
Functional Roles of Homologous Recombination and Non-Homologous End Joining in DNA Damage Response and Microevolution in Cryptococcus neoformans

Abstract
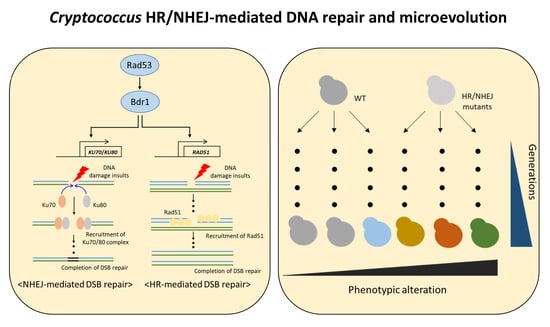
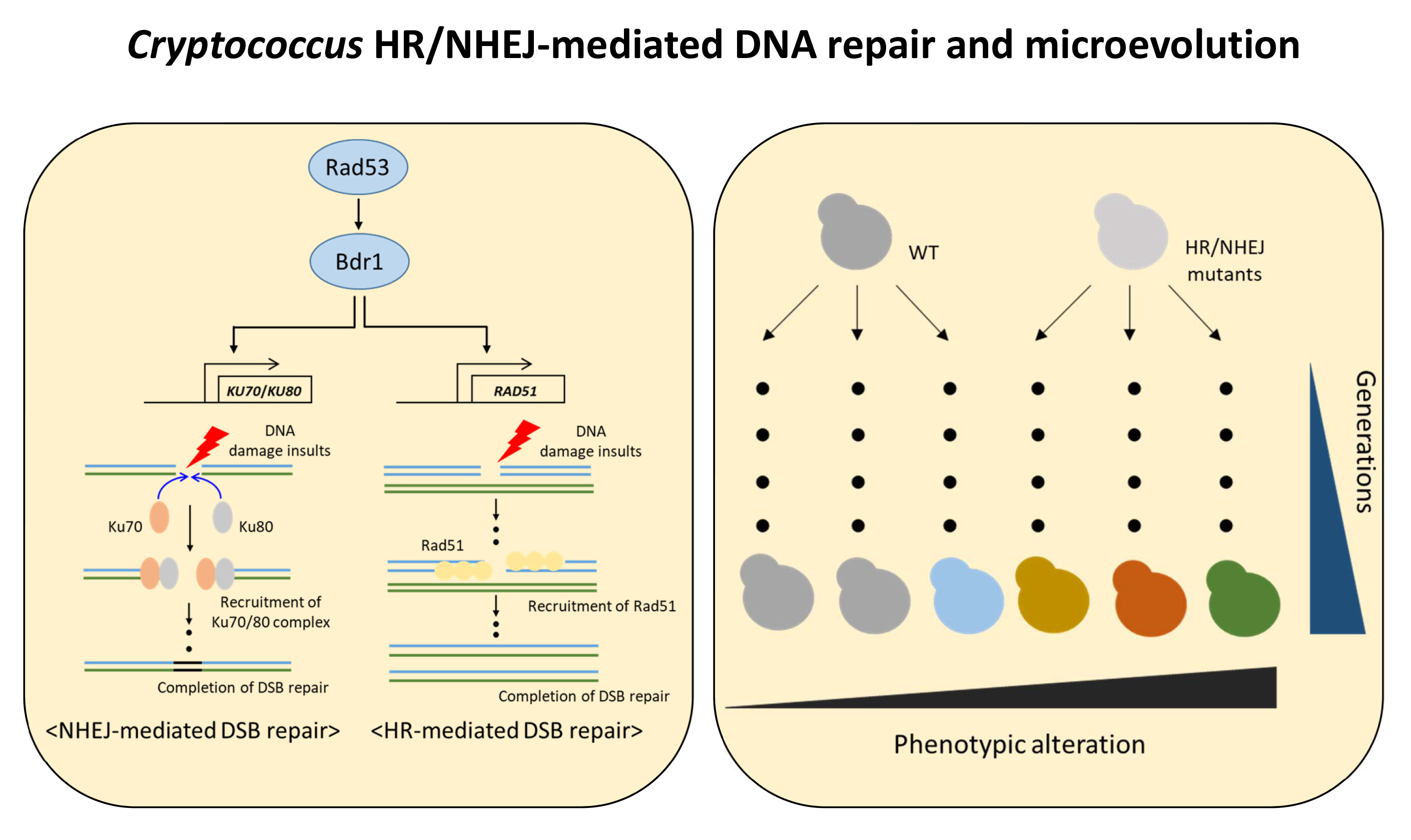
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Strains and Growth Condition
2.2. Construction of Strains
2.3. Construction of KU70, KU80, and RAD51 Complemented Strains
2.4. Spotting Assay
2.5. Total RNA Isolation, cDNA Synthesis, and Real-Time Quantitative PCR (qRT-PCR)
2.6. Mutation Rate Assay
2.7. Fluconazole Resistance Assay
2.8. Phenotypic Characterization of Microevolution of C. neoformans Strains
2.9. Identification of DNA Mutations
3. Results
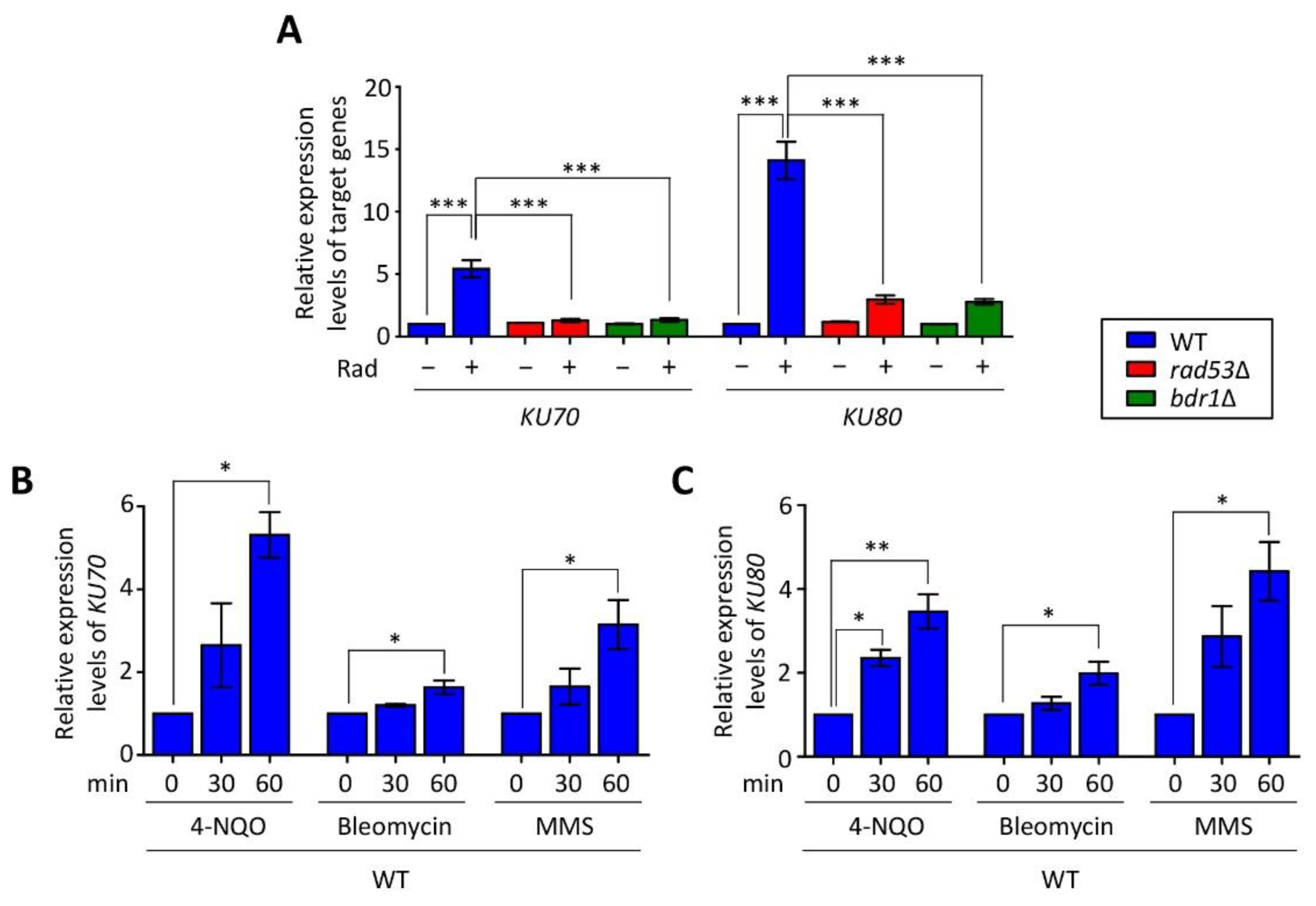
3.1. Expression of KU70 and KU80 Is Induced in Response to DNA Damage Stress in a Rad53–Bdr1 Pathway-Dependent Manner
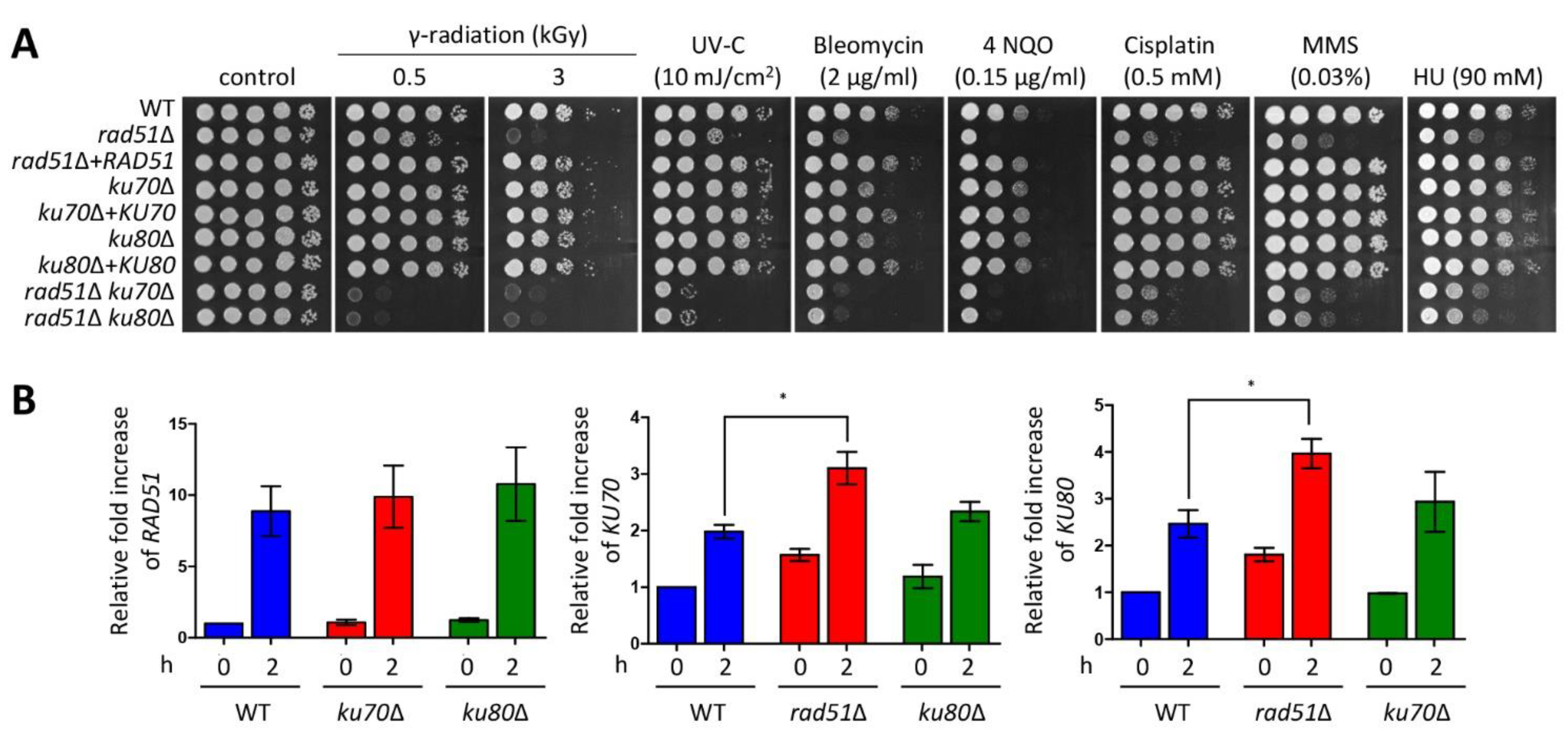
3.2. Ku70/Ku80 and Rad51 Cooperatively Regulate DNA Damage Response
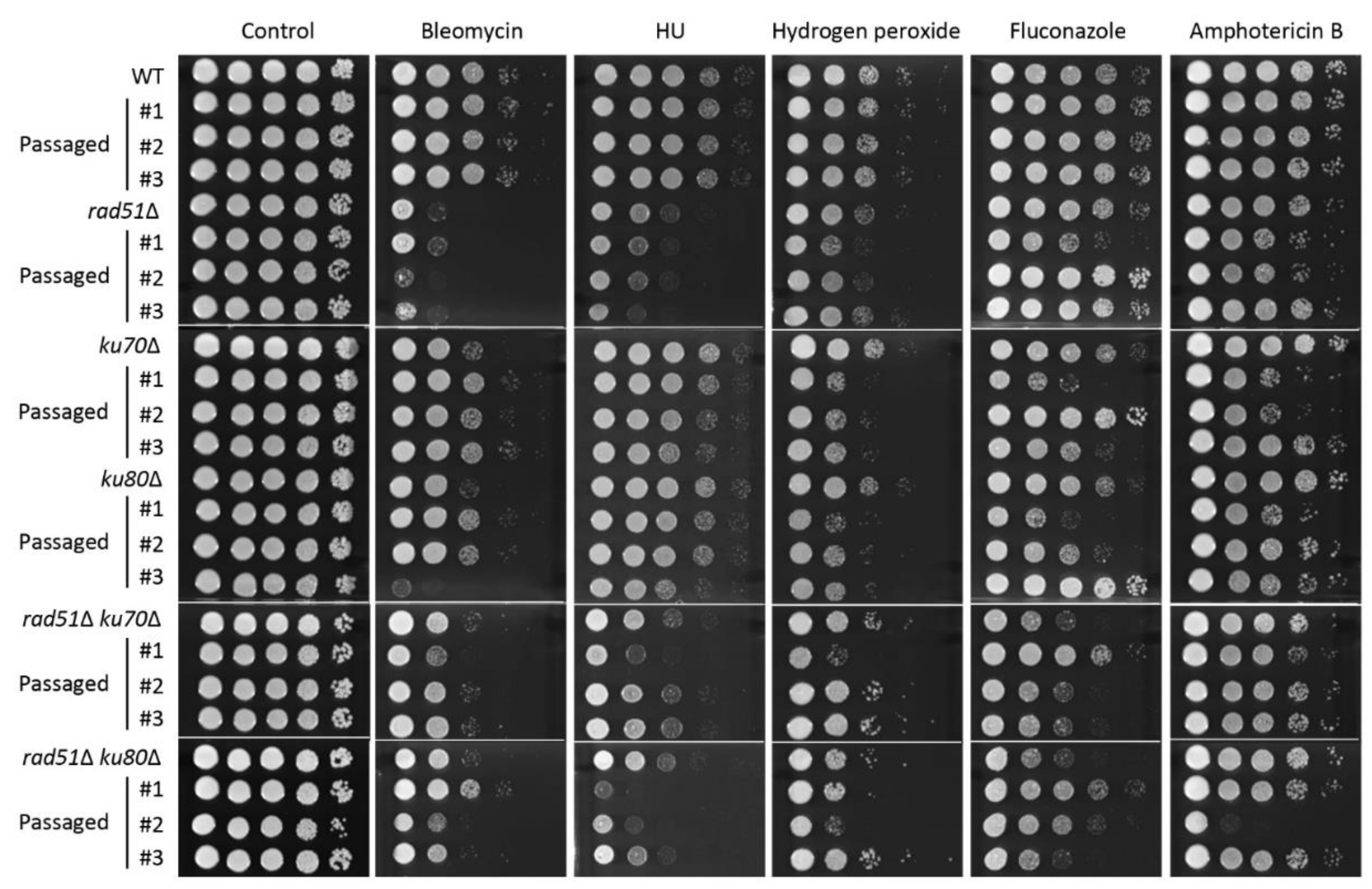
3.3. Ku70/Ku80 and Rad51 Are Involved in the Oxidative Stress Response
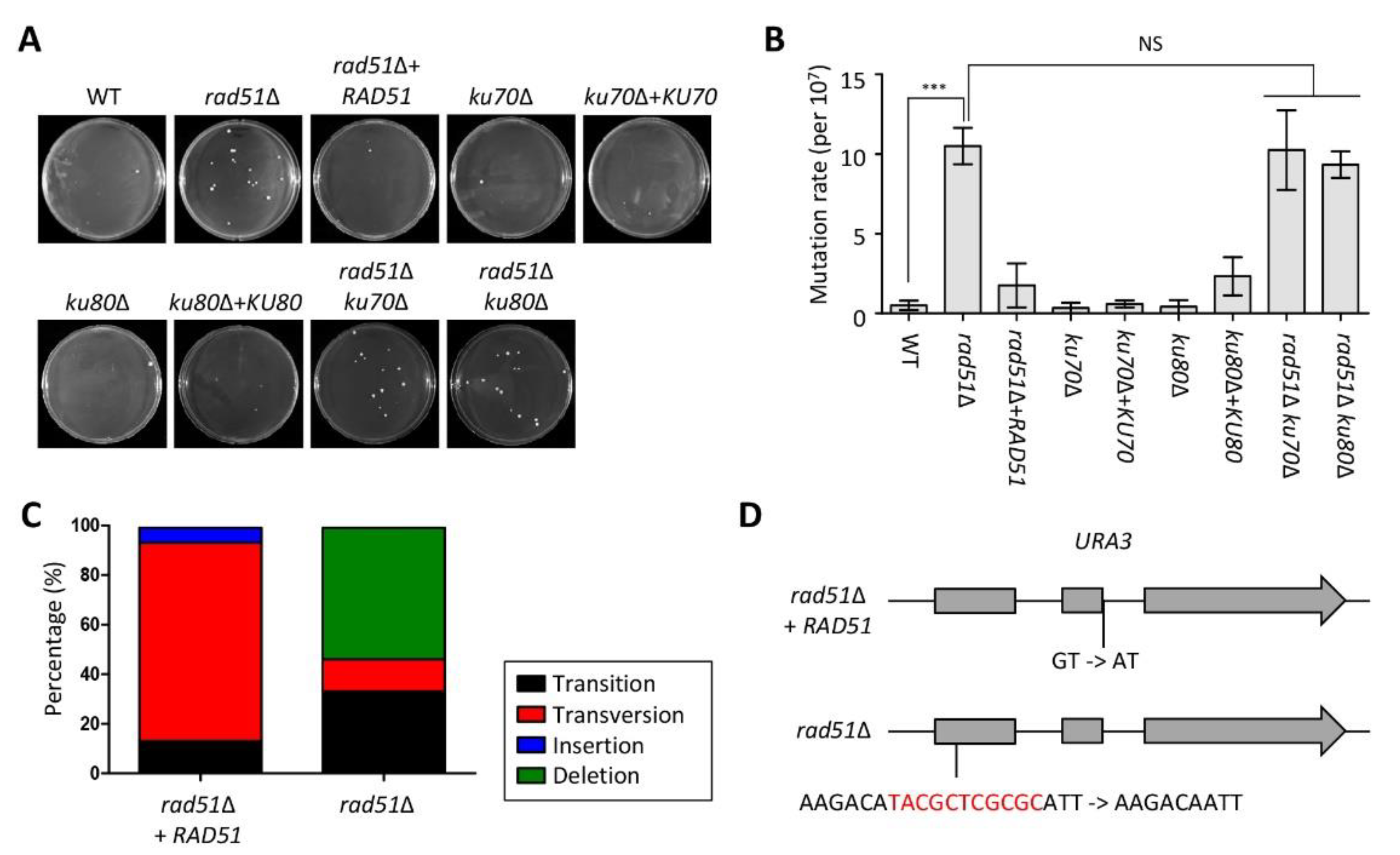
3.4. Functional Role of Accumulation of DNA Mutations and Microevolution Mediated by HR and NHEJ
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lindahl, T. Instability and decay of the primary structure of DNA. Nat. Cell Biol. 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Lindahl, T.; Barnes, D. Repair of Endogenous DNA Damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Standal, R.; Slupphaug, G. DNA glycosylases in the base excision repair of DNA. Biochem. J. 1997, 325, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Boiteux, S.; Guillet, M. Abasic sites in DNA: Repair and biological consequences in Saccharomyces cerevisiae. DNA Repair 2004, 3, 1–12. [Google Scholar] [CrossRef]
