
Genomic and Experimental Investigations of Auriscalpium and Strobilurus Fungi Reveal New Insights into Pinecone Decomposition



1
CAS Key Laboratory for Plant Diversity and Biogeography of East Asia, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, China
2
Yunnan Key Laboratory for Fungal Diversity and Green Development, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650201, China
3
University of Chinese Academy of Sciences, Beijing 100049, China
4
Department of Biology, McMaster University, Hamilton, ON L8S 4K1, Canada
5
College of Life Sciences, Chifeng University, Chifeng 024000, China
*
Author to whom correspondence should be addressed.
J. Fungi 2021, 7(8), 679; https://doi.org/10.3390/jof7080679
Submission received: 13 July 2021
/
Revised: 16 August 2021
/
Accepted: 19 August 2021
/
Published: 23 August 2021
(This article belongs to the Section Fungal Genomics, Genetics and Molecular Biology)
Abstract
:Saprophytic fungi (SPF) play vital roles in ecosystem dynamics and decomposition. However, because of the complexity of living systems, our understanding of how SPF interact with each other to decompose organic matter is very limited. Here we studied their roles and interactions in the decomposition of highly specialized substrates between the two genera Auriscalpium and Strobilurus fungi-colonized fallen pinecones of the same plant sequentially. We obtained the genome sequences from seven fungal species with three pairs: A. orientale-S. luchuensis, A. vulgare-S. stephanocystis and A. microsporum-S. pachcystidiatus/S. orientalis on cones of Pinus yunnanensis, P. sylvestris and P. armandii, respectively, and the organic profiles of substrate during decomposition. Our analyses revealed evidence for both competition and cooperation between the two groups of fungi during decomposition, enabling efficient utilization of substrates with complementary profiles of carbohydrate active enzymes (CAZymes). The Auriscalpium fungi are highly effective at utilizing the primary organic carbon, such as lignin, and hemicellulose in freshly fallen cones, facilitated the invasion and colonization by Strobilurus fungi. The Strobilurus fungi have genes coding for abundant CAZymes to utilize the remaining organic compounds and for producing an arsenal of secondary metabolites such as strobilurins that can inhibit other fungi from colonizing the pinecones.
1. Introduction
Decomposition of organic matter is vitally important in ecosystem processes such as carbon and nitrogen cycling, soil formation and biodiversity maintenance [1,2]. In nature, decomposition is highly dynamic with ever-changing interactions among decomposers and between substrate composition and successive development of microbial communities [3,4]. Fungi are key components of the microbial communities in most natural decompositions with different fungal species interacting with each other in multiple ways, leading to complete degradation of complex organic compounds such as lignocelluloses in wood and plant litter. Indeed, there is emerging evidence showing orderly succession among fungal members of microbial communities through the different stages of lignocellulose decomposition [5].
As one of the most important members of microbial communities in forest ecosystems, saprotrophic fungi (SPF) have diverse degradation mechanisms and play key roles in the degradations of dead organic matters [3,6]. However, due to the limited resources across space and time in most ecological niches and the presence of many (potential) competitors, fungal decomposers have evolved mechanisms to allow them successfully colonizing one to several substrates/ecological niches [7]. Those colonizing only one type of ecological niche are called ecological “specialists” while others capable of colonizing many types of ecological niches are called “generalists” [8]. There are many who are in-between the obligate specialists and broad generalists, including those that are primarily found in one ecological niche but are capable of surviving and growing in other niches [8]. Evidence for ecological specializations in fungi has been recorded since ancient times [3]. In addition, most ecological niches and substrates have successions where different fungal communities may dominate different phases of substrate decomposition [5].
There are many factors that can impact the composition and structure of saprotrophic fungal community. Among these factors, the chemical composition of substrates plays a major role [9,10]. For example, on woody substrates, the decomposition rate of polymeric lignocellulosic components changes through the decomposition process, due to changes in substrate compositions and in the types and relative abundances of different microbes and their enzymes involved in degradations [11]. Traditionally, saprotrophic fungi are broadly classified into two types, namely ligninolytic white-rot (WR) and cellulolytic brown-rot (BR), although there is a continuum between these two types [7,12]. In the process of decomposition, interaction (including competition) among fungi is likely very common, affecting the distribution, abundance, and the order of occurrence among these fungi in natural communities [13].
Different interaction strategies among species can lead to different orders of SPF colonization on substrates. Both biotic and abiotic factors can also influence their order of colonization and interactions [7,14,15]. Fungal competition on substrates is commonly classified into two major functional types: primary resource capture and secondary resource capture [7,16,17]. Success of SPF in primary resource capture mainly depends on the ability to utilize previously uncolonized resources and on their ability to resist plant derived antifungal compounds in those substrates. In contrast, success in secondary resource capture mainly relies on antagonistic mechanisms (i.e., antifungal production etc.), with different species competing with each other to obtain sufficient nutrients for survival and reproduction [7]. Often, changes in microbial communities during decomposition are related to the secretion of antagonistic enzymes and metabolites [7]. On the one hand, SPFs have the ability to secrete various carbohydrate-active enzymes (CAZymes) to decompose and utilize the major constituents such as lignin, cellulose and hemicellulose in wood and plant litter, facilitating nutrient cycling and energy flow in forest ecosystem [18,19,20]. Indeed, the compositions and characteristics of CAZymes often differ among fungi, likely shaped by characteristics of their substrates and the degree of adaptation to the specific environmental conditions [20]. Therefore, to understand decomposition, it is particularly important to study the compositions and characteristics of CAZymes to clarify the potential mechanisms for different nutritional modes, infection and substrates specificity/preference [20,21,22,23,24,25]. On the other hand, fungal secondary metabolites (SMs) are known to play crucial roles in the defense against pathogens and competitors and provide advantages for their producers and/or those who have resistant mechanisms. Along with CAZymes, fungal SMs can provide important information for understanding the chemical basis of niche specialization during decompositions [26,27].
Multiple groups of SPFs are frequently involved in plant litter decomposition. These fungi belong to diverse clades, but some of them are functionally interchangeable [26,28,29], Figure S1. In forest ecosystems, due to its extractive composition and the presence of antifungal compounds such as resin, pinecone is a specialized substrate and a unique habitat for fungi [30,31]. Indeed, only species in a few fungal genera (e.g., Strobilurus, Auriscalpium, Baeospora and Mycena) are known to colonize and decompose pinecones. Among these genera, Auriscalpium and Strobilurus are highly specialized on pinecones [30,31]. Interestingly, species of Auriscalpium and Strobilurus usually share the dead cones of the same plant species in a chronological order, with fruiting bodies of Auriscalpium fungi often appearing on newly fallen cones, while those of Strobilurus typically occurring on highly rotten cones during later stages of decomposition (Figures S2 and S3). At present, the mechanisms for their succession during pinecone decomposition are unknown.
In this study, we investigated three pinecone substrate–fungus pairs from Europe and East Asia to understand the potential mechanisms for substrate specificities and ecological succession during pinecone decomposition. The three fungal pairs as well as their substrates were A. orientale-S. luchuensis on cones of Pinus yunnanensis, A. vulgare-S. stephanocystis on cones of P. sylvestris, and A. microsporum-S. pachcystidiatus/S. orientalis on cones of P. armandii. We obtained the genome sequences of these seven fungal species and quantified the main chemical compounds during pinecone decomposition. Our analyses revealed both shared and unique features in their substrate specificity and ecological successions among these fungal pairs.
2. Materials and Methods
2.1. Material Collections, Greenhouse Planting Experiment, Fungal Strains, Media and Culture Conditions
The records on the geographical distributions and substrate information of A. vulgare and S. stephanocystis were extracted from data deposited in the Global Biodiversity Information Facility (GBIF; https://www.gbif.org/; accessed on 11 April 2019). The information on substrates and distributions of the following four species A. orientale, A. microsporum, S. luchuensis and S. pachycystidiatus was primarily from the published literature and our own observations during field investigations in China. In order to directly observe the characteristics of pinecone decomposition by Auriscalpium and Strobilurus, a planting experiment was conducted in the greenhouse using about 250 cones of P. armandii fully colonized by A. microsporum collected in Yufeng Temple (Lijiang City, Yunnan Province, China) in July 2017 where both Auriscalpium and Strobilurus existed. The reason for the selection of collection site of Yufeng Temple is that Yufeng Temple is the distribution center of fungi in Strobilurus and Auriscalpium, and the climate in this area is close to Kunming City. After we brought cones back without any specific treatment, we put them in the plastic shed and spray water regularly to ensure their humidity. During our experiment, we keep the temperature in the greenhouse consistent with the outdoor temperature of Kunming.
All seven strains used for de novo sequencing were dikaryotic strains isolated directly from the fruiting bodies from wild mushrooms. Among these, the following five species and strains were collected in Yunnan Province, China: A. microsporum (strain AU-H-210), A. orientale (strain AU-Y-A), S. luchuensis (strain Y-Y-2D), S. pachycystidiatus (strain Y-H-6C) and S. orientalis (strain K-1-1). The remaining two A. vulgare (CBS 236.39) and S. stephanocystis (CBS 113577) were obtained from the Westerdijk Fungal Biodiversity Institute (CBS, Fungal Biodiversity Centre in Netherlands). The strains were stored at 4 °C on MEA solid medium (1% malt extract, 0.1% peptone and 1.5% agar). Vegetative mycelia of those strains were cultivated in MEA liquid medium (1% malt extract and 0.1% peptone) in the dark at 23 °C for 7–25 days, and then the mycelia were collected for DNA extraction and whole genome sequencing [32].
2.2. Measurements of Pinecone Compositions
For each type of pinecone, we analyzed the complex organic compounds at two different states: (i) newly fallen cones, and (ii) cones being decomposed by fungi in the genus Auriscalpium and just colonized by fungi in Strobilurus (Figure S3). The contents of cellulose, hemicellulose and lignin were quantified using the automatic fiber analyzer according to the ANKOM 2000i instructions (https://www.ankom.com/technical-support/fiber-analyzer-a2000; accessed on 21 April 2019) in cones of P. armandii, P. yunnanensis and P. sylvestris at KIB (Kunming Institute of Botany, CAS) as references. The pectin contents in these samples were quantified by using the method of sulfuric acid-carbazole colorimetry [33]. Analyses for all samples were carried in decuplicate.
2.3. Genome Sequencing, Assembly and Protein-Coding Gene Predictions
To yield a high-quality genome assembly, the genomes were sequenced using a whole genome shotgun sequencing strategy with a combined strategy that included both the Pacific Biosciences RS II (Pacific Biosciences, Menlo Park, CA, USA) and Illumina MiSeq platforms (Illumina Inc., San Diego, CA, USA) by Biomarker Technologies Co, LTD (Beijing, China). The PacBio long reads were corrected and assembled using Canu for draft genomes [34]. FinisherSC was used to improve the contiguity of draft genomes [35], and pilon was used to polish the draft genomes with collected Illumina data by Musket [36,37]. HaploMerger2 was used to separate the two haploid sub-assemblies from the assembly [38,39]. Ab initio predictions were carried out using the reference protein domains of Peniophora sp. and Armillaria ostoyae [40,41] for Auriscalpium and Strobilurus, respectively. Based on the two high-quality sequencing datasets described above, the protein-coding gene set of genomes were refined following the GETA gene annotation method [42]. BUSCOs using database of fungi_odb9 were applied to our gene predictions.
2.4. Identification of Carbohydrate-Active Enzymes (CAZymes) and Lignocellulolytic Genes and Swiss-Prot Annotation
Carbohydrate-active enzymes (CAZymes) were identified by using a combination of pipelines that included the HMM and BLASTP algorithms [43]. CAZyme annotation by BLASTP algorithms used a cutoff e-value < 1 × 10−5 and coverage >20%. CAZyme annotation by HMM algorithms used a cutoff e-value < 1 × 10−5 for alignments of >80 amino acids, and for alignments of <80 amino acids, we used an e-value of < 1 × 10−3 and coverage >25%. Perl program was used to extract the annotation results that conform to the two methods as the final result. For Swiss-Prot annotation, the BLASTP algorithm was used to align the protein sequences to Swiss-Prot with e-value < 1 × 10−5 and coverage >20%. Lignocellulolytic genes (cellulase, hemicellulase, pectinase, lignin oxidase and lignin degrading auxiliary enzymes) were identified mainly by the Swiss-Prot annotation with keywords as described by Chen et al. [43] in their Table S20. In the following analyses, lignin oxidases and lignin degrading auxiliary enzymes encoded by lignocellulolytic genes were combined into one category called ligninases [43].
2.5. Principal Component Analyses (PCA) and Heatmap Analyses of CAZymes and Lignocellulolytic Genes
In PCA of CAZymes, organisms are clustered with others which have the similar nutrient patterns to determine the rot patterns of fungi. Here, PCA was used to cluster WR and BR fungi based on the diversity of their CAZymes. The CAZymes number matrix of wood decay fungi (WDF) from the following species was used as input: Galerina marginata (abbreviated as “gama”), A. orientale (“auor”), Heterobasidion annosum (“hean”), Stereum hirsutum (“sthi”), A. vulgare (“auvu”), Punctularia strigosozonata (“pust”), A. microsporum (“aumi”), Fomitiporia mediterranea (“fome”), S. pachycystidiatus (“stpa”), S. luchuensis (“stlu”), Dichomitus squalens (“disq”), Trametes versicolor (“trve”), Phanerochaete chrysosporium (“phch”), S. stephanocystis (“stst”), Phanerochaete carnosa (“phca”), S. orientalis (“stor”), Wolfiporia cocos (“woco”), Gloeophyllum trabeum (“gltr”), Fomitopsis pinicola (“fopi”), Dacryopinax primogenitus (“dapr”), Serpula lacrymans (“sela”) and Coniophora puteana (“copu”) [12,44,45,46,47,48,49].
