Safety and Tolerability of SRX246, a Vasopressin 1a Antagonist, in Irritable Huntington’s Disease Patients—A Randomized Phase 2 Clinical Trial

 , , , ,
, , , ,
Abstract
:1. Introduction
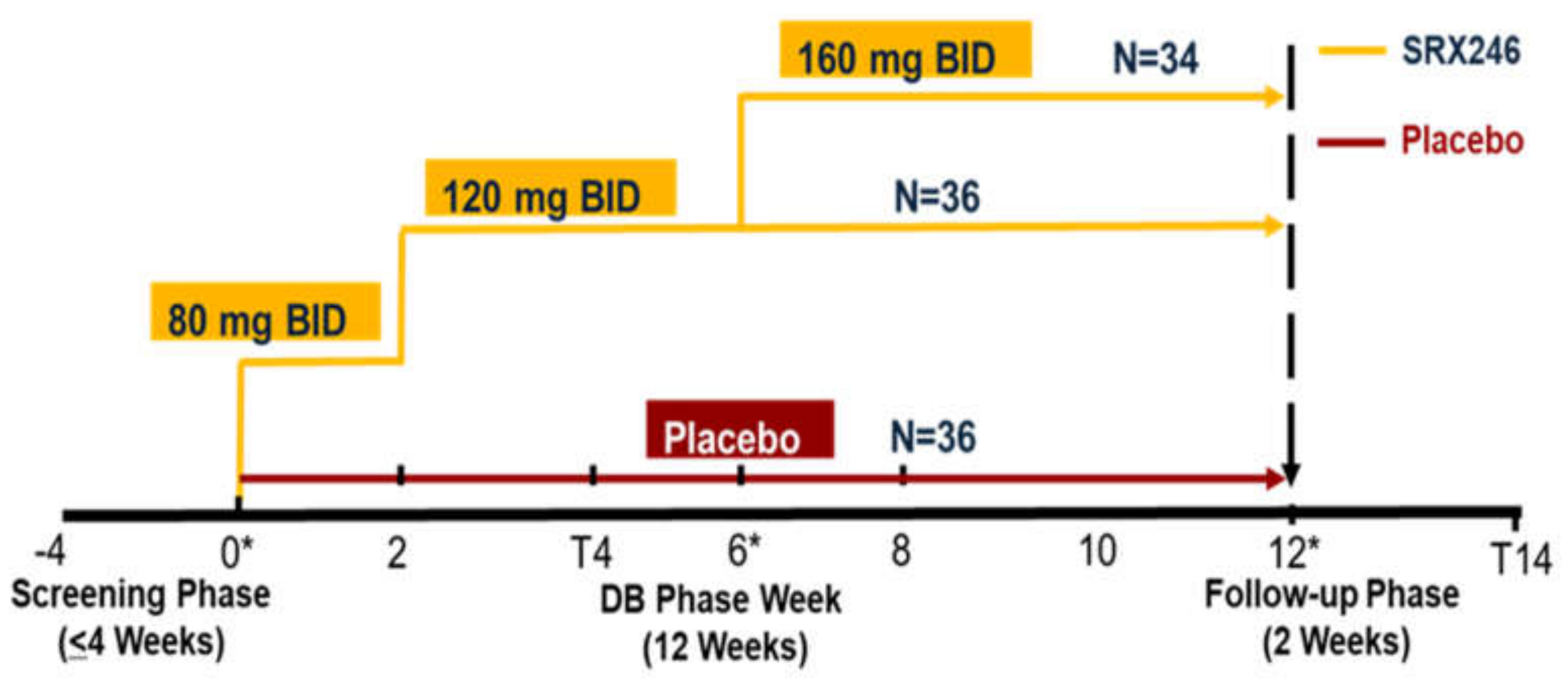
2. Experimental Section
Statistical Analysis
3. Results
3.1. Recruiting
3.2. Demographics
3.3. Primary Aim: Tolerability
3.4. Secondary Aim: Safety
3.5. Apathy
3.6. Suicidality
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ghosh, R.; Tabrizi, S.J. Clinical Features of Huntington’s Disease. In Polyglutamine Disorders. Advances in Experimental Medicine and Biology; Nóbrega, C., Pereira de Almeida, L., Eds.; Springer: Cham, Switzerland, 2018; Volume 1049. [Google Scholar] [CrossRef]
- FDA. The Voice of the Patient: Huntington’s Disease. 2016. Available online: https://www.fda.gov/media/96196.