- Petit, C.; Sancar, A. Nucleotide excision repair: From E. coli to man. Biochimie 1999, 81, 15–25. [Google Scholar] [CrossRef]
- Helena, J.M.; Joubert, A.M.; Grobbelaar, S.; Nolte, E.M.; Nel, M.; Pepper, M.S.; Coetzee, M.; Mercier, A.E. Deoxyribonucleic Acid Damage and Repair: Capitalizing on Our Understanding of the Mechanisms of Maintaining Genomic Integrity for Therapeutic Purposes. Int. J. Mol. Sci. 2018, 19, 1148. [Google Scholar] [CrossRef] [Green Version]
- Prakash, S.; Prakash, L. Nucleotide excision repair in yeast. Mutat. Res. Mol. Mech. Mutagen. 2000, 451, 13–24. [Google Scholar] [CrossRef]
- Modrich, P. Mechanisms and Biological Effects of Mismatch Repair. Annu. Rev. Genet. 1991, 25, 229–253. [Google Scholar] [CrossRef]
- Marsischky, G.T.; Filosi, N.; Kane, M.F.; Kolodner, R. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 1996, 10, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Prolla, T.A.; Pang, Q.; Alani, E.; Kolodner, R.D.; Liskay, R.M. MLH1, PMS1, and MSH2 interactions during the initiation of DNA mismatch repair in yeast. Science 1994, 265, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Tishkoff, D.X.; Boerger, A.L.; Bertrand, P.; Filosi, N.; Gaida, G.M.; Kane, M.F.; Kolodner, R.D. Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proc. Natl. Acad. Sci. USA 1997, 94, 7487–7492. [Google Scholar] [CrossRef] [Green Version]
- Kolodner, R.D.; Marsischky, G.T. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 1999, 9, 89–96. [Google Scholar] [CrossRef]
