Directed Mutational Strategies Reveal Drug Binding and Transport by the MDR Transporters of Candida albicans


 ,
,  ,
,
Abstract
:1. Introduction
2. Historical Background of the MDR Pumps in Yeast
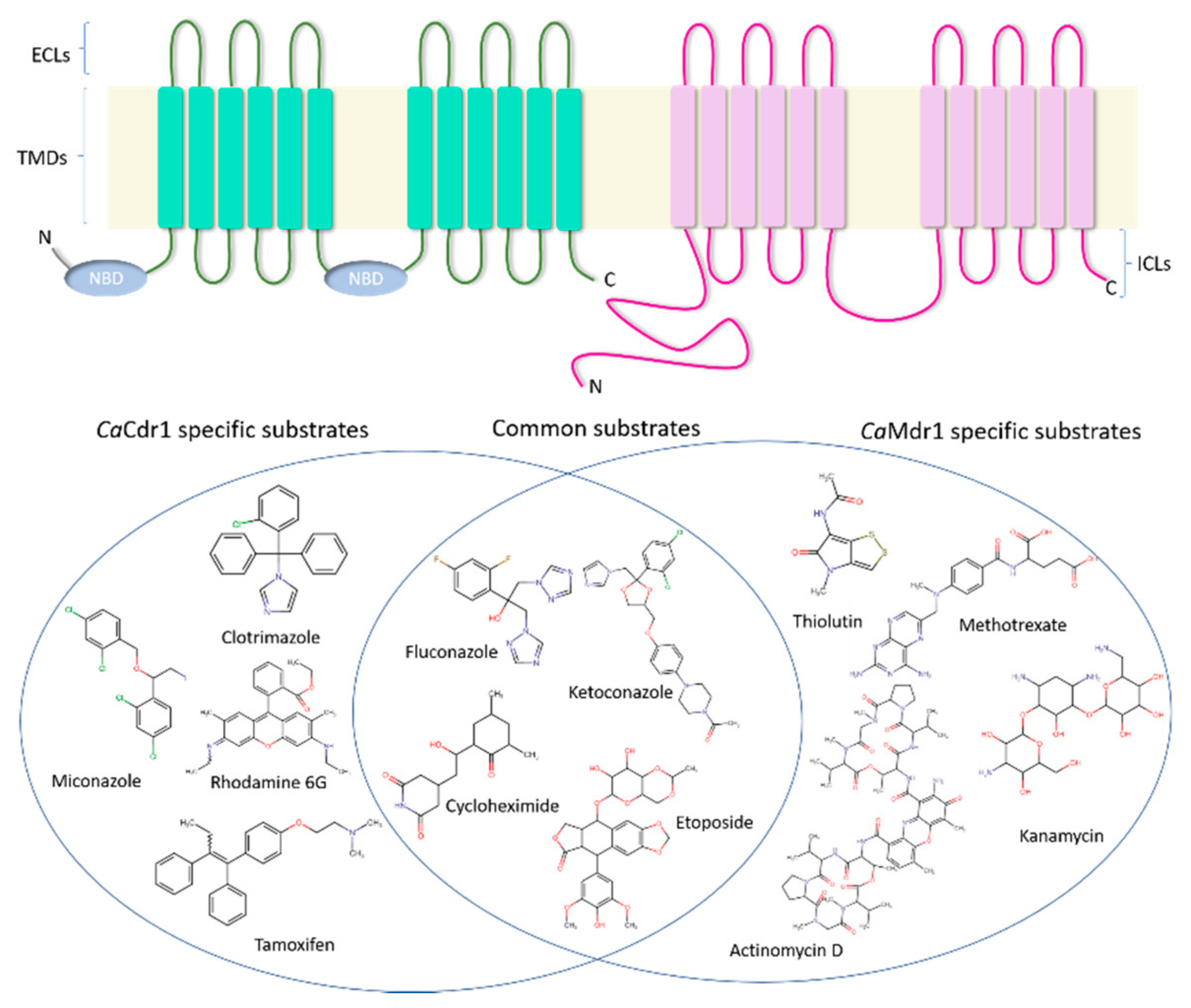
3. Structural Organization of CaCdr1 and CaMdr1
3.1. CaCdr1
3.2. CaMdr1
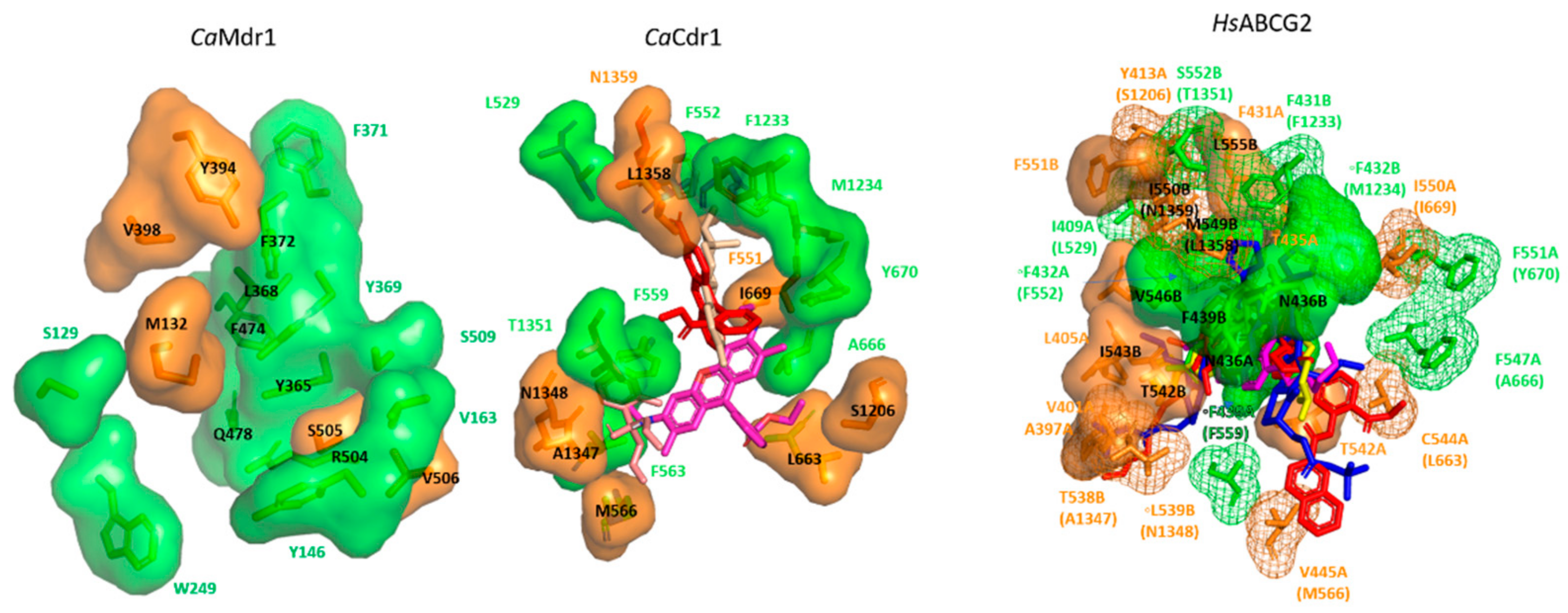
4. Insights into the Drug Binding Pocket of CaCdr1 and CaMdr1
4.1. CaCdr1
4.2. CaMdr1
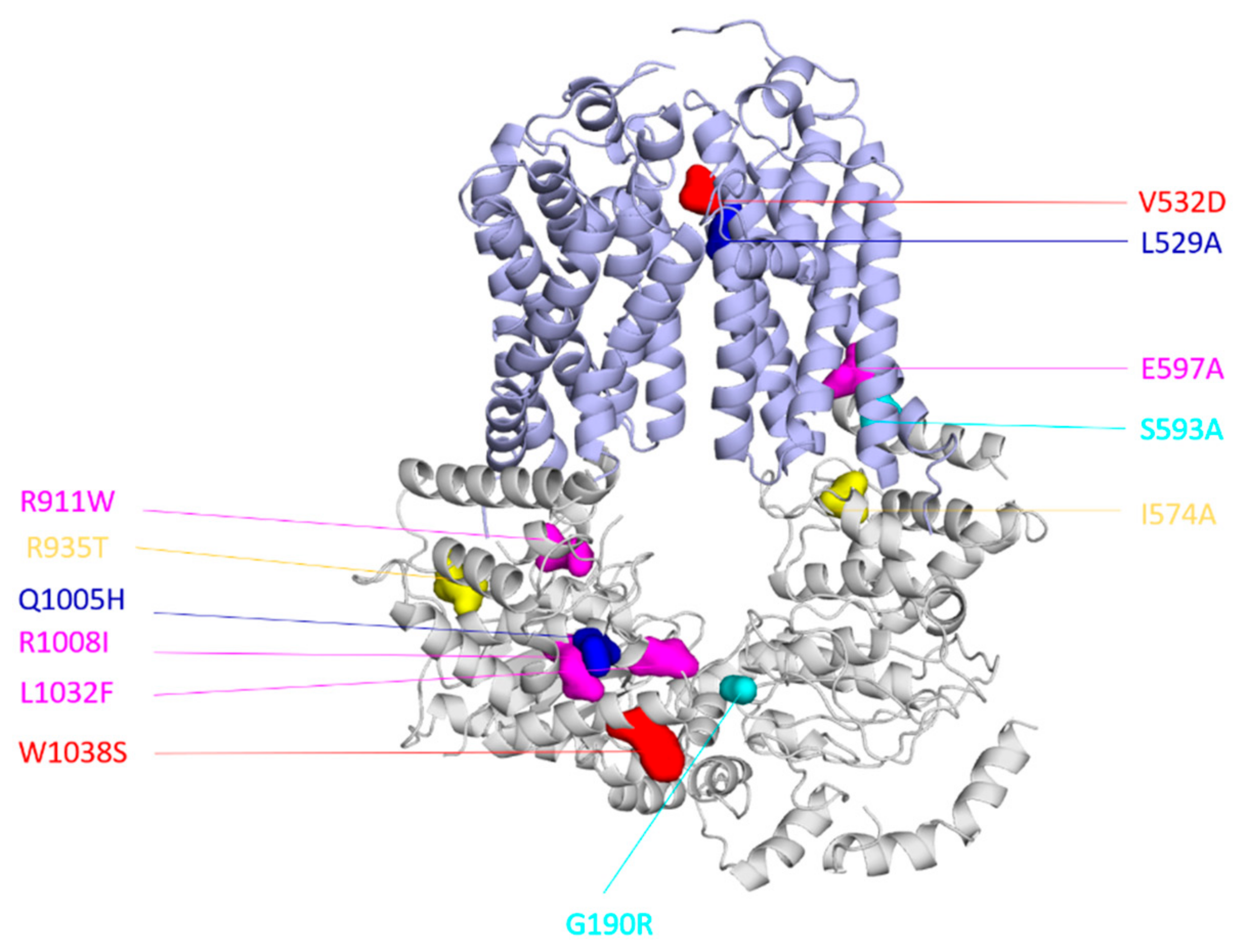
5. Interdomain Crosstalk in CaCdr1 and CaMdr1
6. Challenges Ahead
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sanches, M.D.; Mimura, L.A.N.; Oliveira, L.R.C.; Ishikawa, L.L.W.; Garces, H.G.; Bagagli, E.; Sartori, A.; Kurokawa, C.S.; Fraga-Silva, T.F.C. Differential Behavior of Non-albicans Candida Species in the Central Nervous System of Immunocompetent and Immunosuppressed Mice. Front. Microbiol. 2019, 9, 2968. [Google Scholar] [CrossRef] [PubMed]
- Chowdhary, A.; Sharma, C.; Meis, J.F. Candida auris: A rapidly emerging cause of hospital-acquired multidrug-resistant fungal infections globally. PLoS Pathog 2017, 13, e1006290. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, K.; Woodworth, K.; Walters, M.; Berkow, E.L.; Jackson, B.; Chiller, T.; Vallabhaneni, S. Candida auris: The recent emergence of a multidrug-resistant fungal pathogen. Med. Mycol. 2019, 57, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Taei, M.; Chadeganipour, M.; Mohammadi, R. An alarming rise of non-albicans Candida species and uncommon yeasts in the clinical samples; a combination of various molecular techniques for identification of etiologic agents. BMC Res. Notes 2019, 12, 779. [Google Scholar] [CrossRef] [Green Version]
- Lockhart, S.R.; Etienne, K.A.; Vallabhaneni, S.; Farooqi, J.; Chowdhary, A.; Govender, N.P.; Colombo, A.L.; Calvo, B.; Cuomo, C.A.; Desjardins, C.A.; et al. Simultaneous Emergence of Multidrug-Resistant Candida auris on 3 Continents Confirmed by Whole-Genome Sequencing and Epidemiological Analyses. Clin. Infect. Dis. 2016, 64, 134–140. [Google Scholar] [CrossRef] [Green Version]
- Prasad, R.; Banerjee, A.; Khandelwal, N.K.; Dhamgaye, S. The ABCs of Candida albicans Multidrug Transporter Cdr1. Eukaryot. Cell 2015, 14, 1154–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, R.D.; Lamping, E.; Holmes, A.R.; Niimi, K.; Baret, P.V.; Keniya, M.V.; Tanabe, K.; Niimi, M.; Goffeau, A.; Monk, B.C. Efflux-mediated antifungal drug resistance. Clin. Microbiol. Rev. 2009, 22, 291–321. [Google Scholar] [CrossRef] [Green Version]
- Ksiezopolska, E.; Gabaldón, T. Evolutionary Emergence of Drug Resistance in Candida Opportunistic Pathogens. Genes 2018, 9, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, T.C. Increased mRNA levels of ERG16, CDR, and MDR1 correlate, with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob. Agents Chemother. 1997, 41, 1482–1487. [Google Scholar] [CrossRef] [Green Version]
- Sanglard, D.; Kuchler, K.; Ischer, F.; Pagani, J.L.; Monod, M.; Bille, J. Mechanisms of resistance to azole antifungal agents in Candida albicans isolates from AIDS patients involve specific multidrug transporters. Antimicrob. Agents Chemother. 1995, 39, 2378–2386. [Google Scholar] [CrossRef] [Green Version]