Among these species, phca, phch, hean, sthi, gama, disq, trve, fome and pust belonged to WR fungi, while copu, gltr, fopi, woco, dapr and sela belonged to BR fungi [12,43]. The online OmicShare tools (http://www.omicshare.com/tools; accessed on 14 August 2019) were used to generate the illustrations, heatmaps and other outputs from PCA and statistical analysis. Adobe Photoshop (PS) and Adobe Illustrator (AI) were used for image editing and finalization.
2.6. Analyses of Enzymes on Resin Decomposition
By adding latex to media, Oghenekaro et al. (2020) identified that some genes had elevated expressions in R. microporus. Latex is a complex emulsion that includes a diversity of chemicals such as proteins, alkaloids, carbohydrates, oils, tannins, and resins that coagulate on exposure to air [50,51]. It is commonly produced by plants after tissue injury and serve as a defense against pathogens and pests [50,51]. Here we used the high expressed genes induced by latex treatment in R. microporus to infer the distributions of these genes in the seven fungi analyzed in our study. Using those genes in R. microporus as references, we identified the homologous genes in our genomes through BLASTP algorithms with a cut off e-value < 1 × 10−5 and coverage >20%.
2.7. Secondary Metabolites (SMs) in Auriscalpium and Strobilurus
SMs genes are frequently located in gene clusters of microorganisms [52], and they may have important physiological and ecological significances with antifungal, antibacterial, antitumor, antiviral, antialgal, immune-suppressive and other biological effects [53]. Here, for our seven genomes, SM gene clusters were determined using a web-based analysis platform named AntiSMASH fungal 6.0 (https://fungismash.secondarymetabolites.org/#!/start; accessed on 13 August 2019) [54].
2.8. Species Tree Construction, Divergence Time Estimation and Gene Family Expansions of CAZymes Analysis
Orthologous genes and single-copy orthologous genes among 22 publicly available fungal genomes above were identified by using software of OrthoMCL [55,56] with same parameters of Chen et al. [43]. The generated proteomes and corresponding coding sequences were used as input to phylogenomic and comparative genomic analyses. We constructed a phylogenomic tree included genomes with predicted proteome clusters generated from the comparative analysis of 22 available fungal genomes, with Dacryopinax primogenitus as the outgroup. Phylogenomics analysis was conducted based on the framework proposed by Chen et al. [43] using software of ProtTest [57], MAFFT v7.158b [58], Gblocks 0.91b and RAxML-8.0.26 [59]. For divergence time estimation with r8s v1.80 [60] with same parameters of Chen et al. [43], one fossil calibration points and three secondary calibration point [45] were fixed in the molecule clock analysis: the most recent common ancestor (MRCA) of Serpula lacrymans and Coniophora puteana were diverged at 104.23 million years ago (MYA); the MRCA of Punctularia strigosozonata and Gloeophyllum trabeum were diverged at 170.42 MYA; the MRCA of Stereum hirsutum and Heterobasidion annosum were diverged at 100.94 MYA and the MRCA of Fomitiporia mediterranea and Dacryopinax primogenitus were diverged at 349.72 MYA. The orthologous gene family expansions and contractions were calculated by CAFE with p-values less than 0.05 based on the ultrametric tree [61]. When we got the expanded gene families, we selected the expanded gene families belonging to CAZymes for subsequent analysis.
3. Results
3.1. Ecological Characteristics of Fungi in Auriscalpium and Strobilurus
The temporal and trophic niches as reported in the literature and our own observations for Auriscalpium and Strobilurus fungi in this study are summarized in Figure 1, Figures S2 and S3 and Table S1. The data showed clear differences between these two groups of fungi in their temporal distributions during pinecone decomposition (Figure 1, Figures S2 and S3; Table S1). Specifically, fruiting bodies of A. vulgare are mostly found on newly fallen cones of P. sylvestris, P. pinaster, P. halepensis and P. mugo in September and October, while those of S. stephanocystis mostly appear on highly rotten cones already decomposed by A. vulgare in May. Similarly, A. orientale mainly appears on newly fallen cones of P. yunnanensis, P. tabuliformis, P. densiflora, P. densata, P. massoniana and P. hwangshanensis in August, while S. luchuensis on highly rotten cones already decomposed by A. orientale. In addition, A. microsporum mostly produces fruiting bodies in September on newly fallen cones of P. armandii and with fruiting bodies of S. pachycystidiatus found in June on cones newly decomposed by A. microsporum, and S. orientalis in October on highly decomposed cones by A. microsporum and/or S. pachycystidiatus. Although the time of fruiting was different, the preferences of the two genera for the substrates with different degrees of decay were not affected.
The ecological characteristics of colonization and fruiting by Auriscalpium and Strobilurus fungi on pinecones as observed in the field were similarly found in our greenhouse fruiting experiment (Figure S2). There are hyphae or spores of S. pachcystidiatus and S. orientalis in the cones occupied by A. microsporum collected from the field. Under the greenhouse culture condition, fruiting bodies of A. microsporum formed first from July to September, then S. pachcystidiatus appeared from May to July over the next 3 years, and finally S. orientalis appeared from October to December. Together, these greenhouse observations are consistent with our field observations on the ecological successions of these fungi on pinecones (Figure S2). However, because the environmental humidity in the greenhouse is stable, the fruiting time of fungi in Auriscalpium and Strobilurus is relatively continuous.
3.2. Genome Sequencing, Data Preprocessing, Assembly, General Genome Features, Protein-Coding Gene Prediction and Functional Annotation
The general features of the seven genomes that we sequenced are summarized in Table 1. Raw data were generated with Pacbio sequencing and Illumina sequencing with coverage 85.84–386.08× and 101.04–206.85×, respectively (Table S2). Among the Auriscalpium species, A. vulgare had the largest genome (51.68 Mb), followed by A. orientale (45.40 Mb), and A. microsporum had the smallest genome (43.46 Mb) (Table 1). Among the species of Strobilurus, S. pachycystidiatus had the largest genome (51.82 Mb), followed by S. orientalis (51.25 Mb) and S. luchuensis (46.71 Mb), and S. stephanocystis had the smallest genome (42.38 Mb) (Table 1). The sets of annotated protein-coding genes in the seven assembled genomes were estimated to be 92.4–100% complete (Table S3). Among the Auriscalpium species, A. orientale had the most coding genes (16,958), followed by A. microsporum (15,333), and A. vulgare had the least coding genes (13,636). Among the taxa of Strobilurus, S. orientalis has the most coding genes (18,509), followed by S. pachycystidiatus (18,157) and S. luchuensis (16,796), and S. stephanocystis had the least number of coding genes (16,439).
3.3. Identification of CAZymes and Lignocellulolytic Genes in Fungi of Auriscalpium and Strobilurus
The number of CAZymes in A. vulgare, A. microsporum, A. orientale, S. stephanocystis, S. luchuensis, S. pachycystidiatus and S. orientalis were 450, 425, 464, 532, 542, 583 and 621, respectively (Figure 2b; Table S4). Our statistical analyses revealed that the average number of CAZymes in Auriscalpium fungi is significantly lower than that in Strobilurus such as AA5, CBM18, CBM67, CE12, CE16, CE4, CE8, GH105, GH12, GH127, GH128, GH13, GH135, GH16, GH17, GH18, GH27, GH28, GH29, GH35, GH43, GH45, GH5, GH53, GH55, GH71, GH76, GH93, GT1, GT15, GT17, GT33, GT4, GT8, PL1 and PL4 (Table S5). The average number of CAZymes in Auriscalpium fungi is significantly more than that in Strobilurus only in seven gene families including AA2, GH2, GH3, GH15, GH31, GH109 and PL8 (Table S5). Similarly, in the statistical analyses, the comparison of CAZymes between Auriscalpium and other WR fungi, and the comparison of CAZymes between Strobilurus and other WR fungi are shown in Table S6 and S7, respectively. The average number of CAZymes in Auriscalpium fungi is significantly more than that in other WR fungi only in six gene families, however the average number of CAZymes in Strobilurus fungi is significantly more than that other WR fungi in 31 gene families (Tables S6 and S7).
The number of predicted lignocellulolytic genes in A. vulgare, A. microsporum, A. orientale, S. stephanocystis, S. luchuensis, S. pachycystidiatus and S. orientalis were 112, 97, 111, 107, 106, 122 and 111, respectively (Table S8). Our analyses demonstrated that the number of genes coding for AA2, GH3 and GH7 in Auriscalpium are significantly higher than those in Strobilurus, while the number of genes coding for AA1, CE8, GH12, GH5_5, GH45 and PL1_4 in Strobilurus are significantly higher than those in Auriscalpium (Table S9). For the fungi growing on cones of the same pine species, the proportions of genes encoding ligninases and hemicellulases were higher in Auriscalpium species than those in their corresponding Strobilurus species. In contrast, the proportions of genes encoding cellulases and pectinases were lower in Auriscalpium than those in the Strobilurus. For fungi living on cones of P. armandii, A. microsporum and S. pachycystidiatus living on relatively newly fallen cones also showed a similar pattern but the difference was less obvious than those in other two fungal pairs. However, S. orientalis with a preference of living on more rotten cones showed more obvious differences in their respective enzymes compared with A. microsporum.
3.4. PCA and Heatmap Analyses of CAZymes and Lignocellulolytic Genes
Results of the PCA of CAZymes and lignocellulolytic genes showed a clear separation of WR and BR fungi along PCA1 (Figure 2a and Figure S4a). All the fungi in Auriscalpium and Strobilurus cluster together with other WR fungi, consistent with their potent lignin-decomposing ability (Figure 2a and Figure S4a). The heatmap analyses showed that there are unique enzymes within each of the 22 fungi analyzed, which likely reflect the diversity of their preferred substrates (Figure 2c and Figure S4b). In the separate analyses of the two genera, it is apparent that fungi in the two genera showed complementary profiles of carbohydrate-active enzymes (CAZymes) (Figure 3a and Figure 4g,h). The results clearly illustrate that there are remarkable differences in the overall pattern of genes between two different genera. At the same time, it demonstrates that the type and number of these CAZymes in Auriscalpium and Strobilurus are different among different fungi, which may partly explain the cause of the substrate specificity (Figure S5a,b). The results of the lignocellulolytic genes are overall consistent with the results of CAZymes (Figure S5c,d). These lignocellulolytic genes are also specific among genera, and there are great differences in the type and number of enzymes among different genera, and each has a set of different enzyme system (Figure 4g,h and Figure S5c,d).
3.5. Composition Analyses of Pinecones
Among newly fallen cones, that of P. yunnanensis showed the highest contents of lignin and hemicellulose, but the least amounts of cellulose and pectin. In contrast, the cones of P. armandii showed the highest contents of cellulose and pectin, but the least amount of lignin (Figure 3b; Table S10). In addition to the least amount of hemicellulose, the other components in the cone of P. sylvestris are between those in the cones of the other two species (Figure 3b; Table S10). Compared with the newly fallen cones, the cones already decomposed by corresponding Auriscalpium fungi had reduced relative proportions of lignin and hemicellulose and increased relative proportions of cellulose and pectin (Figure 4a–c; Table S10).
3.6. Enzymes Related to Resin Decompositions
Among the top 10 enzymes showing elevated expressions in the presence of latex in R. microporus, our analyses revealed that four of them had an overall greater numbers of genes coding for these enzymes in Auriscalpium fungi than in Strobilurus in the top No. 1, 2, 6 and 10 in Auriscalpium is more than those in Strobilurus (Table 2). Overall, among all the genes, the number of genes coding for peptidase S8 (No. 1) and peptidase S53 (No. 6) in Auriscalpium were significantly higher than that in Strobilurus (p < 0.05), while the other genes were not significantly different. Of the genes coding for peptidase S8, the average number in Auriscalpium is 20.33, while only 5.4 in Strobilurus (Table 2). In genes coding for peptidase S53, the average numbers in Auriscalpium and Strobilurus were 3.7 and 1.2, respectively (Table 2).
3.7. SM Clusters in Strobilurus and Auriscalpium
Terpenes, NRPS (nonribosomal peptide synthetase)-like compounds, and siderophores are among the main groups of SMs shared by fungi in Auriscalpium and Strobilurus and the putative genes coding for enzymes involved in their syntheses are summarized in Table 3. Our analyses showed that T1PKS (type I polyketide synthases)-NRPS like and betalactone existed solely in Auriscalpium, while T1PKS-terpene, strobilurin, T1PKS, and indole alkaloids were only found in Strobilurus (Table 3). Our results revealed that the average number of gene clusters for SMs in Auriscalpium fungi was fewer (average 18.33) than that in Strobilurus (average 21.25). As expected, each species of Strobilurus harbors one SM clusters of strobilurin (Table 3). Fungi in Auriscalpium contain very few gene clusters for NRPS-like but abundant for terpenes, while fungi in Strobilurus are the opposite (Table 3). In summary, the number of genes predicted for the synthesis of NRPS-like, T1PKS, strobilurin and indole alkaloids in Strobilurus fungi is higher than those in Auriscalpium taxa.
