The Scope for Thalassemia Gene Therapy by Disruption of Aberrant Regulatory Elements

 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Murine Erythroid Leukemia (MEL) HBBIVS Cells
2.2. Indel Characterization in Transgenic Humanized MEL-HBBIVS Cell Lines
2.2.1. Analysis of Indels in Bulk Cells
2.2.2. Analysis of Indels in Disrupted MEL-HBBIVS Clones
2.3. Sequencing
2.4. Assessment of RNA Expression
2.5. Globin Chain Analyses
2.6. Statistical and Bioinformatics Analyses
2.6.1. Statistical Analyses
2.6.2. Retrieval of Target Sequences
2.6.3. In Silico Design and Evaluation of Designer Nucleases
3. Results
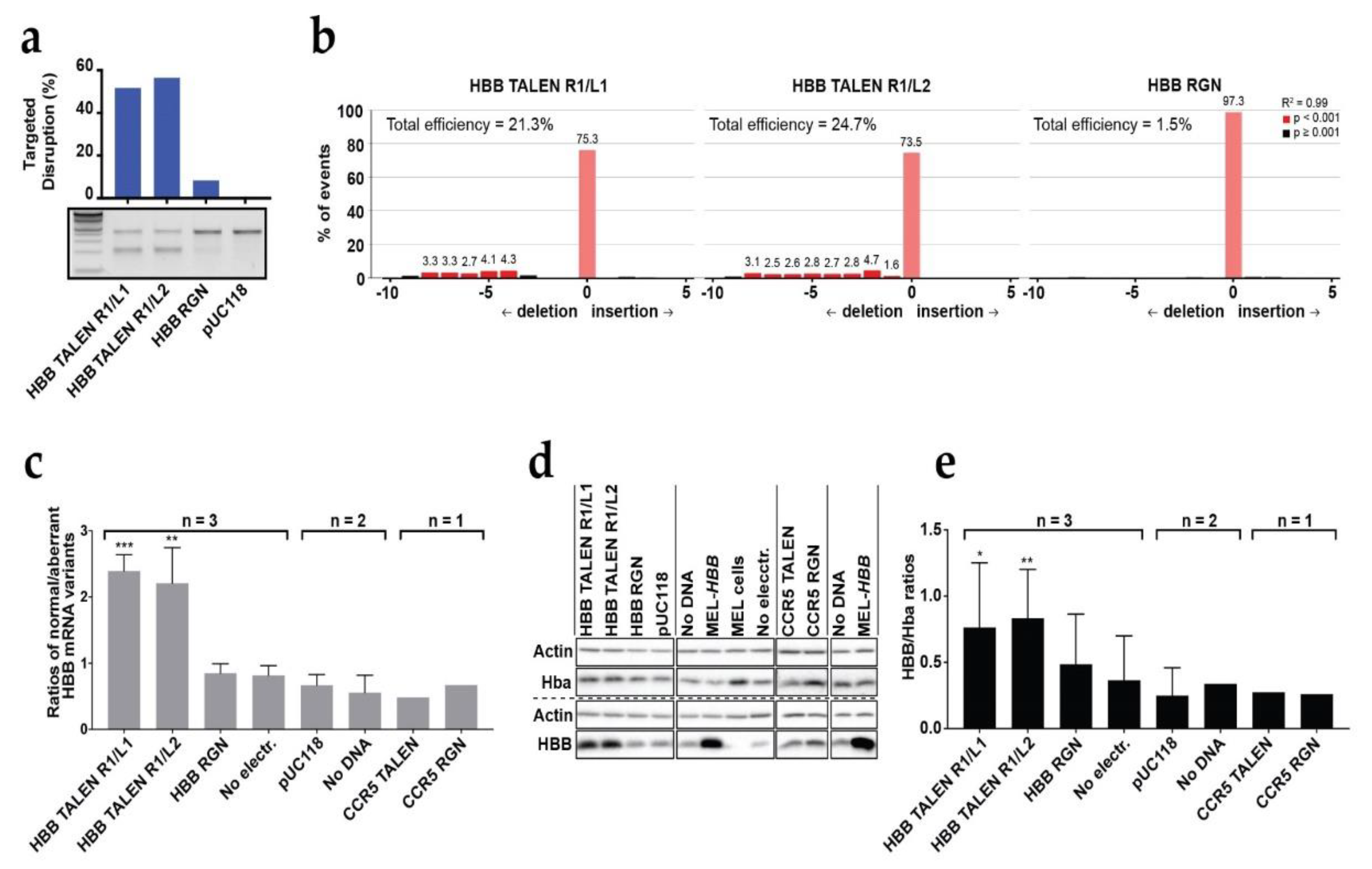
3.1. Analysis of DARE in Bulk Populations of Modified Polyclonal MEL-HBBIVS Cells
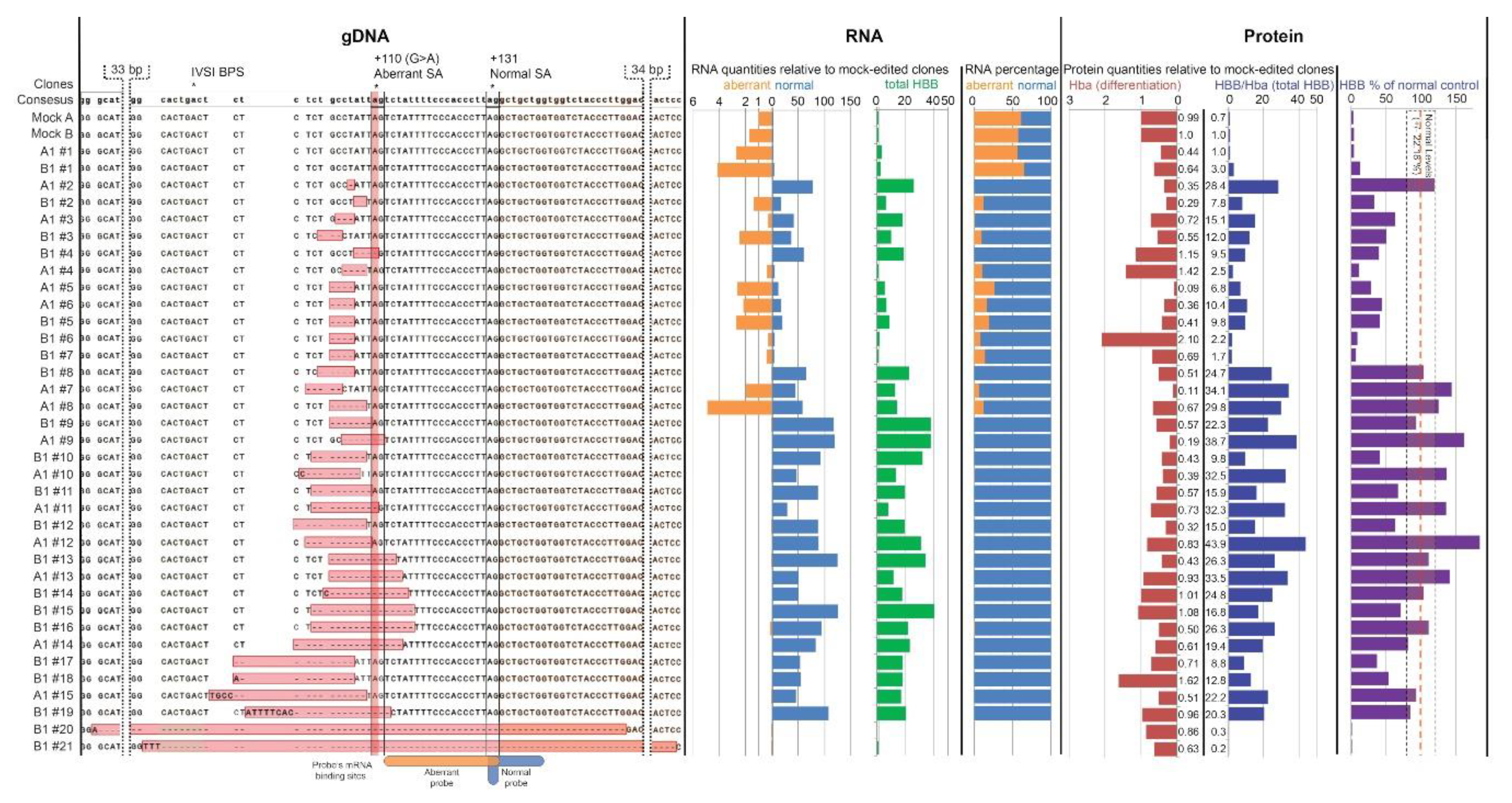
3.2. Analysis of DARE in Cell Clones of Modified Clonal MEL-HBBIVS Cells
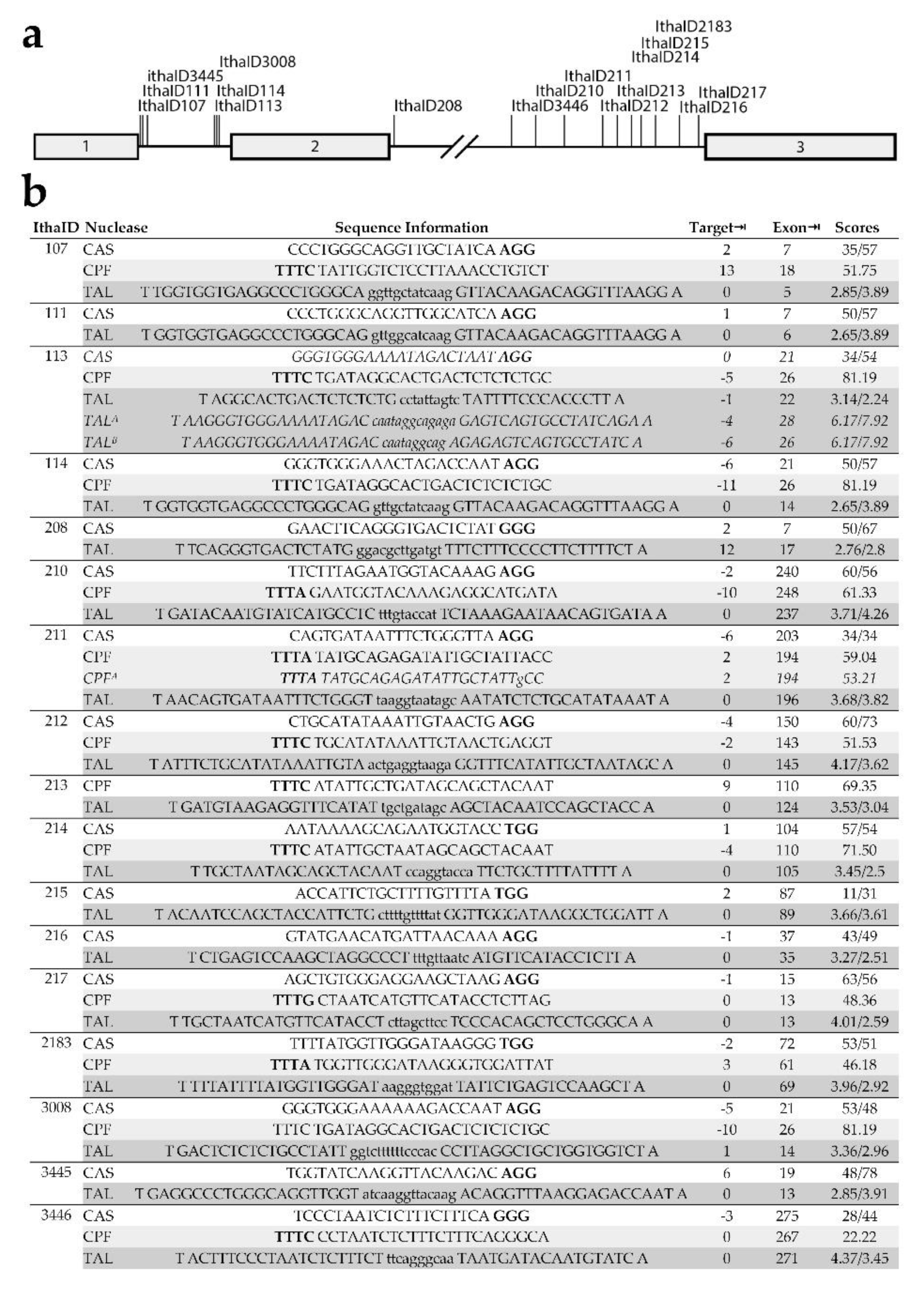
3.3. Analysis of Potential Alternative DARE Targets for β-Thalassemia
3.4. Additional Disease Targets for DARE
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Papasavva, P.; Kleanthous, M.; Lederer, C.W. Rare opportunities: CRISPR/Cas-based therapy development for rare genetic diseases. Mol. Diagn. Ther. 2019, 23, 201–222. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 574, 464–465. [Google Scholar] [CrossRef]
- Sebastiano, V.; Maeder, M.L.; Angstman, J.F.; Haddad, B.; Khayter, C.; Yeo, D.T.; Goodwin, M.J.; Hawkins, J.S.; Ramirez, C.L.; Batista, L.F.; et al. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells 2011, 29, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Mali, P.; Huang, X.; Dowey, S.N.; Cheng, L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood 2011, 118, 4599–4608. [Google Scholar] [CrossRef] [PubMed]
- Patsali, P.; Kleanthous, M.; Lederer, C.W. Disruptive technology: CRISPR/Cas-based tools and approaches. Mol. Diagn. Ther. 2019, 23, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Lederer, C.W.; Kleanthous, M. Beta testing: Preclinical genome editing in β-globin disorders. Cell Gen. Ther. Insights 2015, 1, 231–242. [Google Scholar] [CrossRef]
- Vierstra, J.; Reik, A.; Chang, K.-H.; Stehling-Sun, S.; Zhou, Y.; Hinkley, S.J.; Paschon, D.E.; Zhang, L.; Psatha, N.; Bendana, Y.R.; et al. Functional footprinting of regulatory DNA. Nat. Methods 2015, 12, 927–930. [Google Scholar] [CrossRef]
- Canver, M.C.; Orkin, S.H. Customizing the genome as therapy for the β-hemoglobinopathies. Blood 2016, 127, 2536–2545. [Google Scholar] [CrossRef]
- Chang, K.-H.; Smith, S.E.; Sullivan, T.; Chen, K.; Zhou, Q.; West, J.A.; Liu, M.; Liu, Y.; Vieira, B.F.; Sun, C.; et al. Long-term engraftment and fetal globin induction upon BCL11A gene editing in bone-marrow-derived CD34+ hematopoietic stem and progenitor cells. Mol. Ther. Methods Clin. Dev. 2017, 4, 137–148. [Google Scholar] [CrossRef]