- Symington, L.S.; Gautier, J. Double-Strand Break End Resection and Repair Pathway Choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.E.; Grawunder, U.; Lieber, M. Yeast DNA ligase IV mediates non-homologous DNA end joining. Nat. Cell Biol. 1997, 388, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Gellert, M. The 3′ to 5′ Exonuclease Activity of Mre11 Facilitates Repair of DNA Double-Strand Breaks. Mol. Cell 1998, 1, 969–979. [Google Scholar] [CrossRef]
- Wolner, B.; van Komen, S.; Sung, P.; Peterson, C.L. Recruitment of the Recombinational Repair Machinery to a DNA Double-Strand Break in Yeast. Mol. Cell 2003, 12, 221–232. [Google Scholar] [CrossRef]
- Sugawara, N.; Wang, X.; Haber, J.E. In Vivo Roles of Rad52, Rad54, and Rad55 Proteins in Rad51-Mediated Recombination. Mol. Cell 2003, 12, 209–219. [Google Scholar] [CrossRef]
- Couedel, C.; Mills, K.D.; Barchi, M.; Shen, L.; Olshen, A.; Johnson, R.D.; Nussenzweig, A.; Essers, J.; Kanaar, R.; Li, G.C.; et al. Collaboration of homologous recombination and nonhomologous end-joining factors for the survival and integrity of mice and cells. Genes Dev. 2004, 18, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Bakr, A.; Köcher, S.; Volquardsen, J.; Reimer, R.; Borgmann, K.; Dikomey, E.; Rothkamm, K.; Mansour, W.Y. Functional crosstalk between DNA damage response proteins 53BP1 and BRCA1 regulates double strand break repair choice. Radiother. Oncol. 2016, 119, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Farrer, R.A.; Giamberardino, C.; Sakthikumar, S.; Jones, A.; Yang, T.; Tenor, J.L.; Wagih, O.; Van Wyk, M.; Govender, N.P.; et al. Microevolution of Serial Clinical Isolates of Cryptococcus neoformans var. grubii and C. gattii. mBio 2017, 8, e00166-17. [Google Scholar] [CrossRef] [Green Version]
- Ene, I.V.; Farrer, R.; Hirakawa, M.; Agwamba, K.; Cuomo, C.A.; Bennett, R.J. Global analysis of mutations driving microevolution of a heterozygous diploid fungal pathogen. Proc. Natl. Acad. Sci. USA 2018, 115, E8688–E8697. [Google Scholar] [CrossRef] [Green Version]