3.8. Result of Species Tree Construction, Divergence Time Estimation and Gene Family Expansions of CAZymes Analysis
Maximum-likelihood phylogenetic analysis using concatenated nucleotide sequences of the 1519 single-copy orthologs identified through OrthoMCL analysis present in all species of 22 fungi. Most of the nodes have high support rate, which proves that our phylogenetic tree can well analyze the phylogenetic and evolutionary relationship between species. The result of divergence time estimation is shown in Figure 5a.
Among those expanded Ortholog Cluster Groups (OCGs) examined using CAFE since their most recent common ancestor (MRCA) bifurcated, S. stephanocystis contained 3 OCGs of CAZymes including OCG97 (AA3_3) (gene number (GM) 5), OCG128 (AA7) (GM 4) and OGC281 (GH18) (GM 4), S. luchuensis contained 6 OCGs of CAZymes including CG1483 (AA2) (GM 3), OCG128 (AA7) (GM 2), OCG338 (AA7) (GM 3), OCG509 (AA7) (GM 2), OCG847 (GH11) (GM 4) and OCG73 (GH35) (GM 8), S. pachycystidiatus contained 6 OCGs of CAZymes including OCG24 (AA1_1) (GM 9), OCG128 (AA7) (GM 4), OCG6763 (AA7) (GM 3), OCG 119 (GH7) (GM 8) and OCG489 (GH18) (GM 4), S. orientalis contained 6 OCGs of CAZymes including OCG82 (AA3_2) (GM 6), OCG338 (AA7) (GM 6), OCG665 (AA7) (GM 5), OCG281 (GH18) (GM 6), OCG436 (GH28) (GM 3) and OCG73 (GH35) (GM 10), A. microsporum contained 5 OCGs of CAZymes including OCG14 (AA3_2) (GM 13), OCG97 (AA3_3) (GM 6), OCG338 (AA7) (GM 4), OCG119 (GH7) (GM 6), and OCG847 (GH11) (GM 5), A. vulgare contained 5 OCGs of CAZymes including OCG14 (AA3_2) (GM 19), OCG624 (AA3_2) (GM 6), OCG128 (AA7) (GM 12), OCG509 (AA7) (GM 6), and OCG910 (AA9) (GM 3), and A. orientale contained 3 OCGs of CAZymes including OCG24 (AA1_1) (GM 13), OCG130 (AA4) (GM 12), and OCG259 (GH79) (GM 3) (Figure 5b; Table S11). Those expanded OCGs of other species shown in Figure 5b and Table S11.
4. Discussion
4.1. Successive Decomposition of Pinecones by Fungi of Auriscalpium and Strobilurus
The successive decomposition of substrates by microbial communities is a common phenomenon [28,62]. Often, the microbial community structure, including the relative abundances of saprotrophic fungi, changes significantly during the successive decomposition process [63]. Though occupying the same ecological niche, species in these communities may develop unique but complementary strategies to partition the resources in the substrates, leading to temporal niche differentiation and divergence [64]. Here, part of the resource partition is temporal changes of fungi with different fungi use different sets of nutrients within the pinecone. Similar phenomena have been found in other substrates such as plant litters and deadwoods [3,13,65]. In the process of biodegradation, microbial coordination with different ecological strategies and certain orders are evident [65,66]. For example, successions of fungi in temperate forests were considered to be reflected in sugar utilizing fungi, followed by wood structural decaying fungi and finally residual decaying fungi in some cases [67]. This change may be explained in part by nutrients released by the primary decomposers that enabled the colonization of secondary decomposers [7]. However, the successive decomposition of substrates, such as deadwood and plant litter, requires the action and interaction of many fungi with their fungal community showing a high degree of complexity [3,68]. For example, Zhang and Wei [29] had carried out relevant research on fungi in the same forest, in which different fungi will appear on rotten wood in the same state, or even on the same rotten wood. At the same time, the same kind of fungi can also exist in different periods of rotten wood (Figure S1). Some fungi can only appear in one period, but most fungi can produce fruiting bodies at several stages of rotten wood [29], Figure S1. Similarly, Niemela et al. [28] reported the succession of more than one hundred species of lignicolous Basidiomycetes on fallen trunks in Picea obovata and P. sylvestris. Our study revealed that fungi in Auriscalpium and Strobilurus possess clear differences in the type and number of CAZymes and lignocellulolytic genes (Figure 2c, Figure 3a, Figure 4g,h and Figure S4b). Our results indicate that even though they colonize the same pinecones, there are significant divergence and niche differentiation in the utilization of substrates in pinecones between the fungi of the two genera, which leads to the dynamic changes of their colonization on the pinecones.
During the initial decomposition of pinecones, Auriscalpium fungi are the primary colonizers, likely related to their ability to break down resin and their strong capacity to decompose lignin and hemicellulose (Figure 4a–c; Table 2). Such abilities are common among WR fungi [69]. For example, the fungi of Ceriporiopsis subvermispora, Phellinus pini, Ganoderma australe, and Phlebia tremellosa specifically degrade lignin and hemicellulose among WR fungi [70]. The most compelling evidence supporting the early colonizing ability of Auriscalpium fungi is that their number of peptidases S8 and S53 is far greater than that in Strobilurus fungi. Peptidases S8 and S53 are among the top 10 most up-regulated enzymes in R. microporus in the presence of latex [50], Table 2. The genomic evidence is consistent with Auriscalpium fungi capable of colonizing newly fallen cones and decomposing proteins in resin rapidly. Polo et al. [71] showed that lignin and hemicellulose are in the outermost layer of plant cell wall which prevents the cellulolytic enzymes reaching the cellulose and protect plants from microbes. Our analyses demonstrated that the number of genes coding for lignin oxidases (AA2) and hemicellulase (GH3) in Auriscalpium are significantly higher than those in Strobilurus (Figure 4g,h; Table S9), and these genes may be related to lignin and hemicellulase decompositions in the outermost layer of pinecones. Similarly, in the analysis of gene family expansions, we found that the number of lignin related decomposition gene families was significantly higher in Auriscalpium than those of fungi in Strobilurus. For example, there are six gene families related to lignin decomposition expanded in Auriscalpium: A. microsporum including OCG14 (AA3_2) (GM 13) and OCG97 (AA3_3) (GM 6), A. vulgare including OCG14 (AA3_2) (GM 19) and OCG624 (AA3_2) (GM 6), and A. orientale including OCG24 (AA1_1) (GM 13) and OCG130 (AA4) (GM 12), while only four gene families related to lignin decomposition expanded in Strobilurus: S. stephanocystis including OCG97 (AA3_3) (GM 5), S. luchuensis including CG1483 (AA2) (GM 3), S. pachycystidiatus including OCG24 (AA1_1) (GM 9), and S. orientalis including OCG82 (AA3_2) (GM 6) (Figure 5b; Table S11). Once the outer layer is breached, the condition is now more favorable for the subsequent invasion of Strobilurus fungi. With increasing decay, the nutritional composition, physical structure, chemical composition and other aspects of the pinecones have changed, which result in the succession changes of fungal community.
After decomposition by Auriscalpium fungi, the proportions of lignin and hemicellulose in the pinecone would decrease and those of cellulose and pectin proportionally would increase (Figure 4a–c). The subsequent colonization by Strobilurus fungi relies on the residual components of the cones suitable for their growth and replacing the corresponding fungi of Auriscalpium (Figure 6). Similarly, comparing Auriscalpium and Strobilurus grown on the same pinecone, fungi of Strobilurus show decreasing trends of in the number of genes coding for ligninases and hemicellulases, but with higher number of genes coding for cellulase and pectinase, which is broadly consistent with the changes of cone components (Figure 4a–f). Strobilurus pachcystidiatus and A. microsporum also show the same pattern, but the differences are not particularly evident, which may relate to the fact that both could grow on newly fallen cones. However, S. orientalis grew on the highly rotten cones after decomposition by A. microsporum or S. pachcystidiatus and it showed a more obvious decrease in the number of ligninase-encoding genes and an increase in the number of cellulase-encoding genes than S. pachcystidiatus (Figure 4f). Accordingly, in the analysis of gene family expansions, we found that the number of GH gene families of fungi in Strobilurus was significantly higher than those of fungi in Auriscalpium. For example, there are eight gene families related to GH expanded in Strobilurus: S. stephanocystis including OGC281 (GH18) (GM 4), S. luchuensis including OCG847 (GH11) (GM 4) and OCG73 (GH35) (GM 8), S. pachycystidiatus including OCG 119 (GH7) (GM 8) and OCG489 (GH18) (GM 4), and S. orientalis including OCG281 (GH18) (GM 6), OCG436 (GH28) (GM 3) and OCG73 (GH35) (GM 10), while only three gene families related to GH expanded in Auriscalpium: A. microsporum including OCG119 (GH7) (GM 6), and OCG847 (GH11) (GM 5) and A. orientale including OCG259 (GH79) (GM 3) (Figure 5; Table S11). These gene families of GHs are related to the decomposition of cellulose, hemicellulose and pectin, respectively. For example, GH7 is related to cellulose decomposition, GH11 and GH35 are related to hemicellulose decomposition, and GH28 is related to pectin decomposition. All the above results show that the fungi in Strobilurus may have a good decomposition effect on utilizing the remaining organic compounds of the decomposed cones.
In addition, in the field, we observed that the fungi of Auriscalpium can decompose cones independently, especially in tropical areas, but the successive decomposition of the two genera is more common. However, we did not observe the decomposition of cones by fungi of Strobilurus independently. In each distribution areas of fungi in Strobilurus, fungi in Auriscalpium could be collected in different periods, and the fungi in Strobilurus collected all grow on the cones with high degree of decay. For successive decomposition of P. armandii’s cones, in addition to the most common combination of A. microsporum-S. pachcystidiatus-S. orientalis, we also observed the combinations of A. microsporum-S. pachcystidiatus and A. microsporum-S. orientalis. Therefore, various situations may occur in the field (Figure 6). Although Strobilurus fungi always appear on cones with a high degree of decomposition, however, the results of our field observation show that compared with Auriscalpium fungi, the Strobilurus fungi can occupy the cone for a long time and fully decompose the cone. There have been reports on the positive correlation between the large amount of CAZymes in the genome and the degradation of plant biomass [72], so we speculate that fungi in Strobilurus are the main decomposer with the type and number of CAZymes in Strobilurus being richer than those of Auriscalpium (Figure 2b,c and Figure 3a). In the CAZymes comparisons between the two genera, only seven CAZyme gene families have significantly more genes in the Auriscalpium fungi than in Strobilurus, while the other 36 gene families have more genes in Strobilurus fungi than in Auriscalpium fungi (Table S5), which broadly supports that the subsequent decomposers are main components of substrate decompositions [17].
4.2. Fungal Competition of Auriscalpium and Strobilurus on Pinecones
Successive decomposition may be affected by fungal competition. Fungal competitions of Auriscalpium and Strobilurus on substrates correspond to the two functional types: primary resource capture and secondary resource capture, respectively [7], so that the two genera appear orderly on the cones. On newly fallen cones, there may be hundreds of fungi competing to colonize cones on the surface, which is similar to the situation reported in woods [73]. However, the most obvious obstacles for fungi on newly fallen cones are the lack of easily assimilated nutrient matrixes such as lignin and hemicellulose, and the presences of inhibitory substances such as resin, etc. [74]. Due to their ability to breakdown these resistant substances, Auriscalpium can gain a competitive edge with other microorganisms in microbial community on newly fallen cones.
With the increase of the decomposition levels, the competitive pressures on the cones gradually increase attributed to the disappearance or reduction of antibacterial or antifungal substances. On one hand, the cones decomposed by the Auriscalpium provide suitable conditions for Strobilurus growth. During this period, the fruiting bodies of Strobilurus and Auriscalpium can co-exist on the same cone (Figure S3). On the other hand, fungi of Strobilurus, as aggressive competitors, produce toxins (e.g., strobilurin) to enhance its competitiveness with Auriscalpium and other microorganisms (Figure S3). For nutrients and spaces, the antagonists can either interact directly with the competitors [75], or secrete antibiotics to suppress competitors [76]. Some studies on antifungal SMs and enzymes produced by fungi with antagonists have been conducted extensively in vitro [77,78]. Through SM clusters analyses, we find that the number of SM clusters of the genus Auriscalpium is fewer than that of Strobilurus (Table 3), while all fungi of Strobilurus contain a fungicidal strobilurin cluster whose derivatives have broad spectrum antifungal activity, and, thus, are widely used as biological fungicide [79]. In addition, the number of NRPS-like in the genomes of Strobilurus is higher than those in Auriscalpium (Table 3). Most of the NRPS, PKS and their combinations have antibacterial and antifungal activities [80], which indicates that the fungi in Strobilurus have stronger antibacterial and antifungal activities than those in Auriscalpium. Aside from the SMs, other factors such as microclimate, the size and quality of nutrient sources can shift the balance between fungal decomposer groups [7]. Thus, in the natural environment, the outcomes of interactions between fungi are variable, leading to differences in community structures.