- Traxler, E.; Yao, Y.; Li, C.; Grevet, J.; Huang, P.; Wright, S.; Blobel, G.A.; Weiss, M.J. Genome editing recreates hereditary persistence of fetal hemoglobin in primary human erythroblasts (abstract 640). In Proceedings of the American Society of Hematology Annual Meeting, Orlando, FL, USA, 5–8 December 2015. [Google Scholar]
- Canver, M.C.; Smith, E.C.; Sher, F.; Pinello, L.; Sanjana, N.E.; Shalem, O.; Chen, D.D.; Schupp, P.G.; Vinjamur, D.S.; Garcia, S.P.; et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 2015, 527, 192–197. [Google Scholar] [CrossRef]
- Traxler, E.A.; Yao, Y.; Wang, Y.D.; Woodard, K.J.; Kurita, R.; Nakamura, Y.; Hughes, J.R.; Hardison, R.C.; Blobel, G.A.; Li, C.; et al. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat. Med. 2016, 22, 987–990. [Google Scholar] [CrossRef] [PubMed]
- Van Agtmaal, E.L.; André, L.M.; Willemse, M.; Cumming, S.A.; van Kessel, I.D.G.; van den Broek, W.J.A.A.; Gourdon, G.; Furling, D.; Mouly, V.; Monckton, D.G.; et al. CRISPR/Cas9-induced (CTG⋅CAG)n repeat instability in the myotonic dystrophy type 1 locus: Implications for therapeutic genome editing. Mol. Ther. 2017, 25, 24–43. [Google Scholar] [CrossRef] [PubMed]
- Amoasii, L.; Long, C.; Li, H.; Mireault, A.A.; Shelton, J.M.; Sanchez-Ortiz, E.; McAnally, J.R.; Bhattacharyya, S.; Schmidt, F.; Grimm, D.; et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017, 9, eaan8081. [Google Scholar] [CrossRef] [PubMed]
- Patsali, P.; Turchiano, G.; Papasavva, P.; Romito, M.; Loucari, C.; Stephanou, C.; Sitarou, M.; Mussolino, C.; Cornu, T.I.; Antoniou, M.N.; et al. CRISPR/Cas9-and TALEN-mediated disruption of aberrant regulatory elements restores normal splicing and gene function. In Human Gene Therapy; Mary Ann Liebert, Inc.: New Rochelle, NY, USA, 2018; Volume 29, pp. A100–A101. [Google Scholar]
- Patsali, P.; Turchiano, G.; Papasavva, P.; Romito, M.; Loucari, C.C.; Stephanou, C.; Christou, S.; Sitarou, M.; Mussolino, C.; Cornu, T.I.; et al. Correction of IVS I-110(G>A) β-thalassemia by CRISPR/Cas-and TALEN-mediated disruption of aberrant regulatory elements in human hematopoietic stem and progenitor cells. Haematologica 2019, 104, e497–e501. [Google Scholar] [CrossRef]
- Xu, S.; Luk, K.; Yao, Q.; Shen, A.H.; Zeng, J.; Wu, Y.; Luo, H.-Y.; Brendel, C.; Pinello, L.; Chui, D.H.K.; et al. Editing aberrant splice sites efficiently restores β-globin expression in β-thalassemia. Blood 2019, 133, 2255–2262. [Google Scholar] [CrossRef]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd; Barbas, C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef]
- Patsali, P.; Papasavva, P.; Stephanou, C.; Christou, S.; Sitarou, M.; Antoniou, M.N.; Lederer, C.W.; Kleanthous, M. Short-hairpin RNA against aberrant HBBIVSI-110(G>A) mRNA restores β-globin levels in a novel cell model and acts as mono- and combination therapy for β-thalassemia in primary hematopoietic stem cells. Haematologica 2018, 103, e419–e423. [Google Scholar] [CrossRef]