- Fabio, K.; Guillon, C.; Lu, S.; Heindel, N.; Miller, M.; Ferris, C.; Brownstein, M.J.; Garripa, C.; Steiner, M.; Coccaro, E.; et al. Vasopressin antagonists as anxiolytics and antidepressants: Recent developments. Front. CNS Drug Discov. 2010, 1, 156–183. [Google Scholar]
- Guillon, C.D.; Koppel, G.A.; Brownstein, M.J.; Chaney, M.O.; Ferris, C.F.; Lu, S.F.; Fabio, K.M.; Miller, M.J.; Heindel, N.D.; Hunden, D.C.; et al. Azetidinones as vasopressin V1a antagonists. Bioorg. Med. Chem. 2007, 15, 2054–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferris, C.; Stolberg, T.; Kulkarni, P.; Murugavel, M.; Blanchard, R.; Blanchard, D.C.; Febo, M.; Brevard, M.; Simon, N.G. Imaging the neural circuitry and chemical control of aggressive motivation. BMC Neurosci. 2008, 9, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, N.G.; Guillon, C.; Fabio, K.; Heindel, N.D.; Lu, S.F.; Miller, M.; Ferris, C.F.; Brownstein, M.J.; Garripa, C.; Koppel, G.A. Vasopressin antagonists as anxiolytics and antidepressants: Recent developments. Recent Pat. CNS Drug Discov. 2008, 3, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.J.; Coccaro, E.F.; Cremers, H.; McCarron, R.; Lu, S.F.; Brownstein, M.; Simon, N.G. A novel V1a receptor antagonist blocks vasopressin-induced changes in the CNS response to emotional stimuli: An fMRI study. Front. Syst. Neurosci. 2013, 7, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackwelder, W.C. “Proving the Null Hypothesis” in clinical trials. Control. Clin. Trials 1982, 3, 345–353. [Google Scholar] [CrossRef]
- The Huntington Study Group. Safety and tolerability of the free-radical scavenger opc-14117 in Huntington’s disease. Neurology 1998, 50, 1366–1373. [Google Scholar] [CrossRef] [PubMed]
- The Huntington Study Group. Dosage effects of riluzole in Huntington’s disease. Neurology 2003, 61, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- The Huntington Study Group. Minocycline safety and tolerability in Huntington disease. Neurology 2004, 63, 547–549. [Google Scholar] [CrossRef] [PubMed]
- The Huntington Study Group. Tetrabenazine as antichorea therapy in Huntington disease. Neurology 2006, 66, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Fagerland, M.W.; Lydersen, S.; Laake, P. Recommended confidence intervals for two independent binomial proportions. In Statistical Methods in Medical Research; John Wiley & Sons: Hoboken, NJ, USA, 2011; Volume 24, pp. 224–254. [Google Scholar]
- Wang, W. On construction of the smallest one-sided confidence interval for the difference of two proportions. Ann. Stat. 2010, 38, 1227–1243. [Google Scholar] [CrossRef] [Green Version]
- Shan, G.; Wang, W. ExactCIdiff: Inductive Confidence Intervals for the Difference between Two Proportions. 2013. Available online: https://CRAN.R-project.org/package=ExactCIDiff.
- FDA. Guidance for Industry: Drug-Induced Liver Injury: Premarketing Clinical Evaluation. 2009. Available online: https://www.fda.