5. Conclusions
Our results reveal that there are differentiations of temporal and trophic niches for fungi of Auriscalpium and Strobilurus. The decompositions of pinecones are frequently completed by these two groups of fungi through successive colonization, occupying the same physical niche but at different times. The primary colonizers were the fungi of Aurscalpium and the secondary ones were Strobilurus (Figure 6). The CAZymes of the two groups of fungi are highly unique but also complementary, leading to the complete biodegradation pinecones. For future research directions, we will clearly delineate successional timelines between Auriscalpium and Strobilurus to inform the mechanisms of successional decomposition through controlling expression profiles based on these timelines. Further associating these profiles with realized relative loss in the cones over the time course has the potential to link critical successional dynamics to functional outcomes.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/jof7080679/s1, Figure S1: The distribution characteristics of wood decay fungi in different degrees of decay in Fenglin Nature Reserve (data from Zhang and Wei 2016). For the graphs, the x-axis shows different species and the y-axis shows number of occurrences of collected or observed fruiting bodies on wood in different years in Fenglin Nature Reserve. Different colors represent different levels of decay, and lengths represent numbers, Figure S2: The successive decomposition of Pinus armandii cones by Auriscalpium and Strobilurus. (a–b) A. microsporum fruiting from 2017 to 2019. (c–d) S. pachcystidiatus fruiting from 2018 to 2020. (e–f) S. orientalis fruiting from 2019 to now, Figure S3: The successive decomposition of cones by Auriscalpium and Strobilurus in the field. (a) Auriscalpium (white arrow) and Strobilurus (red arrow) on cone of P. armandii. (b–f) Auriscalpium (white arrow) and Strobilurus (red arrow) on cone of P. subgenus Pinus, Figure S4: The abundance and distributions of lignocellulolytic genes of Auriscalpium and Strobilurus demonstrated by multidimensional clustering approaches. (a) wood decay fungi (WDF) plotted on the first two principal components from principal component analyses (PCA) of lignocellulolytic genes. (b) Heatmap analysis of lignocellulolytic genes Numbers of family members in each genome are shown. Overrepresented (+4 to 0) and underrepresented (0 to −4) numbers are depicted as scores for each line in heatmap. The clustering on the left involves gene families with the same pattern in number. On the right is the name of the gene family, Figure S5: Analyses of carbohydrate-active enzymes (CAZymes) and lignocellulolytic genes within genera of Auriscalpium and Strobilurus derived from heatmap, and principal component analyses (PCA). (a) Heatmap analysis of three fungi of CAZymes in Auriscalpium. (b) Heatmap analysis of four fungi of CAZymes in Strobilurus. (c) Heatmap analysis of three fungi of lignocellulolytic genes in Auriscalpium. (d) Heatmap analysis of four fungi of lignocellulolytic genes in Strobilurus, Table S1: The collection information of fungi in Auriscalpium and Strobilurus, Table S2: Sequencing statistics, Table S3: Assessment of the protein gene set completeness in Auriscalpium and Strobilurus using BUSCO, Table S4: Gene distribution of carbohydrate-active enzymes (CAZymes) in Auriscalpium and Strobilurus fungi and the other 15 fungi, Table S5: Statistical analyses revealed that the average number of carbohydrate-active enzymes (CAZymes) in Auriscalpium and Strobilurus fungi, Table S6: The comparison of carbohydrate-active enzymes (CAZymes) between Auriscalpium and other WR fungi, Table S7: The comparison of carbohydrate-active enzymes (CAZymes) between Strobilurus and other WR fungi, Table S8: Gene distribution of lignocellulolytic genes in Auriscalpium and Strobilurus fungi and the other 15 fungi, Table S9: Statistical analyses revealed that the average number of lignocellulolytic genes in Auriscalpium and Strobilurus fungi, Table S10: Raw data of four major chemical components of cones before and after decomposition by fungi in Auriscalpium, Table S11: Gene family expansion of CAZymes in 22 fungi. Table S12: gff3 files of fungi in Auriscalpium and Strobilurus. Table S13: functional annotation of predicted gene models of fungi in Auriscalpium and Strobilurus based on similarity to annotated genes.
Author Contributions
Z.L.Y. designed the research and revised the manuscript; P.W. performed experiments, analyzed the data and wrote the manuscript; J.X. and G.W. provided help and advice, and revised the manuscript; T.L. provided samples for investigated the chemical compositions of pinecones. All authors have read and agreed to the published version of the manuscript.
Funding
The present research work was supported by the Strategic Priority Research Program of Chinese Academy of Sciences (XDB31000000) and Yunnan Ten-Thousand-Talents Plan-Yunling Scholar Project.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The genomic data of our study were deposited in DDBJ/EMBL/GenBank including the A. vulgare (Accession number: JAHBBC000000000; BioProject: PRJNA728955; BioSample:SAMN19107592), A. microsporum (Accession number: JAHLMG000000000; BioProject: PRJNA735801; BioSample: SAMN19598194), A. orientale (Accession number: JAHLMH000000000; BioProject: PRJNA735804; BioSample: SAMN19598576), S. luchuensis (Accession number: JAHLMJ000000000; BioProject: PRJNA735851; BioSample: SAMN19599093), S. pachycystidiatus (Accession number: JAHLMK000000000; BioProject: PRJNA735855; BioSample: SAMN19599128), S. stephanocystis (Accession number: JAHLMI000000000; BioProject: PRJNA735844; BioSample: SAMN19598582) and S. orientalis (Accession number: JAHLML000000000; BioProject: PRJNA735858; BioSample: SAMN19599131).
Acknowledgments
We are grateful to Fungal Biodiversity Centre in Netherlands (CBS) for providing us cultures of Auriscalpium and Strobilurus. We thank Xiang-Hua Wang, Hong Luo, Yan-Chun Li, Zai-Wei Ge, Feng Bang, Qing Cai, Jiao Qin, Qi Zhao, Xiao-Bin Liu, Xin Xu, Kui Wu, Hu Sun, Sheng-Wen Zhou, Cong Huang, Yi-Feng An, Jian-Yun Wu, Hua Qu, Peng-Cheng Yuan, and Jian-Wei Liu and Mei-Xiang Li, Xiao-Xia Ding (KIB), Xiao-Dan Yu (Shenyang Agricultural University), Tolgor Bau (Jilin Agricultural University), Ping Zhang (Hunan Normal University), Li-Hong Han (Qujing Normal University), Yan-Jia Hao (Anhui Normal University), Shi-Bin Jiao, De-Xian Kong for having provided us samples/specimens, collection information and/or color images. We thank Francis M. Martin (National Research Institute for Agriculture, Food and Environment, France), Li-Hong Han (QuJing Normal University), Hong Luo, Bang Feng, Qing Cai (KIB) and Shao-Xing Chen (Anhui Agricultural University), Xiao-Na Shao (Xishuangbanna Tropical Botanical Garden), Yan-Jia Hao (Anhui Normal University) and Jiao Qin (KIB) for their constructive and illuminating comments, criticisms and valuable suggestions. We appreciate Lian-Fu Chen, Xiao-Bin Liu for valuable information on data analysis.
Conflicts of Interest
The authors declare no conflict of interest.
References
- García-Palacios, P.; McKie, B.G.; Handa, I.T.; Frainer, A.; Hättenschwiler, S. The importance of litter traits and decomposers for litter decomposition: A comparison of aquatic and terrestrial ecosystems within and across biomes. Funct. Ecol. 2016, 30, 819–829. [Google Scholar] [CrossRef] [Green Version]
- Giweta, M. Role of litter production and its decomposition, and factors affecting the processes in a tropical forest ecosystem: A review. J. Ecol. Environ. 2020, 44, 11. [Google Scholar] [CrossRef]
- Baldrian, P.; Zrustová, P.; Tláskal, V.; Davidová, A.; Merhautová, V.; Vrka, T. Fungi associated with decomposing deadwood in a natural beech-dominated forest. Fungal Ecol. 2016, 23, 109–122. [Google Scholar] [CrossRef]
- Katagiri, S.; Shiba, T.; Tohara, H.; Yamaguchi, K.; Hara, K.; Nakagawa, K.; Komatsu, K.; Watanabe, K.; Ohsugi, Y.; Maekawa, S.; et al. Re-initiation of oral food intake following enteral nutrition alters oral and gut microbiota communities. Front. Cell. Infect. Microbiol. 2019, 9, 434. [Google Scholar] [CrossRef] [Green Version]
- Eichlerová, I.; Homolka, L.; Žifčáková, L.; Lisá, L.; Dobiášová, P.; Baldrian, P. Enzymatic systems involved in decomposition reflects the ecology and taxonomy of saprotrophic fungi. Fungal Ecol. 2015, 13, 10–22. [Google Scholar] [CrossRef]
- Rajala, T.; Peltoniemi, M.; Hantula, J.; Mäkipää, R.; Pennanen, T. RNA reveals a succession of active fungi during the decay of Norway spruce logs. Fungal Ecol. 2011, 4, 437–448. [Google Scholar] [CrossRef]
- Boddy, L. Interspecific combative interactions between wood-decaying basidiomycetes. FEMS Microbiol. Ecol. 2000, 31, 185–194. [Google Scholar] [CrossRef]
- Moor, H.; Nordén, J.; Penttil, R.; Siitonen, J.; Snll, T. Long-term effects of colonization-extinction dynamics of generalist versus specialist wood-decaying fungi. J. Ecol. 2020, 109, 491–503. [Google Scholar] [CrossRef]
- Krah, F.S.; Seibold, S.; Brandl, R.; Baldrian, P.; Müller, J.; Bässler, C. Independent effects of host and environment on the diversity of wood-inhabiting fungi. J. Ecol. 2018, 106, 1428–1442. [Google Scholar] [CrossRef]
- Rajala, T.; Peltoniemi, M.; Pennanen, T.; Mäkipää, R. Relationship between wood-inhabiting fungi determined by molecular analysis (DGGE) and quality of decaying logs. Can. J. For. Res. 2010, 40, 2384–2397. [Google Scholar] [CrossRef]
- Šnajdr, J.; Cajthaml, T.; Valášková, V.; Merhautová, V.; Petránková, M.; Spetz, P.; Leppänen, K.; Baldrian, P. Transformation of Quercus petraea litter: Successive changes in litter chemistry are reflected in differential enzyme activity and changes in the microbial community composition. FEMS Microbiol. Ecol. 2011, 75, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Riley, R.; Salamov, A.A.; Brown, D.W.; Nagy, L.G.; Floudas, D.; Held, B.W.; Levasseur, A.; Lombard, V.; Morin, E.; Otillar, R.; et al. Extensive sampling of basidiomycete genomes demonstrate inadequacy of the white-rot/brown-rot paradigm for wood decay fungi. Proc. Natl. Acad. Sci. USA 2014, 111, 9923–9928. [Google Scholar] [CrossRef] [Green Version]
- Edman, M.; Eriksson, A.M. Competitive outcomes between wood-decaying fungi are altered in burnt wood. FEMS Microbiol. Ecol. 2016, 92, fiw068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edman, M.; Fällström, I. An introduced tree species alters the assemblage structure and functional composition of wood-decaying fungi in microcosms. For. Ecol. Manag. 2013, 306, 9–14. [Google Scholar] [CrossRef]
- Fukami, T.; Dickie, I.; Wilkie, J.P.; Paulus, B.C.; Park, D.; Roberts, A.; Buchanan, P.K.; Allen, R.B. Assembly history dictates ecosystem functioning: Evidence from wood decomposer communities. Ecol. Lett. 2010, 13, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Sasha, V.; Bhatnagar, J.M. An evolutionary signal to fungal succession during plant litter decay. FEMS Microbiol. Ecol. 2019, 95, fiz145. [Google Scholar] [CrossRef]
- Song, Z.W.; Vail, A.; Sadowsky, M.J.; Schilling, J.S. Competition between two wood-degrading fungi with distinct influences on residues. FEMS Microbiol. Ecol. 2012, 79, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Kohler, A.; Kuo, A.; Nagy, L.G.; Morin, E.; Barry, K.; Buscot, F.; Canbäck, B.; Choi, C.; Cichocki, N.; Clum, A.; et al. Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat. Genet. 2015, 47, 410–415. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, H.; Wang, C.; Xu, J.R. Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genom. 2013, 14, 274. [Google Scholar] [CrossRef] [Green Version]
- De Wit, P.J.G.M.; Van Der Burgt, A.; Ökmen, B.; Stergiopoulos, I.; Abd-Elsalam, K.A.; Aerts, A.L.; Bahkali, A.H.; Beenen, H.G.; Chettri, P.; Cox, M.P.; et al. The genomes of the fungal plant pathogens Cladosporium fulvum and Dothistroma septosporum reveal adaptation to different hosts and lifestyles but also signatures of common ancestry. PLoS Genet. 2012, 8, e1003088. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Kohler, A.; Murat, C.; Veneault-Fourrey, C.; Hibbett, D.S. Unearthing the roots of ectomycorrhizal symbioses. Nat. Rev. Microbiol. 2016, 14, 760–773. [Google Scholar] [CrossRef]