- Healey, K.; Zhao, Y.; Perez, W.B.; Lockhart, S.R.; Sobel, J.D.; Farmakiotis, D.; Kontoyiannis, D.P.; Sanglard, D.; Taj-Aldeen, S.J.; Alexander, B.D.; et al. Prevalent mutator genotype identified in fungal pathogen Candida glabrata promotes multi-drug resistance. Nat. Commun. 2016, 7, 11128. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.M.; Bennett, R.J.; Anderson, M.Z. The Genome of the Human Pathogen Candida albicans Is Shaped by Mutation and Cryptic Sexual Recombination. mBio 2018, 9, e01205-18. [Google Scholar] [CrossRef] [Green Version]
- Popp, C.; Ramírez-Zavala, B.; Schwanfelder, S.; Krüger, I.; Morschhäuser, J. Evolution of Fluconazole-Resistant Candida albicans Strains by Drug-Induced Mating Competence and Parasexual Recombination. mBio 2019, 10, e02740-18. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Heitman, J. The biology of the Cryptococcus neoformans species complex. Annu. Rev. Microbiol. 2006, 60, 69–105. [Google Scholar] [CrossRef]
- Rajasingham, R.; Smith, R.M.; Park, B.J.; Jarvis, J.N.; Govender, N.P.; Chiller, T.M.; Denning, D.; Loyse, A.; Boulware, D. Global burden of disease of HIV-associated cryptococcal meningitis: An updated analysis. Lancet Infect. Dis. 2017, 17, 873–881. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.-W.; Yang, D.-H.; Kim, M.-K.; Seo, H.S.; Lim, S.; Bahn, Y.-S. Unraveling Fungal Radiation Resistance Regulatory Networks through the Genome-Wide Transcriptome and Genetic Analyses of Cryptococcus neoformans. mBio 2016, 7, e01483-16. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.-W.; Lee, Y.; Huh, E.Y.; Lee, S.C.; Lim, S.; Bahn, Y.