- Nilsson, M.E.; Suryawanshi, S.; Gassmann-Mayer, C.; Dubrava, S.; McSorley, P.; Jiang, K. Columbia–Suicide Severity Rating Scale Scoring and Data Analysis Guide, version 2; Columbia University: New York, NY, USA, 2013. [Google Scholar]
- Nielsen, S.M.; Vinther-Jensen, T.; Nielsen, J.E.; Nørremølle, A.; Hasholt, L.; Hjermind, L.E.; Josefsen, K. Liver function in Huntington’s disease assessed by blood biochemical analyses in a clinical setting. J. Neurol. Sci. 2016, 362, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.; Stüwe, S.H.; Goetze, O.; Banasch, M.; Klotz, P.; Lukas, C.; Tegenthoff, M.; Beste, C.; Orth, M.; Saft, C. Progressive hepatic mitochondrial dysfunction in premanifest Huntington’s disease. Mov. Disord. 2014, 29, 831–834. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Characteristic * | Variable | Placebo (N = 36) | 120 mg (N = 36) | 160 mg (N = 34) | N (% of total N =106) |
|---|---|---|---|---|---|
| Gender | Male | 16 (44.44%) | 18 (50.00%) | 17 (50.00%) | 51 (48.11%) |
| Female | 20 (55.56%) | 18 (50.00%) | 17 (50.00%) | 55 (51.89%) | |
| Missing | 0 | 0 | 0 | 0 | |
| Race | White | 36 (100.0%) | 36 (100.0%) | 33 (97.06%) | 105 (99.06%) |
| Not White | 0 (0.00%) | 0 (0.00%) | 1 (2.94%) | 1 (0.94%) | |
| Unknown/Not Reported | 0 | 0 | 0 | 0 | |
| Ethnicity | Hispanic/Latino | 0 (0.00%) | 2 (5.56%) | 4 (11.76%) | 6 (5.71%) |
| Not Hispanic/Latino | 35 (100.0%) | 34 (94.44%) | 30 (88.24%) | 99 (94.29%) | |
| Missing | 1 | 0 | 0 | 1 | |
| Age | Mean (SD) | 51.7 (10.4) | 51.1 (13.2) | 48.9 (12.7) | 50.6 (12.1) |
| Min.-Max. | 32 - 72 | 19 - 77 | 27 - 78 | 19 - 78 | |
| Missing | 0 | 0 | 0 | 0 | |
| Years of Education | 1–11 years | 1 (2.78%) | 2 (5.56%) | 0 (0.00%) | 3 (2.83%) |
| High school/Assoc./Tech. | 18 (50.00%) | 24 (66.67%) | 20 (58.82%) | 62 (58.49%) | |
| Bachelors or Higher | 17 (47.22%) | 10 (27.78%) | 14 (41.18%) | 41 (38.68%) | |
| Unknown/Missing | 0 | 0 | 0 | 0 | |
| UHDRS Irritability Severity (q30b) | Well Controlled | 1 (3%) | 0 (0%) | 0 (0%) | p = 0.82 |
| Questionable | 0 (0%) | 1 (3%) | 0 (0%) | ||
| Definite but Mild ** | 8 (22%) | 9 (25%) | 13 (38%) | ||
| Moderate | 22 (61%) | 23 (64%) | 19 (56%) | ||
| Severe | 5 (14%) | 3 (8%) | 2 (6%) | ||
| UHDRS Aggression Severity (q31b) | Well Controlled | 8 (22%) | 6 (16%) | 5 (15%) | p= 0.80 |
| Verbal Threats | 9 (25%) | 9 (25%) | 12 (36%) | ||
| Mild Physical ** | 8 (22%) | 14 (39%) | 9 (26%) | ||
| Definite Physical | 9 (25%) | 5 (14%) | 7 (21%) | ||
| Severe Physical | 2 (6%) | 2 (6%) | 1 (3%) |