- Nagy, L.G.; Riley, R.; Tritt, A.; Adam, C.; Daum, C.; Floudas, D.; Sun, H.; Yadav, J.S.; Pangilinan, J.; Larsson, K.-H.; et al. Comparative genomics of early-diverging mushroom-forming fungi provides insights into the origins of lignocellulose decay capabilities. Mol. Biol. Evol. 2016, 33, 959–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.J.; Jeong, Y.U.; Kong, W.S. Genome sequencing and carbohydrate-active enzyme (CAZyme) repertoire of the white rot fungus Flammulina elastica. Int. J. Mol. Sci. 2018, 19, 2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Yan, B.; Mo, S.; Nie, S.; Li, Q.; Ou, Q.; Wu, B.; Jiang, G.; Tang, J.; Li, N.; et al. Carbohydrate metabolism genes dominant in a subtropical marine mangrove ecosystem revealed by metagenomics analysis. J. Microbiol. 2019, 57, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Arfi, Y.; Levasseur, A.; Record, E. Differential gene expression in Pycnoporus coccineus during interspecific mycelial interactions with different competitors. Appl. Environ. Microbiol. 2013, 79, 6626–6636. [Google Scholar] [CrossRef] [Green Version]
- Saha, P.; Roy-Barman, S. The role of the global regulator of secondary metabolism laea in different fungi. Curr. J. Appl. Sci. Technol. 2018, 31, 1–5. [Google Scholar] [CrossRef]
- Niemela, T.; Renvall, P.; Pentilla, R. Interactions of fungi at late stages of wood decomposition. Ann. Bot. Fenn. 1995, 32, 141–152. [Google Scholar]
- Zhang, L.Y.; Wei, Y.L. Species diversity and distribution characters of wood-decaying fungi in Fenglin Nature Reverse. Chin. J. Ecol. 2016, 35, 2745–2751. [Google Scholar] [CrossRef]
- Qin, J.; Horak, E.; Popa, F.; Rexer, K.H.; Yang, Z.L. Species diversity, distribution patterns, and substrate specificity of Strobilurus. Mycologia 2018, 110, 584–604. [Google Scholar] [CrossRef]
- Wang, P.M.; Yang, Z.L. Two new taxa of the Auriscalpium vulgare species complex with substrate preferences. Mycol. Prog. 2019, 18, 641–652. [Google Scholar] [CrossRef]
- Mayjonade, B.; Gouzy, J.; Donnadieu, C.; Pouilly, N.; Marande, W.; Callot, C.; Langlade, N.; Muños, S. Extraction of high-molecular-weight genomic DNA for long-read sequencing of single molecules. Biotechniques 2016, 61, 203–205. [Google Scholar] [CrossRef]
- Zhu, N.; Huang, W.; Wu, D.; Chen, K.; He, Y. Quantitative visualization of pectin distribution maps of peach fruits. Sci. Rep. 2017, 7, 9275. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, K.K.; Labutti, K.; Khalak, A.; Tse, D. FinisherSC: A repeat-aware tool for upgrading de novo assembly using long reads. Bioinformatics 2015, 31, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.C.; Jan, S.; Bertil, S. Musket: A multistage k-mer spectrum-based error corrector for Illumina sequence data. Bioinformatics 2013, 29, 308–315. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chen, Z.; Huang, G.; Yu, T.; Yang, P.; Li, J.; Fu, Y.; Yuan, S.; Chen, S.; Xu, A. HaploMerger: Reconstructing allelic relationships for polymorphic diploid genome assemblies. Genome Res. 2012, 22, 1581–1588. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.F.; Kang, M.J.; Xu, A.L. HaploMerger2: Rebuilding both haploid sub-assemblies from high-heterozygosity diploid genome assembly. Bioinformatics 2017, 33, 2577–2579. [Google Scholar] [CrossRef]
- Sipos, G.; Prasanna, A.N.; Walter, M.C.; O’Connor, E.; Balint, B.; Krizsán, K.; Kiss, B.; Hess, J.; Varga, T.; Slot, J.; et al. Genome expansion and lineage-specific genetic innovations in the forest pathogenic fungi Armillaria. Nat. Ecol. Evol. 2017, 1, 1931–1941. [Google Scholar] [CrossRef] [Green Version]
- Varga, T.; Krizsán, K.; Földi, C.; Dima, B.; Sánchez-García, M.; Sánchez-Ramírez, S.; Szöllősi, G.; Szarkándi, J.G.; Papp, V.; Albert, L.; et al. Megaphylogeny resolves global patterns of mushroom evolution. Nat. Ecol. Evol. 2019, 3, 668–678. [Google Scholar] [CrossRef] [Green Version]
- Li, T.C.; Yu, L.Y.; Song, B.; Song, Y.; Li, L.; Lin, X.; Lin, S.J. Genome improvement and core gene set refinement of Fugacium kawagutii. Microorganisms 2020, 8, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Gong, Y.; Cai, Y.; Liu, W.; Zhou, Y.; Xiao, Y.; Xu, Z.; Liu, Y.; Lei, X.; Wang, G.; et al. Genome sequence of the edible cultivated mushroom Lentinula edodes (shiitake) reveals insights into lignocellulose degradation. PLoS ONE 2016, 11, e0160336. [Google Scholar] [CrossRef]
- Eastwood, D.C.; Floudas, D.; Binder, M.; Majcherczyk, A.; Schneider, P.; Aerts, A.; Asiegbu, F.O.; Baker, S.E.; Barry, K.; Bendiksby, M.; et al. The plant cell wall-decomposing machinery underlies the functional diversity of forest fungi. Science 2011, 333, 762–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floudas, D.; Binder, M.; Riley, R.; Barry, K.; Blanchette, R.A.; Henrissat, B.; Martínez, A.T.; Otillar, R.; Spatafora, J.W.; Yadav, J.S.; et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 2012, 336, 1715–1719. [Google Scholar] [CrossRef] [Green Version]
- Martinez, D.; Larrondo, L.; Putnam, N.; Gelpke, M.D.S.; Huang, K.; Chapman, J.; Helfenbein, K.G.; Ramaiya, P.; Detter, J.C.; Larimer, F.W.; et al. Genome sequence of the lignocellulose degrading fungus Phanerochaete chrysosporium strain RP78. Nat. Biotechnol. 2004, 22, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Ohm, R.A.; Riley, R.; Salamov, A.; Min, B.; Choi, I.G.; Grigoriev, I.V. Genomics of wood-degrading fungi. Fungal Genet. Biol. 2014, 72, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Olson, A.; Aerts, A.; Asiegbu, F.O.; Belbahri, L.; Bouzid, O.; Broberg, A.; Canbäck, B.; Coutinho, P.M.; Cullen, D.; Dalman, K.; et al. Insight into trade-off between wood decay and parasitism from the genome of a fungal forest pathogen. New Phytol. 2012, 194, 1001–1013. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; MacDonald, J.; Syed, K.; Salamov, A.; Hori, C.; Aerts, A.; Henrissat, B.; Wiebenga, A.; Vankuyk, P.A.; Barry, K.; et al. Comparative genomics of the white-rot fungi, Phanerochaete carnosa and P. chrysosporium, to elucidate the genetic basis of the distinct wood types they colonize. BMC Genom. 2012, 13, 444. [Google Scholar] [CrossRef] [Green Version]
- Oghenekaro, A.O.; Kovalchuk, A.; Raffaello, T.; Camarero, S.; Asiegbu, F.O. Genome sequencing of Rigidoporus microporus provides insights on genes important for wood decay, latex tolerance and interspecific fungal interactions. Sci. Rep. 2020, 10, 5250. [Google Scholar] [CrossRef] [Green Version]
- Konno, K. Plant latex and other exudates as plant defense systems: Roles of various defense chemicals and proteins contained therein. Phytochemistry 2011, 72, 1510–1530. [Google Scholar] [CrossRef]
- Keller, N.P.; Turner, G.; Bennett, J.W. Fungal secondary metabolism—From biochemistry to genomics. Nat. Rev. Microbiol. 2005, 3, 937–947. [Google Scholar] [CrossRef]
- Devi, S.; Kiesewalter, H.T.; Kovács, R.; Frisvad, J.C.; Weber, T.; Larsen, T.O.; Kovács, T.; Ding, L. Depiction of secondary metabolites and antifungal activity of Bacillus velezensis DTU001. Synth. Syst. Biotechnol. 2019, 4, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Medema, M.H.; Kazempour, D.; Fischbach, M.A.; Breitling, R.; Takano, E.; Weber, T. antiSMASH 2.0-a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013, 41, W204–W212. [Google Scholar] [CrossRef] [Green Version]
- Matys, V.; Fricke, E.; Geffers, R.; Gößling, E.; Haubrock, M.; Hehl, R.; Hornischer, K.; Karas, D.; Kel, A.E.; Kel-Margoulis, O.V.; et al. TRANSFAC1: Transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003, 31, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Jang, S.; Kim, S.; Kong, S.; Choi, J.; Ahn, K.; Kim, J.; Lee, S.; Park, B.; Jung, K.; et al. FTFD: An informatics pipeline supporting phylogenomic analysis of fungal transcription factors. Bioinformatics 2008, 24, 1024–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abascal, F.; Zardoya, R.; Posada, D. Prottest: Selection of best-fit models of protein evolution. Bioinformatics 2005, 21, 2104–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Sanderson, M.J. r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 2003, 19, 301–302. [Google Scholar] [CrossRef] [Green Version]
- Han, M.V.; Thomas, G.W.; Lugo-Martinez, J.; Hahn, M.W. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 2013, 30, 1987–1997. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.R.; Boddy, L.; Weightman, A.J. Bacteria in decomposing wood and their interactions with wood-decay fungi. FEMS Microbiol. Ecol. 2016, 92, fiw179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukasawa, Y.; Matuoka, S. Communities of wood-inhabiting fungi in dead pine logs along a geographical gradient in Japan. Fungal Ecol. 2015, 18, 75–82. [Google Scholar] [CrossRef]
- Friedemann, G.; Leshem, Y.; Kerem, L.; Shacham, B.; Bar-Massada, A.; McClain, K.M.; Bohrer, G.; Izhaki, I. Multidimensional differentiation in foraging resource use during breeding of two sympatric top predators. Sci. Rep. 2016, 6, 35031. [Google Scholar] [CrossRef] [Green Version]
- Herzog, C.; Hartmann, M.; Frey, B.; Stierli, B.; Brunner, I. Microbial succession on decomposing root litter in a drought-prone scots pine forest. ISME J. 2019, 13, 2346–2362. [Google Scholar] [CrossRef] [Green Version]
- Holmer, L.; Renvall, P.; Stenlid, J. Selective replacement between species of wood-rotting basidiomycetes, a laboratory study. Mycol. Res. 1997, 101, 714–720. [Google Scholar] [CrossRef]
- Stokland, J.N.; Siitonen, J.; Jonsson, B.G. Biodiversity in Dead Wood; Cambridge University Press: Cambridge, UK, 2012. [Google Scholar]
- Voříšková, J.; Baldrian, P. Fungal community on decomposing leaf litter undergoes rapid successional changes. ISME J. 2013, 7, 477–486. [Google Scholar] [CrossRef]
- Floudas, D.; Bentzer, J.; Ahrén, D.; Johansson, T.; Persson, P.; Tunlid, A. Uncovering the hidden diversity of litter-decomposition mechanisms in mushroom-forming fungi. ISME J. 2020, 14, 1–14. [Google Scholar] [CrossRef]
- Weng, C.H.; Peng, X.W.; Han, Y.J. Depolymerization and conversion of lignin to value-added bioproducts by microbial and enzymatic catalysis. Biotechnol. Biofuels 2021, 14, 84. [Google Scholar] [CrossRef] [PubMed]
- Polo, C.C.; Pereira, L.; Mazzafera, P.; Flores-Borges, D.N.A.; Meneau, F. Correlations between lignin content and structural robustness in plants revealed by x-ray ptychography. Sci. Rep. 2020, 10, 6023. [Google Scholar] [CrossRef] [PubMed]
- Adams, A.S.; Jordan, M.S.; Adams, S.M.; Suen, G.; Goodwin, L.A.; Davenport, K.W.; Currie, C.R.; Raffa, K.F. Cellulose-degrading bacteria associated with the invasive woodwasp Sirex noctilio. ISME J. 2011, 5, 1323–1331. [Google Scholar] [CrossRef]
- Coates, D.; Rayner, A.D.M. Fungal population and community development in cut beech logs. III. Spatial dynamics, interactions and strategies. New Phytol. 1985, 101, 153–171. [Google Scholar] [CrossRef]
- Abad, M.; Ansuategui, M.; Bermejo, P. Active antifungal substances from natural sources. Arch. Org. Chem. 2006, 2007, 116–145. [Google Scholar] [CrossRef]
- Lima, G.; Arru, S.; de Curtis, F.; Arras, G. Influence of antagonist, host fruit and pathogen on the biological control of postharvest fungal diseases by yeasts. J. Ind. Microbiol. Biotechnol. 1999, 23, 223–229. [Google Scholar] [CrossRef]
- Wilson, C.L.; Wisniewski, M.E.; Biles, C.L.; McLaughlin, R.; Chalutz, E.; Droby, S. Biological control of post-harvest diseases of fruits and vegetables: Alternatives to synthetic fungicides. Crop Prot. 1991, 10, 172–177. [Google Scholar] [CrossRef]
- Alves, M.J.; Ferreira, I.C.F.R.; Martins, A.; Pintado, M. Antimicrobial activity of wild mushroom extracts against clinical isolates resistant to different antibiotics. J. Appl. Microbiol. 2012, 113, 466–475. [Google Scholar] [CrossRef]
- Jonkers, W.; Rodriguez, E.A.E.; Lee, K.; Breakspear, A.; May, G.; Kistler, H.C. Metabolome and transcriptome of the interaction between Ustilago maydis and Fusarium verticillioides in vitro. Appl. Environ. Microbiol. 2012, 78, 3656–3667. [Google Scholar] [CrossRef] [Green Version]
- Niego, A.G.; Raspé, O.; Thongklang, N.; Charoensup, R.; Lumyong, S.; Stadler, M.; Hyde, K.D. Taxonomy, diversity and cultivation of the Oudemansielloid/Xeruloid taxa Hymenopellis, Mucidula, Oudemansiella, and Xerula with respect to their bioactivities: A review. J. Fungi 2021, 7, 51. [Google Scholar] [CrossRef]
- Fernandes, A.F.; Costa, L.; Sousa, J.R.; Zalocha, J.; Almeida, M.G. Biological activities of marine-derived actinomycetes: Testing the aqueous extracellular phase of Streptomyces aculeolatus. Ann. Med. 2019, 51 (Suppl. 1), 44. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Basidiomata and percentage of monthly occurrences of fruiting bodies of Auriscalpium and Strobilurus on pinecones. The basidiomata of Auriscalpium usually on newly fallen cones on the ground. The basidiomata of Strobilurus usually on highly rotten cones under the ground. For the graphs, the x-axis shows months and the y-axis shows percentage of monthly occurrences of collected or observed fruiting bodies on pinecones in different areas and years in Table S1.