-S. Rad53- and Chk1-Dependent DNA Damage Response Pathways Cooperatively Promote Fungal Pathogenesis and Modulate Antifungal Drug Susceptibility. mBio 2019, 10, e01726-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goins, C.L.; Gerik, K.J.; Lodge, J.K. Improvements to gene deletion in the fungal pathogen Cryptococcus neoformans: Absence of Ku proteins increases homologous recombination, and co-transformation of independent DNA molecules allows rapid complementation of deletion phenotypes. Fungal Genet. Biol. 2006, 43, 531–544. [Google Scholar] [CrossRef]
- Bahn, Y.-S.; Hicks, J.K.; Giles, S.S.; Cox, G.M.; Heitman, J. Adenylyl Cyclase-Associated Protein Aca1 Regulates Virulence and Differentiation of Cryptococcus neoformans via the Cyclic AMP-Protein Kinase A Cascade. Eukaryot. Cell 2004, 3, 1476–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Kim, S.-Y.; Yoon, J.K.; Lee, Y.-W.; Bahn, Y.-S. An efficient gene-disruption method in Cryptococcus neoformans by double-joint PCR with NAT-split markers. Biochem. Biophys. Res. Commun. 2009, 390, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.-W.; Kim, S.-Y.; Okagaki, L.H.; Nielsen, K.; Bahn, Y.-S. Ste50 adaptor protein governs sexual differentiation of Cryptococcus neoformans via the pheromone-response MAPK signaling pathway. Fungal Genet. Biol. 2011, 48, 154–165. [Google Scholar] [CrossRef] [Green Version]
- Ko, Y.J.; Yu, Y.M.; Kim, G.B.; Lee, G.W.; Maeng, P.J.; Kim, S.S.; Floyd, A.; Heitman, J.; Bahn, Y.S. Remodeling of global transcription patterns of Cryptococcus neoformans genes mediated by the stress-activated HOG signaling pathways. Eukaryot. Cell 2009, 8, 1197–1217. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Boyce, K.J.; Wang, Y.; Verma, S.; Shakya, V.; Xue, C.; Idnurm, A. Mismatch Repair of DNA Replication Errors Contributes to Microevolution in the Pathogenic Fungus Cryptococcus neoformans. mBio 2017, 8, e00595-17. [Google Scholar] [CrossRef] [Green Version]