- Mussolino, C.; Alzubi, J.; Fine, E.J.; Morbitzer, R.; Cradick, T.J.; Lahaye, T.; Bao, G.; Cathomen, T. TALENs facilitate targeted genome editing in human cells with high specificity and low cytotoxicity. Nucleic Acids Res. 2014, 42, 6762–6773. [Google Scholar] [CrossRef]
- Loucari, C.C.; Patsali, P.; van Dijk, T.B.; Stephanou, C.; Papasavva, P.; Zanti, M.; Kurita, R.; Nakamura, Y.; Christou, S.; Sitarou, M.; et al. Rapid and sensitive assessment of globin chains for gene and cell therapy of hemoglobinopathies. Hum. Gene Ther. Methods 2018, 29, 60–74. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef]
- Christodoulou, I.; Patsali, P.; Stephanou, C.; Antoniou, M.; Kleanthous, M.; Lederer, C.W. Measurement of lentiviral vector titre and copy number by cross-species duplex quantitative PCR. Gene Ther. 2016, 23, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Funnell, A.P.; Vernimmen, D.; Lim, W.F.; Mak, K.S.; Wienert, B.; Martyn, G.E.; Artuz, C.M.; Burdach, J.; Quinlan, K.G.; Higgs, D.R.; et al. Differential regulation of the α-globin locus by Kruppel-like factor 3 in erythroid and non-erythroid cells. BMC Mol. Biol. 2014, 15, 8. [Google Scholar] [CrossRef] [PubMed]
- Kountouris, P.; Lederer, C.W.; Fanis, P.; Feleki, X.; Old, J.; Kleanthous, M. IthaGenes: An interactive database for haemoglobin variations and epidemiology. PLoS ONE 2014, 9, e103020. [Google Scholar] [CrossRef] [PubMed]
- Martyn, G.E.; Quinlan, K.G.R.; Crossley, M. The regulation of human globin promoters by CCAAT box elements and the recruitment of NF-Y. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef]
- Concordet, J.P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef]
- Doyle, E.L.; Booher, N.J.; Standage, D.S.; Voytas, D.F.; Brendel, V.P.; Vandyk, J.K.; Bogdanove, A.J. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: Tools for TAL effector design and target prediction. Nucleic Acids Res. 2012, 40, 117–122. [Google Scholar] [CrossRef]
- Kim, H.; Kim, J.-S.S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014, 15, 321–334. [Google Scholar] [CrossRef]
- Treisman, R.; Orkin, S.H.; Maniatis, T. Specific transcription and RNA splicing defects in five cloned beta-thalassaemia genes. Nature 1983, 302, 591–596. [Google Scholar] [CrossRef]
- Kazazian, H.H.; Orkin, S.H.; Antonarakis, S.E.; Sexton, J.P.; Boehm, C.D.; Goff, S.C.; Waber, P.G. Molecular characterization of seven beta-thalassemia mutations in Asian Indians. EMBO J. 1984, 3, 593–596. [Google Scholar] [CrossRef]
- Tamagnini, G.P.; Lopes, M.C.; Castanheira, M.E.; Wainscoat, J.S.; Wood, W.G. β+ Thalassaemia—Portuguese type: Clinical, haematological and molecular studies of a newly defined form of β thalassaemia. Br. J. Haematol. 2008, 54, 189–200. [Google Scholar] [CrossRef]