- Holmes, A.R.; Cardno, T.S.; Strouse, J.J.; Ivnitski-Steele, I.; Keniya, M.V.; Lackovic, K.; Monk, B.C.; Sklar, L.A.; Cannon, R.D. Targeting efflux pumps to overcome antifungal drug resistance. Future Med. Chem. 2016, 8, 1485–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barabote, R.D.; Thekkiniath, J.; Strauss, R.E.; Vediyappan, G.; Fralick, J.A.; San Francisco, M.J. Xenobiotic efflux in bacteria and fungi: A genomics update. Adv. Enzymol. Relat. Areas Mol. Biol. 2011, 77, 237–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, E.A.; Heppel, L.A. Different mechanisms of energy coupling for the shock-sensitive and shock-resistant amino acid permeases of Escherichia coli. J. Biol. Chem. 1974, 249, 7747–7755. [Google Scholar] [CrossRef]
- Higgins, C.F.; Haag, P.D.; Nikaido, K.; Ardeshir, F.; Garcia, G.; Ames, G.F. Complete nucleotide sequence and identification of membrane components of the histidine transport operon of S. typhimurium. Nature 1982, 298, 723–727. [Google Scholar] [CrossRef]
- Riordan, J.R.; Deuchars, K.; Kartner, N.; Alon, N.; Trent, J.; Ling, V. Amplification of P-glycoprotein genes in multidrug-resistant mammalian cell lines. Nature 1985, 316, 817–819. [Google Scholar] [CrossRef]
- Rank, G.H.; Bech-Hansen, N.T. Single nuclear gene inherited cross resistance and collateral sensitivity to 17 inhibitors of mitochondrial function in S. cerevisiae. Mol. Gen. Genet. 1973, 126, 93–102. [Google Scholar] [CrossRef]
- Balzi, E.; Chen, W.; Ulaszewski, S.; Capieaux, E.; Goffeau, A. The multidrug resistance gene PDR1 from Saccharomyces cerevisiae. J. Biol. Chem. 1987, 262, 16871–16879. [Google Scholar] [CrossRef]
- Carvajal, E.; van den Hazel, H.B.; Cybularz-Kolaczkowska, A.; Balzi, E.; Goffeau, A. Molecular and phenotypic characterization of yeast PDR1 mutants that show hyperactive transcription of various ABC multidrug transporter genes. Mol. Gen. Genet. 1997, 256, 406–415. [Google Scholar] [CrossRef] [Green Version]
- Leppert, G.; McDevitt, R.; Falco, S.C.; Van Dyk, T.K.; Ficke, M.B.; Golin, J. Cloning by gene amplification of two loci conferring multiple drug resistance in Saccharomyces. Genetics 1990, 125, 13–20. [Google Scholar] [CrossRef]
- Meyers, S.; Schauer, W.; Balzi, E.; Wagner, M.; Goffeau, A.; Golin, J. Interaction of the yeast pleiotropic drug resistance genes PDR1 and PDR5. Curr. Genet. 1992, 21, 431–436. [Google Scholar] [CrossRef]
- Balzi, E.; Wang, M.; Leterme, S.; Van Dyck, L.; Goffeau, A. PDR5, a novel yeast multidrug resistance conferring transporter controlled by the transcription regulator PDR1. J. Biol. Chem. 1994, 269, 2206–2214. [Google Scholar] [CrossRef]
- Prasad, R.; De Wergifosse, P.; Goffeau, A.; Balzi, E. Molecular cloning and characterization of a novel gene of Candida albicans, CDR1, conferring multiple resistance to drugs and antifungals. Curr. Genet. 1995, 27, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Sanglard, D.; Ischer, F.; Monod, M.; Bille, J. Cloning of Candida albicans genes conferring resistance to azole antifungal agents: Characterization of CDR2, a new multidrug ABC transporter gene. Microbiology 1997, 143 Pt 2, 405–416. [Google Scholar] [CrossRef] [Green Version]