- Boyce, K.; Cao, C.; Xue, C.; Idnurm, A. A spontaneous mutation in DNA polymerase POL3 during in vitro passaging causes a hypermutator phenotype in Cryptococcus species. DNA Repair 2020, 86, 102751. [Google Scholar] [CrossRef]
- Jung, K.-W.; Lee, K.-T.; So, Y.-S.; Bahn, Y.-S. Genetic Manipulation of Cryptococcus neoformans. Curr. Protoc. Microbiol. 2018, 50, e59. [Google Scholar] [CrossRef]
- Peng, C.A.; Gaertner, A.A.E.; Henriquez, S.A.; Fang, D.; Colon-Reyes, R.J.; Brumaghim, J.; Kozubowski, L. Fluconazole induces ROS in Cryptococcus neoformans and contributes to DNA damage in vitro. PLOS ONE 2018, 13, e0208471. [Google Scholar] [CrossRef]
- Billmyre, R.B.; Clancey, S.A.; Heitman, J. Natural mismatch repair mutations mediate phenotypic diversity and drug resistance in Cryptococcus deuterogattii. eLife 2017, 6, e28802. [Google Scholar] [CrossRef]
- Kwon-Chung, K.J.; Varma, A.; Edman, J.C.; E Bennett, J. Selection of ura5 and ura3 mutants from the two varieties of Cryptococcus neoformans on 5-fluoroorotic acid medium. J. Med Vet. Mycol. Bi-Mon. Publ. Int. Soc. Hum. Anim. Mycol. 1992, 30, 61–69. [Google Scholar]
- Chico, L.; Ciudad, T.; Hsu, M.; Lue, N.F.; Larriba, G. The Candida albicans Ku70 Modulates Telomere Length and Structure by Regulating Both Telomerase and Recombination. PLOS ONE 2011, 6, e23732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siede, W.; Friedl, A.; Dianova, I.; Eckardt-Schupp, F.; Friedberg, E.C. The Saccharomyces cerevisiae Ku Autoantigen Homologue Affects Radiosensitivity Only in the Absence of Homologous Recombination. Genetics 1996, 142, 91–102. [Google Scholar] [CrossRef]