- Sahoo, S.S.; Biswal, S.; Dixit, M. Distinctive mutation spectrum of the HBB gene in an urban eastern Indian population. Hemoglobin 2014, 38, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Metherall, J.E.; Collins, F.S.; Pan, J.; Weissman, S.M.; Forget, B.G. Beta zero thalassemia caused by a base substitution that creates an alternative splice acceptor site in an intron. EMBO J. 1986, 5, 2551–2557. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.H.; Liang, S.; Su, C.; Nechtman, J.F.; Stoming, T.A. A novel beta-thalassemia mutation [IVS-II-5 (G→C)] in a Chinese family from Guangxi Province, P.R. China. Hemoglobin 1993, 17, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.H.; Liang, S. The β+-thalassemia mutation [IVS-II-5 (G→C)] creates an alternative splicing site in the second intervening sequence. Hemoglobin 1999, 23, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, A.; Gorakshakar, A.; Surve, R.; Sawant, P.; Phanasgaonkar, S.; Nair, S.; Ghosh, K.; Colah, R.B. Hematological and molecular analysis of novel and rare beta-thalassemia mutations in the Indian population. Hemoglobin 2009, 33, 59–65. [Google Scholar] [CrossRef]
- Cheng, T.C.; Orkin, S.H.; Antonarakis, S.E.; Potter, M.J.; Sexton, J.P.; Markham, A.F.; Giardina, P.J.; Li, A.; Kazazian, H.H., Jr. beta-Thalassemia in Chinese: Use of in vivo RNA analysis and oligonucleotide hybridization in systematic characterization of molecular defects. Proc. Natl. Acad. Sci. USA 1984, 81, 2821–2825. [Google Scholar] [CrossRef] [Green Version]
- Dobkin, C.; Pergolizzi, R.G.; Bahre, P.; Bank, A. Abnormal splice in a mutant human beta-globin gene not at the site of a mutation. Proc. Natl. Acad. Sci. USA 1983, 80, 1184–1188. [Google Scholar] [CrossRef] [Green Version]
- Dobkin, C.; Bank, A. Reversibility of IVS 2 missplicing in a mutant human beta-globin gene. J. Biol. Chem. 1985, 260, 16332–16337. [Google Scholar]
- Agouti, I.; Bennani, M.; Ahmed, A.; Barakat, A.; Mohamed, K.; Badens, C. Thalassemia intermedia due to a novel mutation in the second intervening sequence of the beta-globin gene. Hemoglobin 2007, 31, 433–438. [Google Scholar] [CrossRef]
- Henderson, S.J.; Timbs, A.T.; McCarthy, J.; Gallienne, A.E.; Proven, M.; Rugless, M.J.; Lopez, H.; Eglinton, J.; Dziedzic, D.; Beardsall, M.; et al. Ten years of routine α-and β-globin gene sequencing in UK hemoglobinopathy referrals reveals 60 novel mutations. Hemoglobin 2016, 40, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Nuntakarn, L.; Fucharoen, S.; Fucharoen, G.; Sanchaisuriya, K.; Jetsrisuparb, A.; Wiangnon, S. Molecular, hematological and clinical aspects of thalassemia major and thalassemia intermedia associated with Hb E-beta-thalassemia in Northeast Thailand. Blood Cells Mol. Dis. 2009, 42, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Varawalla, N.Y.; Old, J.M.; Weatherall, D.J. Rare beta-thalassaemia mutations in Asian Indians. Br. J. Haematol. 1991, 79, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Patsali, P.; Mussolino, C.; Loucari, C.; Stephanou, C.; Antoniou, M.; Cathomen, T.; Lederer, C.W.; Kleanthous, M. 692. Genome editing for personalized gene therapy of IVSI-110 beta-thalassemia. Mol. Ther. 2016, 24, S274. [Google Scholar] [CrossRef]