- Balan, I.; Alarco, A.M.; Raymond, M. The Candida albicans CDR3 gene codes for an opaque-phase ABC transporter. J. Bacteriol. 1997, 179, 7210–7218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franz, R.; Michel, S.; Morschhauser, J. A fourth gene from the Candida albicans CDR family of ABC transporters. Gene 1998, 220, 91–98. [Google Scholar] [CrossRef]
- Holmes, A.R.; Lin, Y.-H.; Niimi, K.; Lamping, E.; Keniya, M.; Niimi, M.; Tanabe, K.; Monk, B.C.; Cannon, R.D. ABC transporter Cdr1p contributes more than Cdr2p does to fluconazole efflux in fluconazole-resistant Candida albicans clinical isolates. Antimicrob. Agents Chemother. 2008, 52, 3851–3862. [Google Scholar] [CrossRef] [Green Version]
- Moreno, A.; Banerjee, A.; Prasad, R.; Falson, P. PDR-like ABC systems in pathogenic fungi. Res. Microbiol. 2019. [Google Scholar] [CrossRef]
- Gaur, M.; Choudhury, D.; Prasad, R. Complete inventory of ABC proteins in human pathogenic yeast, Candida albicans. J. Mol. Microbiol. Biotechnol. 2005, 9, 3–15. [Google Scholar] [CrossRef]
- Dermauw, W.; Van Leeuwen, T. The ABC gene family in arthropods: Comparative genomics and role in insecticide transport and resistance. Insect Biochem. Mol. Biol. 2014, 45, 89–110. [Google Scholar] [CrossRef]
- Kumari, S.; Kumar, M.; Khandelwal, N.K.; Kumari, P.; Varma, M.; Vishwakarma, P.; Shahi, G.; Sharma, S.; Lynn, A.M.; Prasad, R.; et al. ABC transportome inventory of human pathogenic yeast Candida glabrata: Phylogenetic and expression analysis. PLoS ONE 2018, 13, e0202993. [Google Scholar] [CrossRef] [Green Version]
- Wasi, M.; Khandelwal, N.K.; Moorhouse, A.J.; Nair, R.; Vishwakarma, P.; Bravo Ruiz, G.; Ross, Z.K.; Lorenz, A.; Rudramurthy, S.M.; Chakrabarti, A.; et al. ABC Transporter Genes Show Upregulated Expression in Drug-Resistant Clinical Isolates of Candida auris: A Genome-Wide Characterization of ATP-Binding Cassette (ABC) Transporter Genes. Front. Microbiol. 2019, 10, 1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogra, S.; Krishnamurthy, S.; Gupta, V.; Dixit, B.L.; Gupta, C.M.; Sanglard, D.; Prasad, R. Asymmetric distribution of phosphatidylethanolamine in C. albicans: Possible mediation by CDR1, a multidrug transporter belonging to ATP binding cassette (ABC) superfamily. Yeast 1999, 15, 111–121. [Google Scholar] [CrossRef]
- Krishnamurthy, S.; Dixit, B.L.; Gupta, C.M.; Milewski, S.; Prasad, R. ABC transporters Cdr1p, Cdr2p and Cdr3p of a human pathogen Candida albicans are general phospholipid translocators. Yeast 2002, 19, 303–318. [Google Scholar] [CrossRef]
- Sanglard, D.; Ischer, F.; Monod, M.; Dogra, S.; Prasad, R.; Bille, J. Analysis of the ATP-binding cassette (ABC)-transporter gene CDR4 from Candida albicans (abstr). In Proceedings of the ASM Conference on Candida and Candidiasis, Charleston, SC, USA, 1–4 March 1999. [Google Scholar]
- Yan, N. Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem. Sci. 2013, 38, 151–159. [Google Scholar] [CrossRef]
