from Armillaria mellea. X-ray molecular structure of melledonal C. J. Chem. Soc.-Perkin Trans. 1988, 1, 503–510. [Google Scholar] [CrossRef]
- Arnone, A.; Cardillo, R.; Nasini, G. Structures of melleolides B-D, three antibacterial sesquiterpenoids from Armillaria mellea. Phytochemistry 1986, 25, 471–474. [Google Scholar] [CrossRef]
- Midland, S.L.; Izac, R.R.; Wing, R.M.; Zaki, A.I.; Munnecke, D.E.; Sims, J.J. Melleolide, a New Antibiotic from Armillaria-Mellea. Tetrahedron Lett. 1982, 23, 2515–2518. [Google Scholar] [CrossRef]
- Engels, B.; Heinig, U.; Grothe, T.; Stadler, M.; Jennewein, S. Cloning and characterization of an Armillaria gallica cDNA encoding protoilludene synthase, which catalyzes the first committed step in the synthesis of antimicrobial melleolides. J. Biol. Chem. 2011, 286, 6871–6878. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, D.M.X.; Konishi, T.; Dunne, O.; Cremin, P. Sesquiterpene aryl esters from Armillaria tabescens. Phytochemistry 1997, 44, 1473–1478. [Google Scholar] [CrossRef]
- Bohnert, M.; Miethbauer, S.; Dahse, H.M.; Ziemen, J.; Nett, M.; Hoffmeister, D. In vitro cytotoxicity of melleolide antibiotics: Structural and mechanistic aspects. Bioorg. Med. Chem. Lett. 2011, 21, 2003–2006. [Google Scholar] [CrossRef]
- Coetzee, M.P.A.; Wingfield, B.D.; Harrington, T.C.; Dalevi, D.; Coutinho, T.A.; Wingfield, M.J. Geographical diversity of Armillaria mellea s. s. based on phylogenetic analysis. Mycologia 2019, 92, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Muszynska, B.; Sulkowska-Ziaja, K.; Wolkowska, M.; Ekiert, H. Chemical, Pharmacological, and Biological Characterization of the Culinary-Medicinal Honey Mushroom, Armillaria mellea (Vahl) P. Kumm. (Agaricomycetideae): A Review. Int. J. Med. Mushrooms 2011, 13, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.Y.; Igarashi, T. Armillaria species associated with Gastrodia elata in Japan. For. Pathol. 1995, 25, 319–326. [Google Scholar] [CrossRef]
- Chum, W.W.; Kwan, H.S.; Au, C.H.; Kwok, I.S.; Fung, Y.W. Cataloging and profiling genes expressed in Lentinula edodes fruiting body by massive cDNA pyrosequencing and LongSAGE. Fungal Genet. Biol. 2011, 48, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Ohm, R.A.; de Jong, J.F.; Lugones, L.G.; Aerts, A.; Kothe, E.; Stajich, J.E.; de Vries, R.P.; Record, E.; Levasseur, A.; Baker, S.E.; et al. Genome sequence of the model mushroom Schizophyllum commune. Nat. Biotechnol. 2010, 28, 957–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuratani, M.; Tanaka, K.; Terashima, K.; Muraguchi, H.; Nakazawa, T.; Nakahori, K.; Kamada, T. The dst2 gene essential for photomorphogenesis of Coprinopsis cinerea encodes a protein with a putative FAD-binding-4 domain. Fungal Genet. Biol. 2010, 47, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Boulianne, R.P.; Liu, Y.; Aebi, M.; Lu, B.C.; Kues, U. Fruiting body development in Coprinus cinereus: Regulated expression of two galectins secreted by a non-classical pathway. Microbiology 2000, 146 Pt 8, 1841–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Attribute | Value |
|---|---|
| Assembly summary | |
| No. of contigs | 23 |
| Length of the largest contig (bp) | 3,288,420 |
| Length of the smallest contig (bp) | 15,938 |
| N50 length (bp) | 2,275,160 |
| N90 length (bp) | 1,237,025 |
| Percentage of assembly (%) | |
| Contigs ≥ 500 bp | 100% |
| Contigs ≥ 1 kb | 100% |
| BUSCO analysis (%) | |
| Complete (%) | 89.3 |
| Complete duplicated (%) | 0.3 |
| Fragmented (%) | 3.8 |
| Missing (%) | 6.9 |
| Genomic component analysis | |
| Genome size (bp) | 27,003,024 |
| GC content of genome | 43.54% |
| Gene number | 7068 |
| Gene length | 11,273,474 |
| Average gene length (bp) | 1595 |
| % of genome (genes) | 41.75% |
| GC content of protein-coding genes (%) | 34.93% |
| Average protein length (aa) | 531 |
| Length of largest protein-coding gene, bp | 4023 |
| Length of smallest protein-coding gene, bp | 66 |
| Average no. of exons per gene | 7 |
| Average exon size (bp) | 219 |
| Average no. of introns per gene | 6 |
| Average intron size (bp) | 68 |
| Average size of intergenic regions (bp) | 2225 |
| Gene internal length | 15,729,880 |
| Number of tRNAs | 93 |
| Number of rRNAs | 7 |
| Secondary metabolite biosynthesis gene clusters (BGCs) | 16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Lu, H.; Zhang, X.; Chen, Q. The Genomic and Transcriptomic Analyses of Floccularia luteovirens, a Rare Edible Fungus in the Qinghai–Tibet Plateau, Provide Insights into the Taxonomy Placement and Fruiting Body Formation. J. Fungi 2021, 7, 887. https://doi.org/10.3390/jof7110887
Liu Z, Lu H, Zhang X, Chen Q. The Genomic and Transcriptomic Analyses of Floccularia luteovirens, a Rare Edible Fungus in the Qinghai–Tibet Plateau, Provide Insights into the Taxonomy Placement and Fruiting Body Formation. Journal of Fungi. 2021; 7(11):887. https://doi.org/10.3390/jof7110887
Chicago/Turabian StyleLiu, Zhengjie, Hongyun Lu, Xinglin Zhang, and Qihe Chen. 2021. "The Genomic and Transcriptomic Analyses of Floccularia luteovirens, a Rare Edible Fungus in the Qinghai–Tibet Plateau, Provide Insights into the Taxonomy Placement and Fruiting Body Formation" Journal of Fungi 7, no. 11: 887. https://doi.org/10.3390/jof7110887