- E Critchlow, S.; Jackson, S.P. DNA end-joining: From yeast to man. Trends Biochem. Sci. 1998, 23, 394–398. [Google Scholar] [CrossRef]
- Ferreira, M.E.D.S.; Kress, M.R.V.Z.; Savoldi, M.; Goldman, M.H.S.; Härtl, A.; Heinekamp, T.; Brakhage, A.A.; Goldman, G. The akuBKU80 Mutant Deficient for Nonhomologous End Joining Is a Powerful Tool for Analyzing Pathogenicity in Aspergillus fumigatus. Eukaryot. Cell 2006, 5, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, X.; Yao, Z.; Zhou, Y.; Shang, J.; Lin, H.; Nuss, D.L.; Chen, B. Deletion of the cpku80 gene in the chestnut blight fungus, Cryphonectria parasitica, enhances gene disruption efficiency. Curr. Genet. 2007, 53, 59–66. [Google Scholar] [CrossRef]
- Villalba, F.; Collemare, J.; Landraud, P.; Lambou, K.; Brozek, V.; Cirer, B.; Morin, D.; Bruel, C.; Beffa, R.; Lebrun, M.-H. Improved gene targeting in Magnaporthe grisea by inactivation of MgKU80 required for non-homologous end joining. Fungal Genet. Biol. 2008, 45, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.J.; Hu, P.; Han, M.; Ellis, N.; Jasin, M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001, 15, 3237–3242. [Google Scholar] [CrossRef] [Green Version]
- Kafri, R.; Bar-Even, A.; Pilpel, Y. Transcription control reprogramming in genetic backup circuits. Nat. Genet. 2005, 37, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Terashima, H.; Yabuki, N.; Arisawa, M.; Hamada, K.; Kitada, K. Up-regulation of genes encoding glycosylphosphatidylinositol (GPI)-attached proteins in response to cell wall damage caused by disruption of FKS1 in Saccharomyces cerevisiae. Mol. Genet. Genom. 2000, 264, 64–74. [Google Scholar] [CrossRef]