- Santacroce, R.; Santoro, R.; Sessa, F.; Iannaccaro, P.; Sarno, M.; Longo, V.; Gallone, A.; Vecchione, G.; Muleo, G.; Margaglione, M. Screening of mutations of hemophilia A in 40 Italian patients: A novel G-to-A mutation in intron 10 of the F8 gene as a putative cause of mild hemophilia A in southern Italy. Blood Coagul. Fibrinolysis 2008, 19, 197–202. [Google Scholar] [CrossRef]
- Glaser, B.; Blech, I.; Krakinovsky, Y.; Ekstein, J.; Gillis, D.; Mazor-Aronovitch, K.; Landau, H.; Abeliovich, D. ABCC8 mutation allele frequency in the Ashkenazi Jewish population and risk of focal hyperinsulinemic hypoglycemia. Genet. Med. 2011, 13, 891–894. [Google Scholar] [CrossRef]
- Nestorowicz, A.; Wilson, B.A.; Schoor, K.P.; Inoue, H.; Glaser, B.; Landau, H.; Stanley, C.A.; Thornton, P.S.; Clement, J.P., IV; Bryan, J.; et al. Mutations in the sulfonylurea receptor gene are associated with familial hyperinsulinism in Ashkenazi Jews. Hum. Mol. Genet. 1996, 5, 1813–1822. [Google Scholar] [CrossRef]
- Garanto, A.; Duijkers, L.; Collin, R.W.J. Species-dependent splice recognition of a cryptic exon resulting from a recurrent intronic CEP290 mutation that causes congenital blindness. Int. J. Mol. Sci. 2015, 16, 5285–5298. [Google Scholar] [CrossRef] [Green Version]
- Perrault, I.; Delphin, N.; Hanein, S.; Gerber, S.; Dufier, J.L.; Roche, O.; Defoort-Dhellemmes, S.; Dollfus, H.; Fazzi, E.; Munnich, A.; et al. Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum. Mutat. 2007, 28, 416. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Jacobson, S.G.; Drack, A.V.; Ho, A.C.; Charng, J.; Garafalo, A.V.; Roman, A.J.; Sumaroka, A.; Han, I.C.; Hochstedler, M.D.; et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat. Med. 2019, 25, 225–228. [Google Scholar] [CrossRef]
- Bauwens, M.; de Zaeytijd, J.; Weisschuh, N.; Kohl, S.; Meire, F.; Dahan, K.; Depasse, F.; de Jaegere, S.; De Ravel, T.; de Rademaeker, M.; et al. An augmented abca4 screen targeting noncoding regions reveals a deep intronic founder variant in Belgian stargardt patients. Hum. Mutat. 2015, 36, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Garanto, A.; Duijkers, L.; Tomkiewicz, T.Z.; Collin, R.W.J. Antisense oligonucleotide screening to optimize the rescue of the splicing defect caused by the recurrent deep-intronic ABCA4 variant c.4539+2001G>A in stargardt disease. Genes 2019, 10, 452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominov, J.A.; Uyan, Ö.; McKenna-Yasek, D.; Nallamilli, B.R.R.; Kergourlay, V.; Bartoli, M.; Levy, N.; Hudson, J.; Evangelista, T.; Lochmuller, H.; et al. Correction of pseudoexon splicing caused by a novel intronic dysferlin mutation. Ann. Clin. Transl. Neurol. 