| Group | |||
|---|---|---|---|
| Placebo | 120 mg | 160 mg | |
| Sample Size | 36 | 36 | 34 |
| Number of completers (%) | 30 (83%) | 25 (69%) | 27 (79%) |
| One-Sided 97.5% CI of Proportion Difference | (−1, 0.35) | (−1, 0.27) | |
| Null Hypothesis Decision | Reject | Reject | |
| Group | |||
|---|---|---|---|
| Placebo | 120 mg | 160 mg | |
| Sample Size | 36 | 36 | 34 |
| Number of subjects with AEs (%) | 26 (72%) | 29 (81%) | 30 (88%) |
| One-Sided 97.5% CI of Proportion Difference | (−1, 0.28) | (−1, 0.36) | |
| Null Hypothesis Decision | Reject | Reject | |
| Group | |||
|---|---|---|---|
| Placebo | 120 mg | 160 mg | |
| Sample Size | 36 | 36 | 34 |
| Number of subjects with SAEs (%) | 1 (3%) | 6 (17%) | 2 (6%) |
| One-Sided 97.5% CI of Proportion Difference | (−1, 0.03) | (−1, 0.12) | |
| Null Hypothesis Decision | Reject | Reject | |
| Group | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
System Organ Class | Placebo | 120 mg | 160 mg | RR (95% CI) | ||||||||
| N = 36 | N = 36 | N = 34 | ||||||||||
| # (%) of | # of | Rate | # (%) of | # of | Rate | # (%) of | # of | Rate | Placebo vs. 120 mg | Placebo vs 160 mg | 120 mg vs. 160 mg | |
| Subjects | Events | Subjects | Events | Subjects | Events | |||||||
| Blood | 0 (0.0%) | 0 | 0.000 | 1 (2.8%) | 1 | 0.009 | 0 (0.0%) | 0 | 0.000 | N/A | N/A | N/A |
| Cardiac | 2 (5.6%) | 2 | 0.018 | 1 (2.8%) | 1 | 0.009 | 1 (2.9%) | 1 | 0.009 | 0.50 (0.05, 5.27) | 1.89 (0.18, 19.89) | 0.94 (0.06, 14.51) |
| Gastrointestinal | 4 (11.1%) | 4 | 0.035 | 7 (19.4%) | 13 | 0.117 | 9 (26.5%) | 14 | 0.122 | 1.75 (0.56, 5.46) | 0.42 (0.14, 1.24) | 0.73 (0.31, 1.75) |
| General | 2 (5.6%) | 2 | 0.018 | 3 (8.3%) | 3 | 0.027 | 5 (14.7%) | 7 | 0.061 | 1.50 (0.27, 8.45) | 0.38 (0.08, 1.82) | 0.57 (0.15, 2.19) |
| Infections | 9 (25.0%) | 11 | 0.097 | 6 (16.7%) | 7 | 0.063 | 4 (11.8%) | 4 | 0.035 | 0.67 (0.26, 1.68) | 2.13 (0.72, 6.26) | 1.42 (0.44, 4.59) |
| Injury, Poisoning and Procedural Comp. | 5 (13.9%) | 5 | 0.044 | 4 (11.1%) | 6 | 0.054 | 4 (11.8%) | 7 | 0.061 | 0.80 (0.23, 2.74) | 1.18 (0.35, 4.03) | 0.94 (0.26, 3.48) |
| Investigations | 6 (16.7%) | 8 | 0.071 | 8 (22.2%) | 8 | 0.072 | 8 (23.5%) | 9 | 0.078 | 1.33 (0.51, 3.46) | 0.71 (0.27, 1.83) | 0.94 (0.40, 2.23) |
| Metabolic | 1 (2.8%) | 1 | 0.009 | 1 (2.8%) | 1 | 0.009 | 0 (0.0%) | 0 | 0.000 | 1.00 (0.07, 15.38) | N/A | N/A |
| Musculoskeletal | 1 (2.8%) | 1 | 0.009 | 5 (13.9%) | 5 | 0.045 | 5 (14.7%) | 6 | 0.052 | 5.00 (0.61, 40.70) | 0.19 (0.02, 1.54) | 0.94 (0.30, 2.98) |