- Yan, N. Structural Biology of the Major Facilitator Superfamily Transporters. Annu. Rev. Biophys. 2015, 44, 257–283. [Google Scholar] [CrossRef] [PubMed]
- Gaur, M.; Puri, N.; Manoharlal, R.; Rai, V.; Mukhopadhayay, G.; Choudhury, D.; Prasad, R. MFS transportome of the human pathogenic yeast Candida albicans. BMC Genom. 2008, 9, 579. [Google Scholar] [CrossRef] [Green Version]
- Fling, M.E.; Kopf, J.; Tamarkin, A.; Gorman, J.A.; Smith, H.A.; Koltin, Y. Analysis of a Candida albicans gene that encodes a novel mechanism for resistance to benomyl and methotrexate. Mol. Gen. Genet. MGG 1991, 227, 318–329. [Google Scholar] [CrossRef]
- Goldway, M.; Teff, D.; Schmidt, R.; Oppenheim, A.B.; Koltin, Y. Multidrug resistance in Candida albicans: Disruption of the BENr gene. Antimicrob. Agents Chemother. 1995, 39, 422–426. [Google Scholar] [CrossRef] [Green Version]
- Ben-Yaacov, R.; Knoller, S.; Caldwell, G.A.; Becker, J.M.; Koltin, Y. Candida albicans gene encoding resistance to benomyl and methotrexate is a multidrug resistance gene. Antimicrob. Agents Chemother. 1994, 38, 648–652. [Google Scholar] [CrossRef] [Green Version]
- Redhu, A.K.; Shah, A.H.; Prasad, R. MFS transporters of Candida species and their role in clinical drug resistance. FEMS Yeast Res. 2016, 16. [Google Scholar] [CrossRef] [Green Version]
- Prasad, R.; Banerjee, A.; Shah, A.H. Resistance to antifungal therapies. Essays Biochem. 2017, 61, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locher, K.P. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat. Struct. Mol. Biol. 2016, 23, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, A.; Moreno, A.; Pata, J.; Falson, P.; Prasad, R. ABCG: A New Fold of ABC Exporters and a Whole New Bag of Riddles! Academic Press: New York, NY, USA, 2020. [Google Scholar]
- Lamping, E.; Baret, P.V.; Holmes, A.R.; Monk, B.C.; Goffeau, A.; Cannon, R.D. Fungal PDR transporters: Phylogeny, topology, motifs and function. Fungal Genet. Biol. 2010, 47, 127–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, N.; Prakash, O.; Manoharlal, R.; Sharma, M.; Ghosh, I.; Prasad, R. Analysis of physico-chemical properties of substrates of ABC and MFS multidrug transporters of pathogenic Candida albicans. Eur. J. Med. Chem. 2010, 45, 4813–4826. [Google Scholar] [CrossRef]
- Ernst, R.; Kueppers, P.; Stindt, J.; Kuchler, K.; Schmitt, L. Multidrug efflux pumps: Substrate selection in ATP-binding cassette multidrug efflux pumps--first come, first served? FEBS J. 2010, 277, 540–549. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef] [Green Version]
- Golin, J.; Ambudkar, S.V. V The multidrug transporter Pdr5 on the 25th anniversary of its discovery: An important model for the study of asymmetric ABC transporters. Biochem. J. 2015, 467, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Moreno, A.; Khan, M.F.; Nair, R.; Sharma, S.; Sen, S.; Mondal, A.K.; Pata, J.; Orelle, C.; Falson, P.; et al. Cdr1p highlights the role of the non-hydrolytic ATP-binding site in driving drug translocation in asymmetric ABC pumps. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183131. [Google Scholar] [CrossRef]