2019, 6, 642–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, B.C.; Ki, C.S.; Kim, J.W.; Cho, A.; Kim, M.J.; Hwang, H.; Kim, K.J.; Hwang, Y.S.; Park, W.Y.; Lim, Y.J.; et al. Fukutin mutations in congenital muscular dystrophies with defective glycosylation of dystroglycan in Korea. Neuromuscul. Disord. 2010, 20, 524–530. [Google Scholar] [CrossRef]
- Oustric, V.; Manceau, H.; Ducamp, S.; Soaid, R.; Karim, Z.; Schmitt, C.; Mirmiran, A.; Peoc’H, K.; Grandchamp, B.; Beaumont, C.; et al. Antisense oligonucleotide-based therapy in human erythropoietic protoporphyria. Am. J. Hum. Genet. 2014, 94, 611–617. [Google Scholar] [CrossRef] [Green Version]
- Balwani, M.; Doheny, D.; Bishop, D.F.; Nazarenko, I.; Yasuda, M.; Dailey, H.A.; Anderson, K.E.; Bissell, D.M.; Bloomer, J.; Bonkovsky, H.L.; et al. Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and X-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-lin. Mol. Med. 2013, 19, 26–35. [Google Scholar] [CrossRef]
- Chillón, M.; Dörk, T.; Casals, T.; Giménez, J.; Fonknechten, N.; Will, K.; Ramos, D.; Nunes, V.; Estivill, X.; Chillon, M.; et al. A novel donor splice site in intron 11 of the CFTR gene, created by mutation 1811+1.6kbA→G, produces a new exon: High frequency in Spanish cystic fibrosis chromosomes and association with severe phenotype. Am. J. Hum. Genet. 1995, 56, 623–629. [Google Scholar]
- Sanz, D.J.; Hollywood, J.A.; Scallan, M.F.; Harrison, P.T. Cas9/gRNA targeted excision of cystic fibrosis-causing deep-intronic splicing mutations restores normal splicing of CFTR mRNA. PLoS ONE 2017, 12, e0184009. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.Y.; Chong, K.W.; Hsu, J.H.; Yu, H.C.; Shih, C.C.; Huang, C.H.; Lin, S.J.; Chen, C.H.; Chiang, C.C.; Ho, H.J.; et al. High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population. Circ. Cardiovasc. Genet. 2009, 2, 450–456. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.H.; Niu, D.M.; Lu, C.Y.; Lin, S.Y.; Liu, T.C.; Chang, J.G. Modulation the alternative splicing of GLA (IVS4+919G>A) in Fabry disease. PLoS ONE 2017, 12, e0175929. [Google Scholar] [CrossRef]
- Anczukow, O.; Buisson, M.; Leone, M.; Coutanson, C.; Lasset, C.; Calender, A.; Sinilnikova, O.M.; Mazoyer, S.; Anczuków, O.; Buisson, M.; et al. BRCA2 deep intronic mutation causing activation of a cryptic exon: Opening toward a new preventive therapeutic strategy. Clin. Cancer Res. 2012, 18, 4903–4909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, E.; Bladen, C.L.; Mayhew, A.; James, M.; Bettinson, K.; Moore, U.; Smith, F.E.; Rufibach, L.; Cnaan, A.; Goebel, D.X.B.; et al. The clinical outcome study for dysferlinopathy an international multicenter study. Neurol. Genet. 2016, 2, e89. [Google Scholar] [CrossRef] [PubMed]