| Neoplasms | 0 (0.0%) | 0 | 0.000 | 1 (2.8%) | 1 | 0.009 | 1 (2.9%) | 1 | 0.009 | N/A | N/A | 0.94 (0.06, 14.51) |
| Nervous System | 8 (22.2%) | 9 | 0.080 | 6 (16.7%) | 7 | 0.063 | 4 (11.8%) | 6 | 0.052 | 0.75 (0.29, 1.94) | 1.89 (0.63, 5.70) | 1.42 (0.44, 4.59) |
| Psychiatric | 4 (11.1%) | 6 | 0.053 | 8 (22.2%) | 11 | 0.099 | 8 (23.5%) | 9 | 0.078 | 2.00 (0.66, 6.06) | 0.47 (0.16, 1.43) | 0.94 (0.40, 2.23) |
| Renal | 1 (2.8%) | 1 | 0.009 | 2 (5.6%) | 2 | 0.018 | 2 (5.9%) | 2 | 0.017 | 2.00 (0.19, 21.09) | 0.47 (0.04, 4.97) | 0.94 (0.14, 6.33) |
| Reproductive | 0 (0.0%) | 0 | 0.000 | 1 (2.8%) | 1 | 0.009 | 0 (0.0%) | 0 | 0.000 | N/A | N/A | N/A |
| Respiratory | 1 (2.8%) | 1 | 0.009 | 3 (8.3%) | 5 | 0.045 | 4 (11.8%) | 4 | 0.035 | 3.00 (0.33, 27.50) | 0.24 (0.03, 2.01) | 0.71 (0.17, 2.94) |
| Skin | 1 (2.8%) | 1 | 0.009 | 2 (5.6%) | 2 | 0.018 | 1 (2.9%) | 1 | 0.009 | 2.00 (0.19, 21.09) | 0.94 (0.06, 14.51) | 1.89 (0.18, 19.89) |
| Vascular | 0 (0.0%) | 0 | 0.000 | 1 (2.8%) | 1 | 0.009 | 2 (5.9%) | 2 | 0.017 | N/A | N/A | 0.47 (0.04, 4.97) |
| Total | 26 (72.2%) | 52 | 0.460 | 29 (80.6%) | 75 | 0.676 | 30 (88.2%) | 73 | 0.635 | 1.12 (0.86, 1.44) | 0.82 (0.65, 1.04) | 0.91 (0.75, 1.12) |
| Group | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
System Organ Class | Placebo | 120 mg | 160 mg | RR (95% CI) | ||||||||
| N = 36 | N = 36 | N = 34 | ||||||||||
| # (%) of | # of | Rate | # (%) of | # of | Rate | # (%) of | # of | Rate | Placebo vs. 120 mg | Placebo vs. 160 mg | 120 mg vs. 160 mg | |
| Subjects | Events | Subjects | Events | Subjects | Events | |||||||
| Cardiac | ||||||||||||
| Angina pectoris | 0 (0.0%) | 0 | 0.000 | 1 (2.8%) | 1 | 0.009 | 0 (0.0%) | 0 | 0.000 | N/A | N/A | N/A |
| Infections | ||||||||||||
| Staphylococcal skin infection | 1 (2.8%) | 1 | 0.009 | 0 (0.0%) | 0 | 0.000 | 0 (0.0%) | 0 | 0.000 | N/A | N/A | N/A |
| Injury, Poisoning and Procedural Comp. | ||||||||||||
| Ankle fracture | 0 (0.0%) | 0 | 0.000 | 1 (2.8%) | 1 | 0.009 | 0 (0.0%) | 0 | 0.000 | N/A | N/A | N/A |
| Musculoskeletal | ||||||||||||
| Muscular weakness | 0 (0.0%) | 0 | 0.000 | 1 (2.8%) | 1 | 0.009 | 0 (0.0%) | 0 | 0.000 | N/A | N/A | N/A |
| Neoplasms | ||||||||||||
| Renal neoplasm | 0 (0.0%) | 0 | 0.000 | 1 (2.8%) | 1 | 0.009 | 0 (0.0%) | 0 | 0.000 | N/A | N/A | N/A |
| Psychiatric | ||||||||||||
| Aggression | 0 (0.0%) | 0 | 0.000 | 0 (0.0%) | 0 | 0.000 | 1 (2.9%) | 1 | 0.009 | N/A | N/A | N/A |
| Irritability | 0 (0.0%) | 0 | 0.000 | 1 (2.8%) | 1 | 0.009 | 0 (0.0%) | 0 | 0.000 | N/A | N/A | N/A |