- Sorum, B.; Torocsik, B.; Csanady, L. Asymmetry of movements in CFTR’s two ATP sites during pore opening serves their distinct functions. Elife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Abele, R.; Tampé, R. Functional non-equivalence of ATP-binding cassette signature motifs in the transporter associated with antigen processing (TAP). J. Biol. Chem. 2004, 279, 46073–46081. [Google Scholar] [CrossRef] [Green Version]
- Procko, E.; Ferrin-O’Connell, I.; Ng, S.-L.; Gaudet, R. Distinct structural and functional properties of the ATPase sites in an asymmetric ABC transporter. Mol. Cell 2006, 24, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.-F.; Jih, K.-Y.; Shimizu, H.; Li, M.; Hwang, T.-C. Optimization of the degenerated interfacial ATP binding site improves the function of disease-related mutant cystic fibrosis transmembrane conductance regulator (CFTR) channels. J. Biol. Chem. 2010, 285, 37663–37671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, P.; Gaur, N.A.; Prasad, R. Chimeras of the ABC drug transporter Cdr1p reveal functional indispensability of transmembrane domains and nucleotide-binding domains, but transmembrane segment 12 is replaceable with the corresponding homologous region of the non-drug transporter Cdr3p. Microbiology 2006, 152, 1559–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.P.; Kueppers, P.; Hanekop, N.; Schmitt, L. Generating symmetry in the asymmetric ATP-binding cassette (ABC) transporter Pdr5 from Saccharomyces cerevisiae. J. Biol. Chem. 2014, 289, 15272–15279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furman, C.; Mehla, J.; Ananthaswamy, N.; Arya, N.; Kulesh, B.; Kovach, I.; Ambudkar, S.V.; Golin, J. The deviant ATP-binding site of the multidrug efflux pump Pdr5 plays an active role in the transport cycle. J. Biol. Chem. 2013, 288, 30420–30431. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Haseeb, A.; Kumari, A.; Moreno, A.; Falson, P. W1038 near D-loop of NBD2 is a focal point for inter-domain communication in multidrug transporter Cdr1 of Candida albicans. BBA Biomembr. 2018, 1860, 965–972. [Google Scholar] [CrossRef]
- Rawal, M.K.; Khan, M.F.; Kapoor, K.; Goyal, N.; Sen, S.; Saxena, A.K.; Lynn, A.M.; Tyndall, J.D.A.; Monk, B.C.; Cannon, R.D.; et al. Insight into pleiotropic drug resistance ATP-binding cassette pump drug transport through mutagenesis of Cdr1p transmembrane domains. J. Biol. Chem. 2013, 288, 24480–24493. [Google Scholar] [CrossRef] [Green Version]
- Pasrija, R.; Banerjee, D.; Prasad, R. Structure and function analysis of CaMdr1p, a major facilitator superfamily antifungal efflux transporter protein of Candida albicans: Identification of amino acid residues critical for drug/H+ transport. Eukaryot. Cell 2007, 6, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Mandal, A.; Kumar, A.; Singh, A.; Lynn, A.M.; Kapoor, K.; Prasad, R. A key structural domain of the Candida albicans Mdr1 protein. Biochem. J. 2012, 445, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Decottignies, A.; Grant, A.M.; Nichols, J.W.; de Wet, H.; McIntosh, D.B.; Goffeau, A. ATPase and Multidrug Transport Activities of the Overexpressed Yeast ABC Protein Yor1p. J. Biol. Chem. 1998, 273, 12612–12622. [Google Scholar] [CrossRef] [Green Version]