- Van den Hurk, J.A.; van de Pol, D.J.; Wissinger, B.; van Driel, M.A.; Hoefsloot, L.H.; de Wijs, I.J.; van den Born, L.I.; Heckenlively, J.R.; Brunner, H.G.; Zrenner, E.; et al. Novel types of mutation in the choroideremia (CHM) gene: A full-length L1 insertion and an intronic mutation activating a cryptic exon. Hum. Genet. 2003, 113, 268–275. [Google Scholar] [CrossRef] [PubMed]
- El-Beshlawy, A.; Mostafa, A.; Youssry, I.; Gabr, H.; Mansour, I.M.; El-Tablawy, M.; Aziz, M.; Hussein, I.R. Correction of aberrant pre-mRNA splicing by antisense oligonucleotides in β-thalassemia Egyptian patients with IVSI-110 mutation. J. Pediatr. Hematol. Oncol. 2008, 30, 281–284. [Google Scholar] [CrossRef]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef]
- Buratti, E.; Chivers, M.; Hwang, G.; Vorechovsky, I. DBASS3 and DBASS5: Databases of aberrant 3′-and 5′-splice sites. Nucleic Acids Res. 2011, 39, D86–D91. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.M.; Musunuru, K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J. Clin. Investig. 2014, 124, 4154–4161. [Google Scholar] [CrossRef] [Green Version]
- De Conti, L.; Baralle, M.; Buratti, E. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip. Rev. RNA 2013, 4, 49–60. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [Green Version]
- Taggart, A.J.; DeSimone, A.M.; Shih, J.S.; Filloux, M.E.; Fairbrother, W.G. Large-scale mapping of branchpoints in human pre-mRNA transcripts in vivo. Nat. Struct. Mol. Biol. 2012, 19, 719–721. [Google Scholar] [CrossRef]
- Komor, A.C.; Badran, A.H.; Liu, D.R. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell 2017, 168, 20–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| IthaID1 | Common Name | HGVS2 Name | Type of Mutation | Region | Exon⇥3 | References |
|---|---|---|---|---|---|---|
| 107 | IVS I-5 G>A | HBB:c.92+5G>C | Activation of cSD4; partial SD5 LoF6 | SD-proximal | 5 nt7 | [31,32] |
| 111 | IVS I-6 T>C | HBB:c.92+6T>C | Activation of cSD; partial SD LoF | SD-proximal | 6 nt | [33] |
| 3445 | IVSI-13G | HBB:c.92+13 | cSD activated by IthaID 101-1038, 107, 111 | Intron | 13 nt | [31] |
| 113 | IVS I-110 G>A | HBB:c.93-21G>A | Confirmed target; GG>GA (aSA9) | aSA | 21 nt | [16,17] |
| 3008 | IVS I-115 A>T | HBB:c.93-16A>T | AT>TT (effect unclear) | Intron | 16 nt | [34] |
| 114 | IVS I-116 T>G | HBB:c.93-15T>G | TT>GT (potential aSD10) | aSD | 15 nt | [35] |
| 208 | IVS II-5 G>C | HBB:c.315+5G>C | Activation of cSD; partial LoF | Intron | 5 nt | [36,37] |
| 3446 | IVS II-579G | HBB:c.316-272 | cSA11 activated by IthaID 214 | Intron | 270 nt | [31] |
| 210 | IVS II-613 C>T | HBB:c.316-238C>T | Activation of cryptic splice site | Intron | 238 nt | [38] |