Figure 1.
Basidiomata and percentage of monthly occurrences of fruiting bodies of Auriscalpium and Strobilurus on pinecones. The basidiomata of Auriscalpium usually on newly fallen cones on the ground. The basidiomata of Strobilurus usually on highly rotten cones under the ground. For the graphs, the x-axis shows months and the y-axis shows percentage of monthly occurrences of collected or observed fruiting bodies on pinecones in different areas and years in Table S1.

Figure 2.
The distributions of genes encoding carbohydrate-active enzymes (CAZymes) in Auriscalpium and Strobilurus fungi. (a) wood decay fungi (WDF) plotted on the first two principal components from principal component analyses (PCA) of CAZymes. (b) Comparative analyses of CAZymes associated with lignocellulose decomposition. AAs: Auxiliary Activities; CBMs: Carbohydrate-Binding Modules; CEs: Carbohydrate Esterases; GHs: Glycoside Hydrolases; GTs: Glycosyl Transferases; PLs: Polysaccharide Lyases. (c) Heatmap analysis of CAZymes showing the distributions of CAZymes among different fungi. Numbers of family members in each genome are demonstrated. Overrepresented (+4 to 0) and underrepresented (0 to −4) numbers are depicted as scores for each line in heatmap. The clustering on the left involves gene families with the same pattern in number. On the right is the name of the gene family.
Figure 2.
The distributions of genes encoding carbohydrate-active enzymes (CAZymes) in Auriscalpium and Strobilurus fungi. (a) wood decay fungi (WDF) plotted on the first two principal components from principal component analyses (PCA) of CAZymes. (b) Comparative analyses of CAZymes associated with lignocellulose decomposition. AAs: Auxiliary Activities; CBMs: Carbohydrate-Binding Modules; CEs: Carbohydrate Esterases; GHs: Glycoside Hydrolases; GTs: Glycosyl Transferases; PLs: Polysaccharide Lyases. (c) Heatmap analysis of CAZymes showing the distributions of CAZymes among different fungi. Numbers of family members in each genome are demonstrated. Overrepresented (+4 to 0) and underrepresented (0 to −4) numbers are depicted as scores for each line in heatmap. The clustering on the left involves gene families with the same pattern in number. On the right is the name of the gene family.

Figure 3.
Comparisons of the carbohydrate-active enzymes (CAZymes) in Auriscalpium and Strobilurus, the proportions of four major chemical components in different cones, and the proportions of different enzymes encoded by lignocellulolytic genes in the two fungi groups. (a) Heatmap analysis of seven fungi in Auriscalpium and Strobilurus of CAZymes. (b) Four components of three different newly fallen cones. A, B, C and D in x-axis represent lignin, cellulose, hemicellulose and pectin, respectively. The y-axis represents the proportion of the four components. (c) The proportions of lignocellulolytic genes in different fungi of Auriscalpium. A’, B’, C’, D’ in x-axis represent ligninase, cellulose, hemicellulose, pectinase, respectively. The y-axis represents the proportion of the four types of enzymes; the same below. (d) The proportions of lignocellulolytic genes in different fungi of Strobilurus.
Figure 3.
Comparisons of the carbohydrate-active enzymes (CAZymes) in Auriscalpium and Strobilurus, the proportions of four major chemical components in different cones, and the proportions of different enzymes encoded by lignocellulolytic genes in the two fungi groups. (a) Heatmap analysis of seven fungi in Auriscalpium and Strobilurus of CAZymes. (b) Four components of three different newly fallen cones. A, B, C and D in x-axis represent lignin, cellulose, hemicellulose and pectin, respectively. The y-axis represents the proportion of the four components. (c) The proportions of lignocellulolytic genes in different fungi of Auriscalpium. A’, B’, C’, D’ in x-axis represent ligninase, cellulose, hemicellulose, pectinase, respectively. The y-axis represents the proportion of the four types of enzymes; the same below. (d) The proportions of lignocellulolytic genes in different fungi of Strobilurus.

Figure 4.
The changes of four major chemical components of cones before and after decomposition, and the relationship to differences of the numbers of lignocellulolytic genes between Strobilurus and Auriscalpium fungi colonizing the same pinecones. (a) The proportion of four components in cones of P. yunnanensis before and after the decomposition by A. orientale. A, B, C and D in x-axis represent lignin, cellulose, hemicellulose and pectin, respectively. The y-axis represents the proportion of the four components. White represents undecomposed cones, while gray represents decomposed cones. the same below. (b) The proportion of four components in cones of P. sylvestris before and after the decomposition by A. vulgare. (c) The content changes of four components in cones of P. armandii before and after being decomposed by A. microsporum. (d) Comparison of lignocellulolytic genes. A’, B’, C’ and D’ in x-axis represent ligninase, cellulose, hemicellulose and pectinase, respectively. The y-axis represents the proportion of the four types of enzymes in A. orientalis and S. luchuensis grown on cones of P. yunnanensis. The same below. (e) Comparison of lignocellulolytic genes in A. vulgare and S. stephanocystis on cones of P. sylvestris. (f) Comparisons of lignocellulolytic genes of A. microsporum, S. pachcystidiatus and S. orientalis grown on cones of P. armandii. (g) Heatmap analysis of lignocellulolytic genes of four fungi on cones of P. subgenus Pinus. (h) Heatmap analysis of lignocellulolytic genes of three fungi on cones of P. subgenus Strobus.
Figure 4.
The changes of four major chemical components of cones before and after decomposition, and the relationship to differences of the numbers of lignocellulolytic genes between Strobilurus and Auriscalpium fungi colonizing the same pinecones. (a) The proportion of four components in cones of P. yunnanensis before and after the decomposition by A. orientale. A, B, C and D in x-axis represent lignin, cellulose, hemicellulose and pectin, respectively. The y-axis represents the proportion of the four components. White represents undecomposed cones, while gray represents decomposed cones. the same below. (b) The proportion of four components in cones of P. sylvestris before and after the decomposition by A. vulgare. (c) The content changes of four components in cones of P. armandii before and after being decomposed by A. microsporum. (d) Comparison of lignocellulolytic genes. A’, B’, C’ and D’ in x-axis represent ligninase, cellulose, hemicellulose and pectinase, respectively. The y-axis represents the proportion of the four types of enzymes in A. orientalis and S. luchuensis grown on cones of P. yunnanensis. The same below. (e) Comparison of lignocellulolytic genes in A. vulgare and S. stephanocystis on cones of P. sylvestris. (f) Comparisons of lignocellulolytic genes of A. microsporum, S. pachcystidiatus and S. orientalis grown on cones of P. armandii. (g) Heatmap analysis of lignocellulolytic genes of four fungi on cones of P. subgenus Pinus. (h) Heatmap analysis of lignocellulolytic genes of three fungi on cones of P. subgenus Strobus.

Figure 5.
Phylogenetic tree of fungi in Auriscalpium and Strobilurus with other 15 fungal species and gene family expansions of CAZymes using CAFE. (a) The topology of the phylogenetic tree was constructed by the maximum likelihood method and divergence time estimation using r8s. Their divergence time was marked on the phylogenetic tree with time scale being shown by MYA (million years ago). (b) gene family expansions of CAZymes in 22 fungi with p-values less than 0.05. “O” in the first row of the table represents OCG (Ortholog Cluster Group); The second row of the table represents the gene family information of CAZymes. The length of the red rectangle represents the quantity; “--“ represents that the gene family has not been significantly expanded in this species.
Figure 5.
Phylogenetic tree of fungi in Auriscalpium and Strobilurus with other 15 fungal species and gene family expansions of CAZymes using CAFE. (a) The topology of the phylogenetic tree was constructed by the maximum likelihood method and divergence time estimation using r8s. Their divergence time was marked on the phylogenetic tree with time scale being shown by MYA (million years ago). (b) gene family expansions of CAZymes in 22 fungi with p-values less than 0.05. “O” in the first row of the table represents OCG (Ortholog Cluster Group); The second row of the table represents the gene family information of CAZymes. The length of the red rectangle represents the quantity; “--“ represents that the gene family has not been significantly expanded in this species.

Figure 6.
Types of successive decomposition of pinecones observed in the field. (a,b) The decomposition of cones of P. subgenus Pinus. (a) The cones decomposed by A. vulgare or A. orientale independently. (b) Successive decomposition and competition of A. orientale/S. luchuensis and A. vulgare/S. stephanocystis on cones of P. subgenus Pinus. The yellow brown pileus represents S. luchuensis or S. stephanocystis. (c–f) The decomposition of cones of P. subgenus Strobus. (c) The cones decomposed by A. microsporum independently. (d) Successive decomposition and competition of A. microsporum and S. orientalis on cones of P. subgenus Strobus. The gray brown pileus represents S. orientalis. (e) Successive decomposition and competition of A. microsporum and S. pachcystidiatus on cones of P. subgenus Strobus. The yellow pileus represents S. pachcystidiatus. (f) Successive decomposition and competition of A. microsporum, S. pachcystidiatus and S. orientalis on cones of P. subgenus Strobus.
Figure 6.
Types of successive decomposition of pinecones observed in the field. (a,b) The decomposition of cones of P. subgenus Pinus. (a) The cones decomposed by A. vulgare or A. orientale independently. (b) Successive decomposition and competition of A. orientale/S. luchuensis and A. vulgare/S. stephanocystis on cones of P. subgenus Pinus. The yellow brown pileus represents S. luchuensis or S. stephanocystis. (c–f) The decomposition of cones of P. subgenus Strobus. (c) The cones decomposed by A. microsporum independently. (d) Successive decomposition and competition of A. microsporum and S. orientalis on cones of P. subgenus Strobus. The gray brown pileus represents S. orientalis. (e) Successive decomposition and competition of A. microsporum and S. pachcystidiatus on cones of P. subgenus Strobus. The yellow pileus represents S. pachcystidiatus. (f) Successive decomposition and competition of A. microsporum, S. pachcystidiatus and S. orientalis on cones of P. subgenus Strobus.