- Lamping, E.; Monk, B.C.; Niimi, K.; Holmes, A.R.; Tsao, S.; Tanabe, K.; Niimi, M.; Uehara, Y.; Cannon, R.D. Characterization of three classes of membrane proteins involved in fungal azole resistance by functional hyperexpression in Saccharomyces cerevisiae. Eukaryot. Cell 2007, 6, 1150–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, N.; Gaur, M.; Sharma, M.; Shukla, S.; Ambudkar, S.V.; Prasad, R. The amino acid residues of transmembrane helix 5 of multidrug resistance protein CaCdr1p of Candida albicans are involved in substrate specificity and drug transport. Biochim. Biophys. Acta 2009, 1788, 1752–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, P.; Prasad, T.; Gaur, N.A.; Shukla, S.; Jha, S.; Komath, S.S.; Khan, L.A.; Haq, Q.M.R.; Prasad, R. Alanine scanning of transmembrane helix 11 of Cdr1p ABC antifungal efflux pump of Candida albicans: Identification of amino acid residues critical for drug efflux. J. Antimicrob. Chemother. 2005, 56, 77–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-Y.; Kinch, L.N.; Borek, D.M.; Wang, J.; Wang, J.; Urbatsch, I.L.; Xie, X.-S.; Grishin, N.V.; Cohen, J.C.; Otwinowski, Z.; et al. Crystal structure of the human sterol transporter ABCG5/ABCG8. Nature 2016, 533, 561–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.; Aller, S.G.; Beis, K.; Carpenter, E.P.; Chang, G.; Chen, L.; Dassa, E.; Dean, M.; Duong Van Hoa, F.; Ekiert, D.; et al. Structural and functional diversity calls for a new classification of ABC transporters. FEBS Lett. 2020. [Google Scholar] [CrossRef]
- Taylor, N.M.I.; Manolaridis, I.; Jackson, S.M.; Kowal, J.; Stahlberg, H.; Locher, K.P. Structure of the human multidrug transporter ABCG2. Nature 2017, 546, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Nim, S.; Lobato, L.G.; Moreno, A.; Chaptal, V.; Rawal, M.K.; Falson, P.; Prasad, R. Atomic modelling and systematic mutagenesis identify residues in multiple drug binding sites that are essential for drug resistance in the major Candida transporter Cdr1. Biochim. Biophys. Acta 2016, 1858, 2858–2870. [Google Scholar] [CrossRef]
- Baghel, P.; Rawal, M.K.; Khan, M.F.; Sen, S.; Siddiqui, M.H.; Chaptal, V.; Falson, P.; Prasad, R. Multidrug ABC transporter Cdr1 of Candida albicans harbors specific and overlapping binding sites for human steroid hormones transport. Biochim. Biophys. Acta 2017, 1859, 1778–1789. [Google Scholar] [CrossRef]
- Kapoor, K.; Rehan, M.; Kaushiki, A.; Pasrija, R.; Lynn, A.M.; Prasad, R. Rational mutational analysis of a multidrug MFS transporter CaMdr1p of Candida albicans by employing a membrane environment based computational approach. PLoS Comput. Biol. 2009, 5, e1000624. [Google Scholar] [CrossRef]
- Kapoor, K.; Rehan, M.; Lynn, A.M.; Prasad, R. Employing information theoretic measures and mutagenesis to identify residues critical for drug-proton antiport function in mdr1p of Candida albicans. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [Green Version]