| 211 | IVS II-654 C>T | HBB:c.316-197C>T | Confirmed target; GC>GT (aSD) | Intron | 197 nt | [17,39] |
| 212 | IVS II-705 T>G | HBB:c.316-146T>G | Activation of cryptic splice site | Intron | 146 nt | [40,41] |
| 213 | IVS II-726 A>G | HBB:c.316-125A>G | Likely block of RNA processing | Intron | 125 nt | [42] |
| 214 | IVS II-745 C>G | HBB:c.316-106C>G | aSD, activating cSA IthaID 3446 | Intron | 106 nt | [31] |
| 215 | IVS II-761 A>G | HBB:c.316-90A>G | AT>GT (potential aSD) | Intron | 90 nt | ITHANET12 |
| 2183 | IVS II-781 C>G | HBB:c.316-70C>G | CT>GT (potential aSD) | Intron | 70 nt | [43] |
| 216 | IVS II-815 C>T | HBB:c.316-36C>T | CT>GT (potential aSD) | Intron | 36 nt | [44] |
| 217 | IVS II-837 T>G | HBB:c.316-14T>G | AT>AG (potential aSA) | Intron | 14 nt | [45] |
| Primarily Affected | Exemplary | Exemplary Mutations | References | |||
|---|---|---|---|---|---|---|
| Organ System | Disorders | Gene | dbSNP ID | Effect1 | Frequency2 | |
| Circulatory | Poikilocytic anemia | SPTA1 | rs757147440 | aSA | 25%3 | [47] |
| Endocrine | Hyperinsulinemic hypoglycemia | ABCC8 | rs151344623 | aSA | 68.8%4 | [48,49] |
| Nervous | Leber congenital amaurosis | CEP290 | rs281865192 | cSD activation | 43%5 | [50,51,52] |
| Sensory | Stargardt disease | ABCA4 | rs1457937638 | cSD activation | 7.5%6 | [53,54] |
| Muscular | Miyoshi myopathy | DYSF | rs1285082850 | cSD activation | 17 families7 | [55] |
| Congenital muscular dystrophy | FKTN | rs1554754182 | cSD activation | 20.8%8 | [56] | |
| Integumentary | Erythropoietic protoporphyria | FECH | rs2272783 | cSA activation | 42.6%9 | [57,58] |
| Respiratory | Cystic fibrosis | CFTR | rs397508266 | aSD | 2.0%10 | [59,60] |
| Multisystemic | Fabry disease | GLA | rs199473684 | cSD activation | 41.1%11 | [61,62] |
| Cancer | Breast cancer | BRCA2 | rs191253965 | cSD activation | 0.2%12 | [63] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patsali, P.; Mussolino, C.; Ladas, P.; Floga, A.; Kolnagou, A.; Christou, S.; Sitarou, M.; Antoniou, M.N.; Cathomen, T.; Lederer, C.W.; et al. The Scope for Thalassemia Gene Therapy by Disruption of Aberrant Regulatory Elements. J. Clin. Med. 2019, 8, 1959. https://doi.org/10.3390/jcm8111959
Patsali P, Mussolino C, Ladas P, Floga A, Kolnagou A, Christou S, Sitarou M, Antoniou MN, Cathomen T, Lederer CW, et al. The Scope for Thalassemia Gene Therapy by Disruption of Aberrant Regulatory Elements. Journal of Clinical Medicine. 2019; 8(11):1959. https://doi.org/10.3390/jcm8111959
Chicago/Turabian StylePatsali, Petros, Claudio Mussolino, Petros Ladas, Argyro Floga, Annita Kolnagou, Soteroula Christou, Maria Sitarou, Michael N. Antoniou, Toni Cathomen, Carsten Werner Lederer, and et al. 2019. "The Scope for Thalassemia Gene Therapy by Disruption of Aberrant Regulatory Elements" Journal of Clinical Medicine 8, no. 11: 1959. https://doi.org/10.3390/jcm8111959