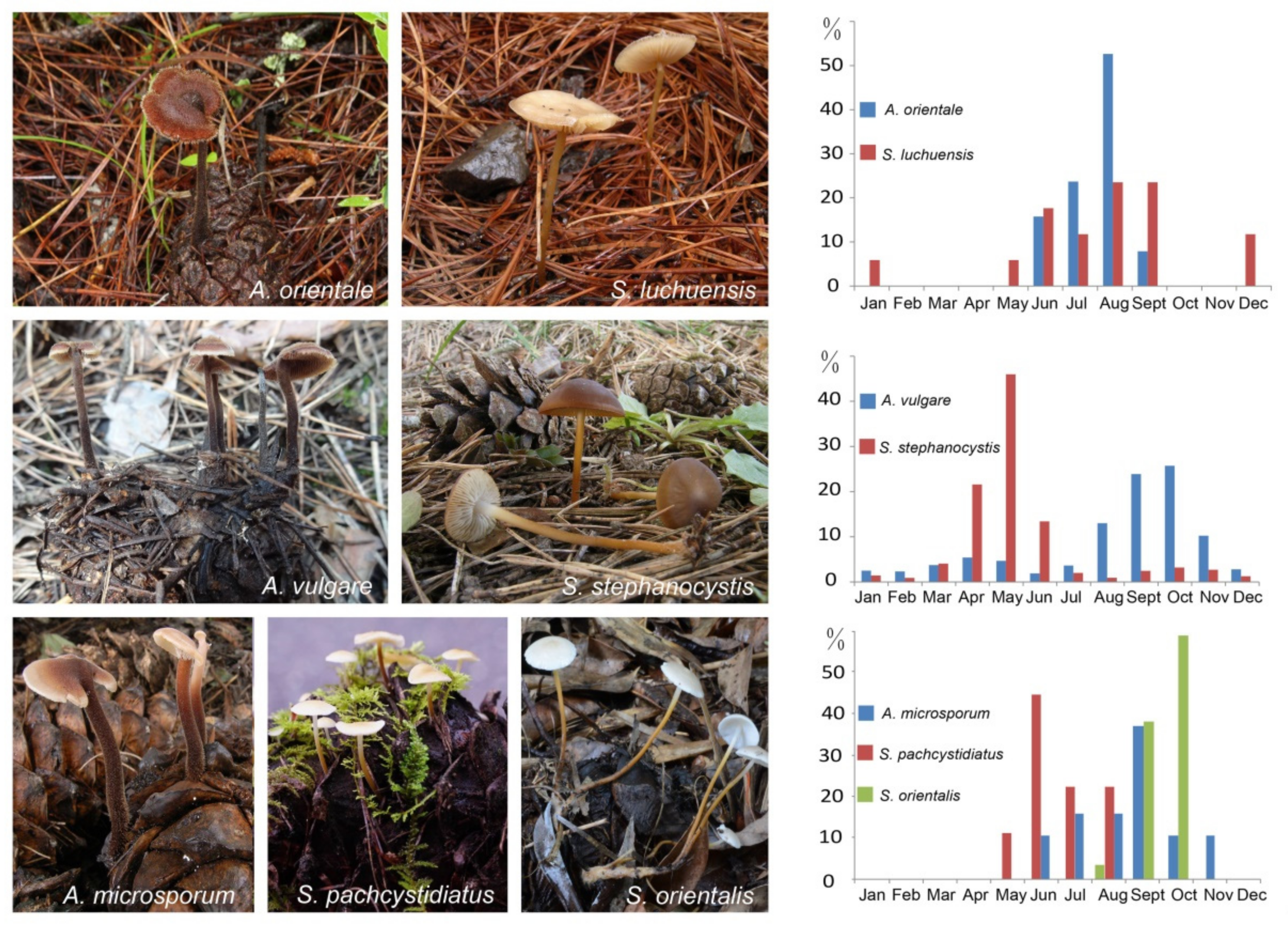{kind=link}
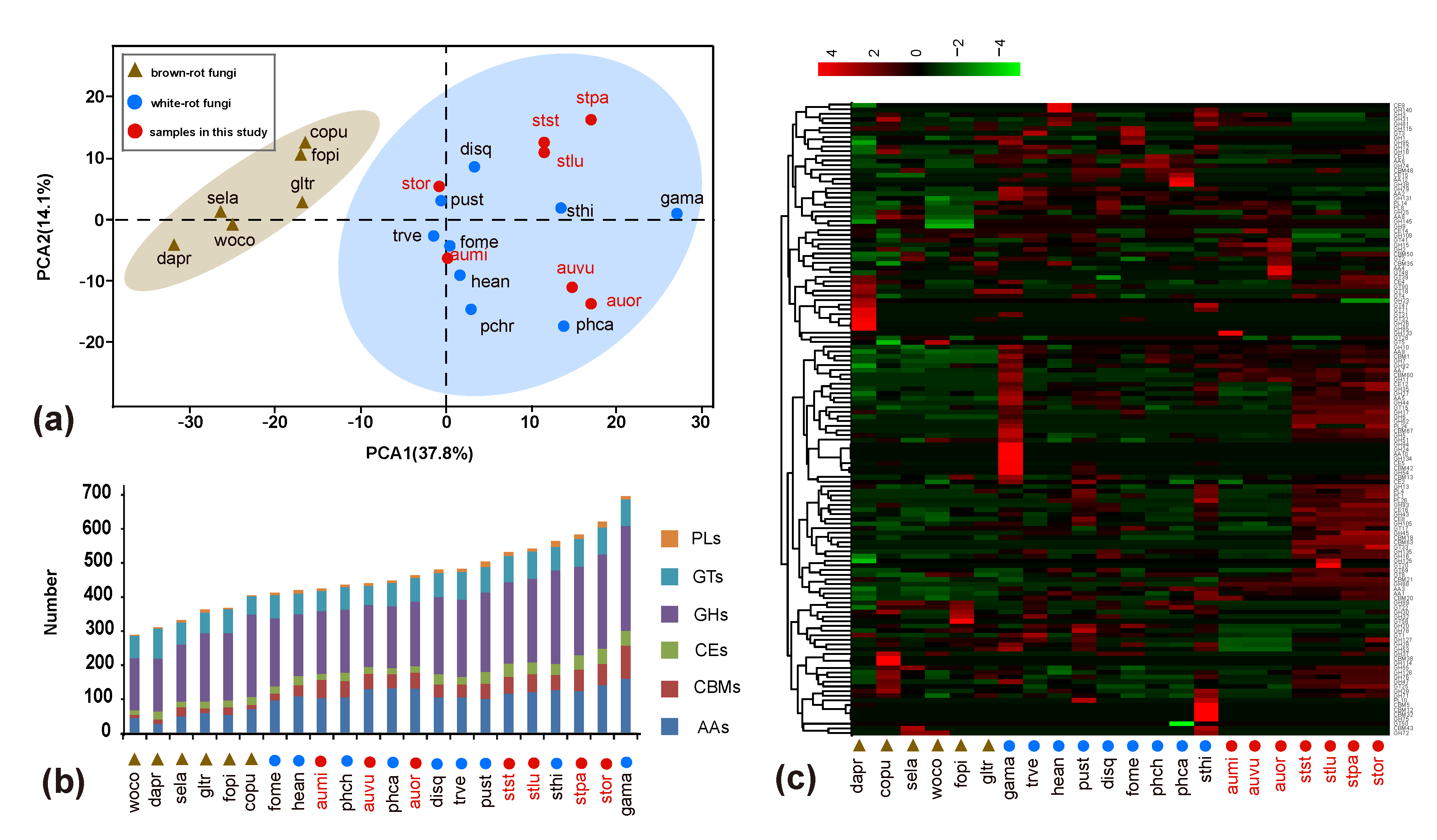{kind=link}
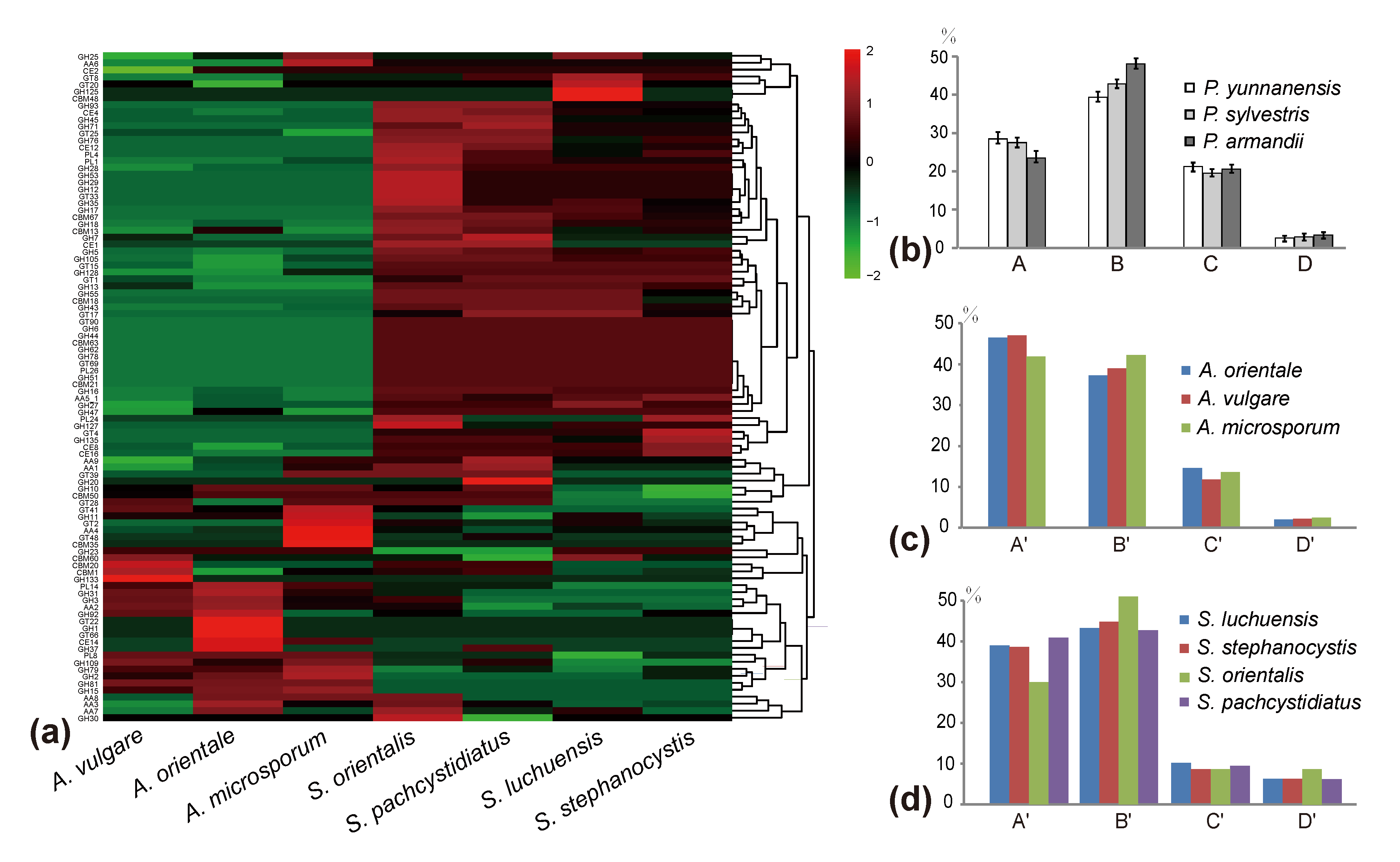{kind=link}
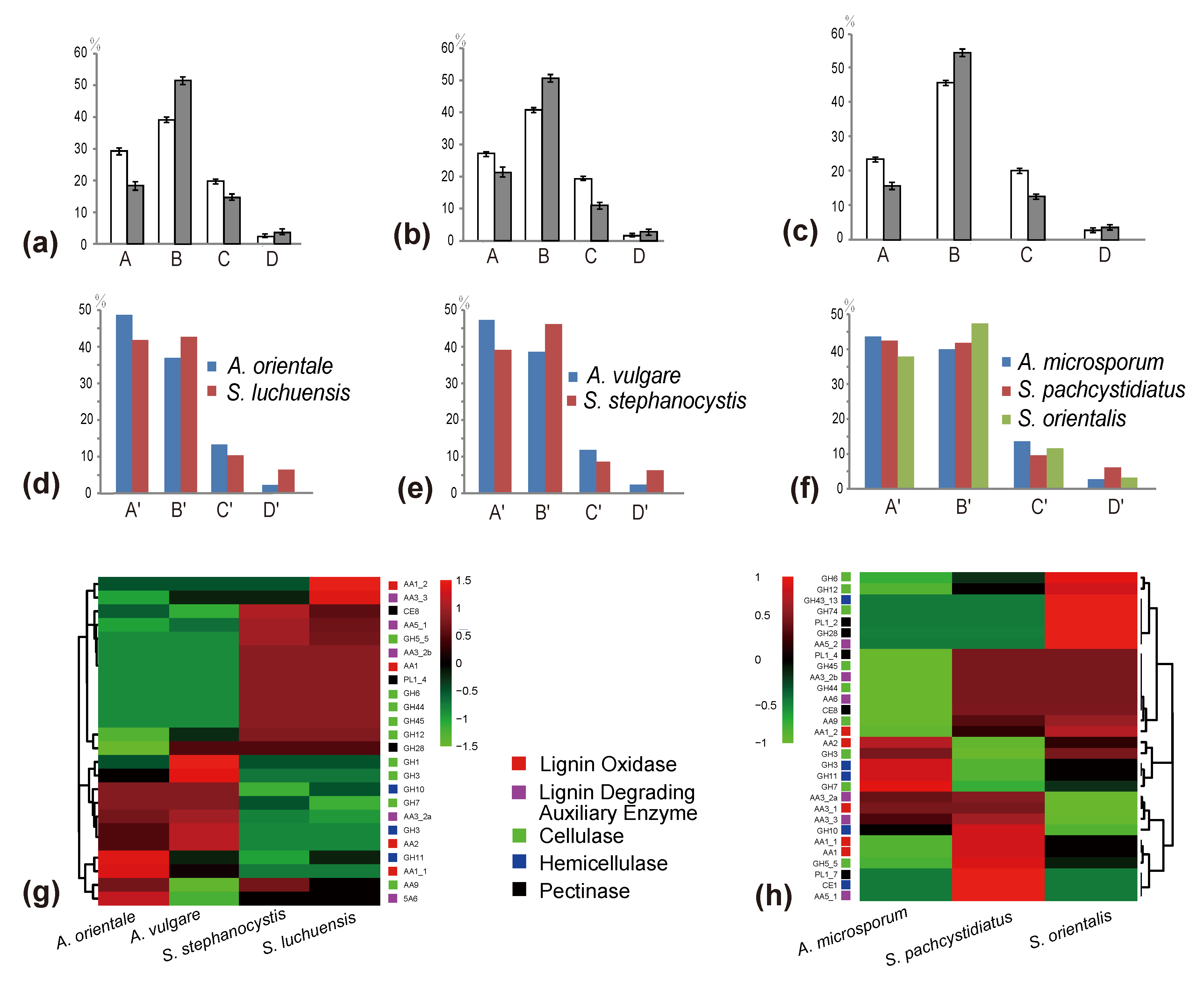{kind=link}
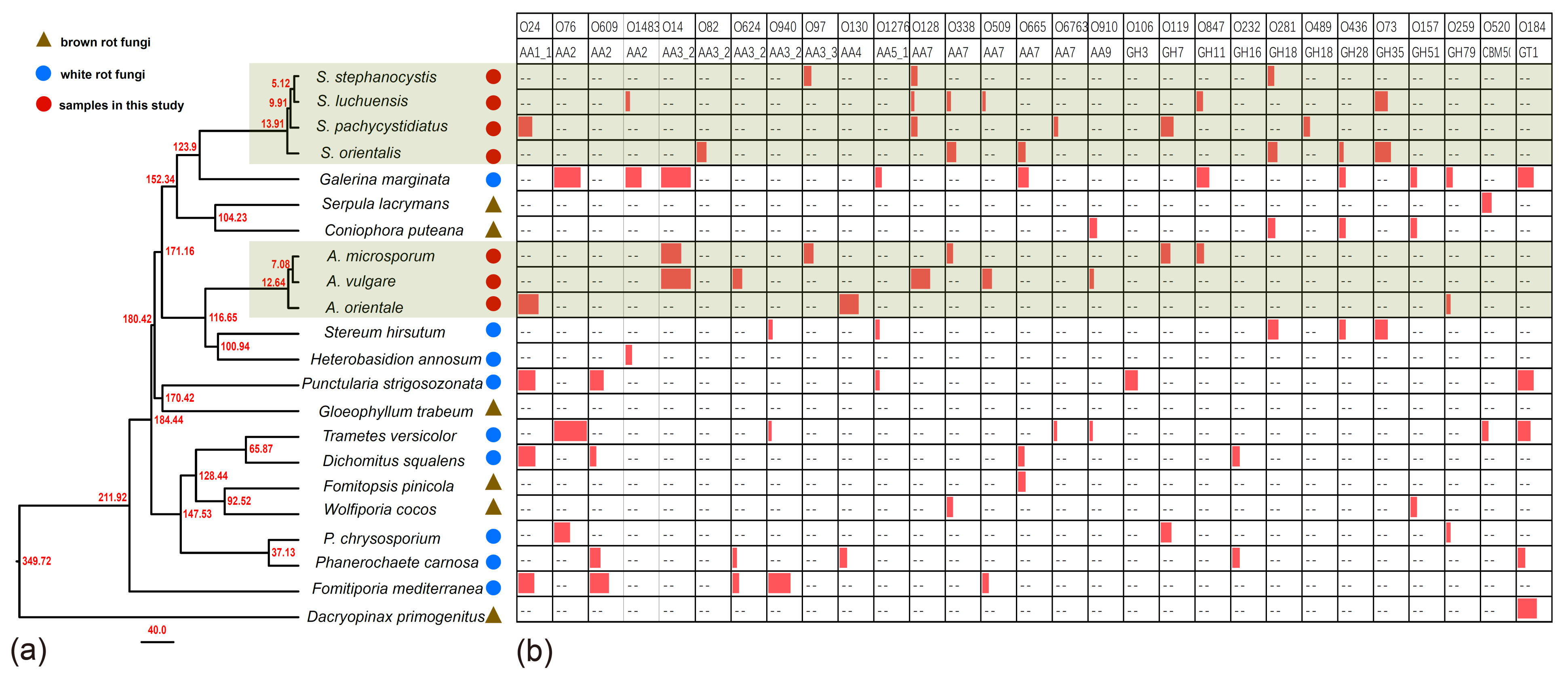{kind=link}
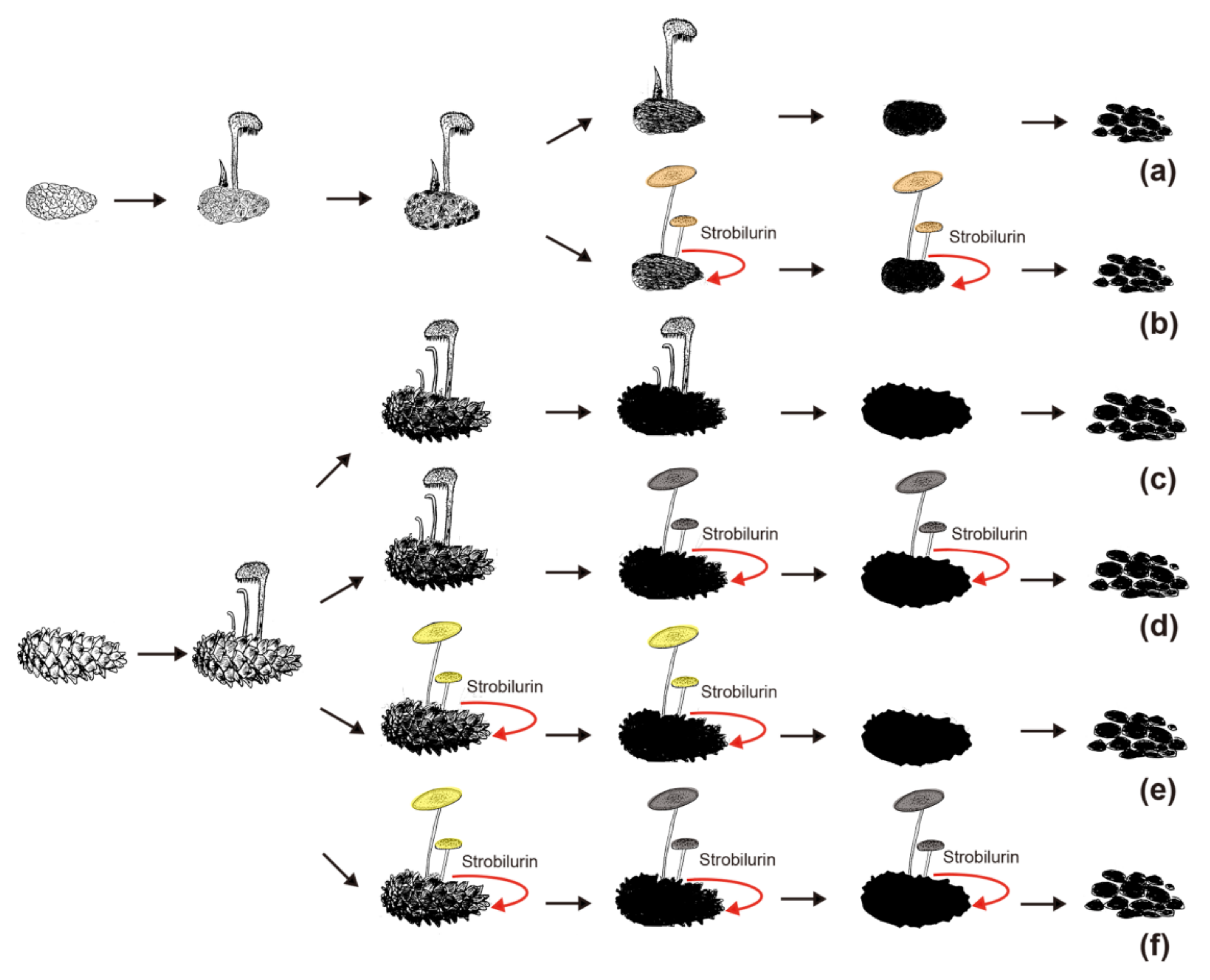{kind=link}
Table 1.
The characteristics of the assembly scaffold and genomes of three Auriscalpium species and four Strobilurus species.
Table 1.
The characteristics of the assembly scaffold and genomes of three Auriscalpium species and four Strobilurus species.
| A. vulgare | A. microsporum | A. orientale | S. stephanocystis | S. pachycystidiatus | S. luchuensis | S. orientalis | |
|---|---|---|---|---|---|---|---|
| Strain No. | CBS 236.39 | A-H-210-14 | A-Y-A | CBS 113577 | Y-H-6C | Y-Y-2D | K-1-1 |
| Number of Contigs | 104 | 54 | 38 | 38 | 71 | 58 | 43 |
| Length of the genome assembly (Mb) | 51.68 | 43.46 | 45.40 | 42.38 | 51.82 | 46.71 | 51.25 |
| Contig N50 (Mb) | 1.721 | 2.449 | 1.814 | 2.875 | 2.269 | 2.554 | 3.397 |
| GC content (%) | 56.39 | 56.13 | 56.68 | 52.13 | 51.75 | 51.76 | 51.53 |
| Number of protein-coding genes | 13,639 | 15,333 | 16,958 | 16,439 | 18,157 | 16,796 | 18,509 |
| Median/Average gene length (bp) | 1716/2202 | 1862/2246 | 1886/2300 | 1702/2033 | 1736/2139 | 1732/2116 | 1747/2146 |
| Median/Average coding sequence size (bp) | 1392/1675 | 1508/1801 | 1535/1867 | 1441/1721 | 1446/1754 | 1467/1761 | 1471/1787 |
| Median/Average number of exons per gene | 5/5.8 | 6/7.1 | 6/7.3 | 5/6.7 | 5/6.7 | 5/6.7 | 5/6.8 |
| Median/Average exon size (bp) | 172/288 | 144/252 | 140/257 | 143/256 | 148/263 | 150/265 | 146/262 |
| Median/Average intron size (bp) | 55/109 | 55/71 | 55/67 | 52/53 | 52/67 | 52/62 | 52/60 |
| Median/Average size of intergenic regions (bp) | 674/1544 | 399/1085 | 386/953 | 396/893 | 454/1086 | 462/1065 | 449/1046 |
| Gene density (genes/Mbp) | 264 | 353 | 374 | 388 | 350 | 360 | 361 |
Table 2.
Abundance and distribution of the top 10 groups of overexpressed genes in the presence of latex among the seven sequenced genomes in this study.
Table 2.
Abundance and distribution of the top 10 groups of overexpressed genes in the presence of latex among the seven sequenced genomes in this study.
| InterPro Hit ID | InterPro Hit Description | S. luchuensis | S. pachycystidiatus | S. stephanocystis | S. orientalis | A. microsporum | A. orientale | A. vulgare | Strobilurus_mean | Auriscalpium_mean | Padj † | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | IPR000209 | Peptidase S8 | 6 | 4 | 5 | 4 | 19 | 23 | 20 | 4.75 | 20.67 | 0.021 * |
| 2 | IPR001461 | Aspartic peptidase | 17 | 17 | 16 | 19 | 18 | 34 | 28 | 17.25 | 26.67 | 0.432 |