- Legrand, M.; Chan, C.L.; Jauert, P.A.; Kirkpatrick, D.T. Role of DNA Mismatch Repair and Double-Strand Break Repair in Genome Stability and Antifungal Drug Resistance in Candida albicans. Eukaryot. Cell 2007, 6, 2194–2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legrand, M.; Chan, C.L.; Jauert, P.A.; Kirkpatrick, D.T. Analysis of base excision and nucleotide excision repair in Candida albicans. Microbiology 2008, 154, 2446–2456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoot, S.J.; Zheng, X.; Potenski, C.J.; White, T.C.; Klein, H.L. The role of Candida albicans homologous recombination factors Rad54 and Rdh54 in DNA damage sensitivity. BMC Microbiol. 2011, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, G.B.; Ross, Z.K.; Gow, N.A.R.; Lorenz, A. Pseudohyphal Growth of the Emerging Pathogen Candida auris Is Triggered by Genotoxic Stress through the S Phase Checkpoint. mSphere 2020, 5. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, K.-W.; Jung, J.-H.; Park, H.-Y. Functional Roles of Homologous Recombination and Non-Homologous End Joining in DNA Damage Response and Microevolution in Cryptococcus neoformans. J. Fungi 2021, 7, 566. https://doi.org/10.3390/jof7070566
Jung K-W, Jung J-H, Park H-Y. Functional Roles of Homologous Recombination and Non-Homologous End Joining in DNA Damage Response and Microevolution in Cryptococcus neoformans. Journal of Fungi. 2021; 7(7):566. https://doi.org/10.3390/jof7070566
Chicago/Turabian StyleJung, Kwang-Woo, Jong-Hyun Jung, and Ha-Young Park. 2021. "Functional Roles of Homologous Recombination and Non-Homologous End Joining in DNA Damage Response and Microevolution in Cryptococcus neoformans" Journal of Fungi 7, no. 7: 566. https://doi.org/10.3390/jof7070566