| Psychotic disorder | 0 (0.0%) | 0 | 0.000 | 1 (2.8%) | 1 | 0.009 | 0 (0.0%) | 0 | 0.000 | N/A | N/A | N/A |
| Suicide attempt | 0 (0.0%) | 0 | 0.000 | 0 (0.0%) | 0 | 0.000 | 1 (2.9%) | 1 | 0.009 | N/A | N/A | N/A |
| Total | 1 (2.8%) | 1 | 0.009 | 6 (16.7%) | 6 | 0.054 | 2 (5.9%) | 2 | 0.017 | 6.00 (0.76, 47.36) | 0.47 (0.04, 4.97) | 2.83 (0.61, 13.09) |
| Time Point | Statistic | Placebo (N = 36) | SRX246 120 mg BID (N = 36) | SRX246 160 mg BID (N = 34) |
|---|---|---|---|---|
| Visit 2, Baseline | n | 35 | 36 | 34 |
| Mean (SD) | 3.5 (3.91) | 4.0 (4.37) | 3.4 (5.01) | |
| Median | 2.0 | 2.0 | 1.0 | |
| Min, Max 95% CI | 0, 16 (2.1, 4.8) | 0, 12 (2.5, 5.5) | 0, 16 (1.7, 5.2) | |
| Visit 4, Week 6 | n | 35 | 31 | 33 |
| Mean (SD) | 2.9 (4.38) | 2.9 (3.61) | 3.4 (4.64) | |
| Median | 1.0 | 2.0 | 1.0 | |
| Min, Max 95% CI | 0, 16 (1.4, 4.4) | 0, 12 (1.6, 4.2) | 0, 16 (1.7, 5.0) | |
| Visit 7, End of Treatment | n | 34 | 31 | 33 |
| Mean (SD) | 3.0 (4.17) | 3.4 (4.27) | 3.8 (4.77) | |
| Median | 1.0 | 2.0 | 2.0 | |
| Min, Max 95% CI | 0, 16 (1.5, 4.5) | 0, 16 (1.8, 5.0) | 0, 16 (2.1, 5.0) |
| Placebo N = 36 | Up to 120 mg N = 36 | Up to 160 mg N = 34 | |
|---|---|---|---|
| Visit | Yes | Yes | Yes |
| 1 * | 10 27.8% | 14 38.9% | 11 32.4% |
| 2 | 4 | 3 | 0 |
| 11.1% | 8.3% | 0.0% | |
| 7 | 0 | 1 | 1 |
| 0.0% | 2.8% | 2.9% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brownstein, M.J.; Simon, N.G.; Long, J.D.; Yankey, J.; Maibach, H.T.; Cudkowicz, M.; Coffey, C.; Conwit, R.A.; Lungu, C.; Anderson, K.E.; et al. Safety and Tolerability of SRX246, a Vasopressin 1a Antagonist, in Irritable Huntington’s Disease Patients—A Randomized Phase 2 Clinical Trial. J. Clin. Med. 2020, 9, 3682. https://doi.org/10.3390/jcm9113682
Brownstein MJ, Simon NG, Long JD, Yankey J, Maibach HT, Cudkowicz M, Coffey C, Conwit RA, Lungu C, Anderson KE, et al. Safety and Tolerability of SRX246, a Vasopressin 1a Antagonist, in Irritable Huntington’s Disease Patients—A Randomized Phase 2 Clinical Trial. Journal of Clinical Medicine. 2020; 9(11):3682. https://doi.org/10.3390/jcm9113682
Chicago/Turabian StyleBrownstein, Michael J., Neal G. Simon, Jeffrey D. Long, Jon Yankey, Hilda T. Maibach, Merit Cudkowicz, Christopher Coffey, Robin A. Conwit, Codrin Lungu, Karen E. Anderson, and et al. 2020. "Safety and Tolerability of SRX246, a Vasopressin 1a Antagonist, in Irritable Huntington’s Disease Patients—A Randomized Phase 2 Clinical Trial" Journal of Clinical Medicine 9, no. 11: 3682. https://doi.org/10.3390/jcm9113682