| 3 | IPR008972 | Cupredoxin | 8 | 8 | 7 | 7 | 9 | 5 | 7 | 7.5 | 7 | 0.889 |
| 4 | IPR001128 | Cytochrome P450 | 92 | 94 | 97 | 96 | 95 | 121 | 98 | 94.75 | 104.67 | 0.584 |
| 5 | IPR018487 | Hemopexin-like repeats | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| 6 | IPR015366 | Peptidase S53 | 1 | 1 | 2 | 1 | 3 | 4 | 4 | 1.25 | 3.67 | 0.021 * |
| 7 | IPR001338 | Hydrophobin | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| 8 | IPR001128 | Cytochrome P450 | 31 | 22 | 27 | 37 | 20 | 23 | 28 | 29.25 | 23.67 | 0.432 |
| 9 | IPR001338 | Hydrophobin | 4 | 9 | 4 | 11 | 6 | 6 | 6 | 7 | 6 | 0.876 |
| 10 | IPR011701 | Major facilitator superfamily | 37 | 42 | 41 | 49 | 44 | 55 | 53 | 42.25 | 50.67 | 0.387 |
* significance at the level of p < 0.05. † adjusted p-value based on the False Discovery Rate (FDR) method for multiple testing correction.
Table 3.
Comparison of secondary metabolisms of fungi in Auriscalpium and Strobilurus.
| Secondary metabolisms | S. stephanocystis | S. luchuensis | S. pachycystidiatus | S. orientalis | A. vulgare | A. orientale | A. microsporum | Strobilurus_mean | Auriscalpium_mean |
|---|---|---|---|---|---|---|---|---|---|
| Terpene | 7 | 8 | 8 | 9 | 13 | 13 | 8 | 8 | 11.33 |
| T1PKS †-Terpene | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0.25 | 0 |
| T1PKS †-NRPS-like | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 |
| NRPS ‡-like | 10 | 9 | 9 | 7 | 6 | 1 | 4 | 8.75 | 3.67 |
| Siderophore | 2 | 1 | 1 | 1 | 1 | 2 | 1 | 1.25 | 1.33 |
| betalactone | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 |
| Strobilurin | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 0 |
| T1PKS † | 1 | 1 | 0 | 2 | 0 | 0 | 0 | 1 | 0 |
| indole | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 0 |
| total | 22 | 21 | 21 | 21 | 22 | 18 | 15 | 21.25 | 18.33 |
† type I polyketide synthases. ‡ nonribosomal peptide synthetase.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, P.; Xu, J.; Wu, G.; Liu, T.; Yang, Z.L. Genomic and Experimental Investigations of Auriscalpium and Strobilurus Fungi Reveal New Insights into Pinecone Decomposition. J. Fungi 2021, 7, 679. https://doi.org/10.3390/jof7080679
AMA Style
Wang P, Xu J, Wu G, Liu T, Yang ZL. Genomic and Experimental Investigations of Auriscalpium and Strobilurus Fungi Reveal New Insights into Pinecone Decomposition. Journal of Fungi. 2021; 7(8):679. https://doi.org/10.3390/jof7080679
Chicago/Turabian StyleWang, Panmeng, Jianping Xu, Gang Wu, Tiezhi Liu, and Zhu L. Yang. 2021. "Genomic and Experimental Investigations of Auriscalpium and Strobilurus Fungi Reveal New Insights into Pinecone Decomposition" Journal of Fungi 7, no. 8: 679. https://doi.org/10.3390/jof7080679
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.