- Redhu, A.K.; Khandelwal, N.K.; Banerjee, A.; Moreno, A.; Falson, P.; Prasad, R. pHluorin enables insights into the transport mechanism of antiporter Mdr1: R215 is critical for drug/H+ antiport. Biochem. J. 2016, 473, 3127–3145. [Google Scholar] [CrossRef] [PubMed]
- Redhu, A.K.; Banerjee, A.; Shah, A.H.; Moreno, A.; Rawal, M.K.; Nair, R.; Falson, P.; Prasad, R. Molecular Basis of Substrate Polyspecificity of the Candida albicans Mdr1p Multidrug/H(+) Antiporter. J. Mol. Biol. 2018, 430, 682–694. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Taylor, N.M.I.; Bause, M.; Bauer, S.; Bartholomaeus, R.; Bernhardt, G.; Koenig, B.; et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 2018. [Google Scholar] [CrossRef]
- Manolaridis, I.; Jackson, S.M.; Taylor, N.M.I.; Kowal, J.; Stahlberg, H.; Locher, K.P. Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature 2018. [Google Scholar] [CrossRef] [Green Version]
- Orlando, B.J.; Liao, M. ABCG2 transports anticancer drugs via a closed-to-open switch. Nat. Commun. 2020, 11, 2264. [Google Scholar] [CrossRef] [PubMed]
- Sauna, Z.E.; Bohn, S.S.; Rutledge, R.; Dougherty, M.P.; Cronin, S.; May, L.; Xia, D.; Ambudkar, S.V.; Golin, J. Mutations define cross-talk between the N-terminal nucleotide-binding domain and transmembrane helix-2 of the yeast multidrug transporter Pdr5: Possible conservation of a signaling interface for coupling ATP hydrolysis to drug transport. J. Biol. Chem. 2008, 283, 35010–35022. [Google Scholar] [CrossRef] [Green Version]
- Ananthaswamy, N.; Rutledge, R.; Sauna, Z.E.; Ambudkar, S.V.; Dine, E.; Nelson, E.; Xia, D.; Golin, J. The signaling interface of the yeast multidrug transporter Pdr5 adopts a cis conformation, and there are functional overlap and equivalence of the deviant and canonical Q-loop residues. Biochemistry 2010, 49, 4440–4449. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.H.; Rawal, M.K.; Dhamgaye, S.; Komath, S.S.; Saxena, A.K.; Prasad, R. Mutational Analysis of Intracellular Loops Identify Cross Talk with Nucleotide Binding Domains of Yeast ABC Transporter Cdr1p. Sci. Rep. 2015, 5, 11211. [Google Scholar] [CrossRef] [Green Version]
- Dawson, R.J.P.; Locher, K.P. Structure of a bacterial multidrug ABC transporter. Nature 2006, 443, 180–185. [Google Scholar] [CrossRef]
- Niimi, K.; Harding, D.R.K.; Holmes, A.R.; Lamping, E.; Niimi, M.; Tyndall, J.D.A.; Cannon, R.D.; Monk, B.C. Specific interactions between the Candida albicans ABC transporter Cdr1p ectodomain and a D-octapeptide derivative inhibitor. Mol. Microbiol. 2012, 85, 747–767. [Google Scholar] [CrossRef] [Green Version]
- Johnson, Z.L.; Chen, J. ATP Binding Enables Substrate Release from Multidrug Resistance Protein 1. Cell 2018, 172, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, F.; Chen, J. Conformational Changes of CFTR upon Phosphorylation and ATP Binding. Cell 2017, 170, 483–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Liu, F.; Chen, J. Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc. Natl. Acad. Sci. USA 2018, 115, 12757–12762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, R.; Kueppers, P.; Klein, C.M.; Schwarzmueller, T.; Kuchler, K.; Schmitt, L. A mutation of the H-loop selectively affects rhodamine transport by the yeast multidrug ABC transporter Pdr5. Proc. Natl. Acad. Sci. USA 2008, 105, 5069–5074. [Google Scholar] [CrossRef] [Green Version]
- Kolaczkowski, M.; Środa-Pomianek, K.; Kolaczkowska, A.; Michalak, K. A conserved interdomain communication pathway of pseudosymmetrically distributed residues affects substrate specificity of the fungal multidrug transporter Cdr1p. Biochim. Biophys. Acta Biomembr. 2013, 1828, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Vishwakarma, P.; Kumar, A.; Lynn, A.M.; Prasad, R. Information theoretic measures and mutagenesis identify a novel linchpin residue involved in substrate selection within the nucleotide-binding domain of an ABCG family exporter Cdr1p. Arch. Biochem. Biophys. 2019, 663, 143–150. [Google Scholar] [CrossRef]
- Tanabe, K.; Bonus, M.; Tomiyama, S.; Miyoshi, K.; Nagi, M.; Niimi, K.; Chindamporn, A.; Gohlke, H.; Schmitt, L.; Cannon, R.D.; et al. FK506 Resistance of Saccharomyces cerevisiae Pdr5 and Candida albicans Cdr1 Involves Mutations in the Transmembrane Domains and Extracellular Loops. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Holmes, A.R.; Keniya, M.V.; Ivnitski-Steele, I.; Monk, B.C.; Lamping, E.; Sklar, L.A.; Cannon, R.D. The monoamine oxidase A inhibitor clorgyline is a broad-spectrum inhibitor of fungal ABC and MFS transporter efflux pump activities which reverses the azole resistance of Candida albicans and Candida glabrata clinical isolates. Antimicrob. Agents Chemother. 2012, 56, 1508–1515. [Google Scholar] [CrossRef] [Green Version]
- Keniya, M.V.; Fleischer, E.; Klinger, A.; Cannon, R.D.; Monk, B.C. Inhibitors of the Candida albicans Major Facilitator Superfamily Transporter Mdr1p Responsible for Fluconazole Resistance. PLoS ONE 2015, 10, e0126350. [Google Scholar] [CrossRef]
- Monk, B.C.; Keniya, M.V. Using Yeast to Discover Inhibitors of Multidrug Efflux in Candida Albicans. In Candida Albicans: Cellular and Molecular Biology; Prasad, R., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 491–543. ISBN 978-3-319-50409-4. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| MOTIF | Catalytic (Consensus) NBS ^ | Non-Catalytic (Deviant) NBS ¥ |
|---|---|---|
| Walker A | GKTT | GCST |
| Walker B | LDE | WDN |
| Q-loop | Q | E |
| Signature sequence | SGG | NVE |
| D-loop | GLD | GLD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banerjee, A.; Pata, J.; Sharma, S.; Monk, B.C.; Falson, P.; Prasad, R. Directed Mutational Strategies Reveal Drug Binding and Transport by the MDR Transporters of Candida albicans. J. Fungi 2021, 7, 68. https://doi.org/10.3390/jof7020068
Banerjee A, Pata J, Sharma S, Monk BC, Falson P, Prasad R. Directed Mutational Strategies Reveal Drug Binding and Transport by the MDR Transporters of Candida albicans. Journal of Fungi. 2021; 7(2):68. https://doi.org/10.3390/jof7020068
Chicago/Turabian StyleBanerjee, Atanu, Jorgaq Pata, Suman Sharma, Brian C. Monk, Pierre Falson, and Rajendra Prasad. 2021. "Directed Mutational Strategies Reveal Drug Binding and Transport by the MDR Transporters of Candida albicans" Journal of Fungi 7, no. 2: 68. https://doi.org/10.3390/jof7020068