Mitochondrial Structural Changes in the Pathogenesis of Diabetic Retinopathy

1
Department of Medicine, Boston University School of Medicine, Boston, MA 02118, USA
2
Department of Ophthalmology, Boston University School of Medicine, Boston, MA 02118, USA
*
Author to whom correspondence should be addressed.
J. Clin. Med. 2019, 8(9), 1363; https://doi.org/10.3390/jcm8091363
Submission received: 8 August 2019
/
Revised: 27 August 2019
/
Accepted: 29 August 2019
/
Published: 1 September 2019
(This article belongs to the Special Issue Diabetic Retinopathy: Biomolecules and Pathophysiology)
Abstract
:At the core of proper mitochondrial functionality is the maintenance of its structure and morphology. Physical changes in mitochondrial structure alter metabolic pathways inside mitochondria, affect mitochondrial turnover, disturb mitochondrial dynamics, and promote mitochondrial fragmentation, ultimately triggering apoptosis. In high glucose condition, increased mitochondrial fragmentation contributes to apoptotic death in retinal vascular and Müller cells. Although alterations in mitochondrial morphology have been detected in several diabetic tissues, it remains to be established in the vascular cells of the diabetic retina. From a mechanistic standpoint, our current work supports the notion that increased expression of fission genes and decreased expression of fusion genes are involved in promoting excessive mitochondrial fragmentation. While mechanistic insights are only beginning to reveal how high glucose alters mitochondrial morphology, the consequences are clearly seen as release of cytochrome c from fragmented mitochondria triggers apoptosis. Current findings raise the prospect of targeting excessive mitochondrial fragmentation as a potential therapeutic strategy for treatment of diabetic retinopathy. While biochemical and epigenetic changes have been reported to be associated with mitochondrial dysfunction, this review focuses on alterations in mitochondrial morphology, and their impact on mitochondrial function and pathogenesis of diabetic retinopathy.
1. Introduction
Time-lapse videography of live cells has documented spectacular mitochondrial movement within a cell where each mitochondrion movement occurs in the direction of its longitudinal axis. The speed at which this movement takes place can vary substantially from 2 to 30 µm/min along the direction of its long axis. Long gone are the days when mitochondria were believed to be static organelles. In fact, movement among mitochondria facilitates connectivity between mitochondria resulting in a dynamic mitochondrial network. Subtle changes in mitochondrial shape seen as local narrowing/contractions and thickening/relaxations reflect a “worm-like swimming” movement coupled with a remarkable phenomenon of fission and fusion, orchestrating the dynamic nature of the mitochondrial network. These changes in mitochondrial structure are important to the normal function of cells. Indeed, mitochondrial shape change can rapidly flip from facilitating normal cell function to promoting apoptotic cell death. The ability of high glucose (HG) condition to promote mitochondrial fragmentation has captured the interest of several labs including ours. Studies from our lab have shown that disturbances to the intrinsic dynamic changes in mitochondrial network can negatively impact cell survival. In HG condition, mitochondrial fragmentation led to compromised metabolic changes, ultimately promoting apoptosis [1,2,3].
2. Consequences of Mitochondrial Dysfunction in Microangiopathy
To maintain proper functionality, the mitochondrial network undergoes constant dynamic reorganization through fission and fusion. In diabetes, mitochondrial dysfunction is attributable, at least in part, to altered mitochondrial morphology, specifically mitochondrial fragmentation. Changes in mitochondrial dynamics have been implicated in diabetic retinopathy [1,3], nephropathy [4,5], and neuropathy [6]. Table 1 shows a list of mitochondrial morphology changes in various tissues affected by diabetes [1,2,3,7,8,9,10,11,12,13,14,15,16,17,18,19,20].
Although evidence suggests that HG-induced changes in mitochondrial structure contribute to cellular dysfunction, the exact molecular mechanism(s) underlying these changes are still under intense investigation. Altered mitochondrial dynamics negatively impact mitochondrial respiration, membrane potential, induce cytochrome c release, and promote apoptosis [24,25,26,27]. Taken together, current findings indicate that HG-induced mitochondrial morphological changes play a key role in the development of retinal lesions associated with diabetic retinopathy (Figure 1), as well as diabetic microangiopathy in general.
3. Changes in Mitochondrial Morphology in Diabetic Retinopathy
Mitochondrial morphological changes have been associated with alterations in normal cellular homeostasis such as energy production [28], calcium production, mitochondrial DNA distribution, apoptosis, and mitophagy [29]. The fine balance between fission and fusion dictates normal cellular homeostasis. Studies have shown that ultrastructural changes in the shape and size of mitochondria is linked to the metabolic state of the cell. Studies examining the relationship between mitochondrial morphology and cellular metabolic states indicate mitochondrial size to be smaller in energetically inactive or quiescent cells than in energetically active cells [30,31].
Changes in mitochondrial morphology such as increased mitochondrial fragmentation have been documented in coronary endothelial cells in diabetic animal models [14]. Similarly, reduced mitochondrial size in skeletal muscle fiber [13,32], and mitochondrial swelling in hepatocytes [33] have been identified in patients with type 2 diabetes [13,32,33]. Alteration in mitochondrial morphology and its impact on retinal function has gained significant attention in studies of the pathogenesis of diabetic retinopathy; clinical trials are currently underway investigating the effects of diabetes and hyperglycemia on mitochondrial fragmentation.
4. Alterations in Mitochondrial Structure: Fission, Fusion, and Fragmentation
Mitochondria are dynamic organelles which undergo constant modifications in their shape and size. This process may vary among different cell types. The maintenance of mitochondrial structure involves a delicate balance between fission and fusion events [34]. The fission and fusion machinery operates continuously, resulting in upkeep of the dynamic mitochondrial network. Hence, fission and fusion cycles are necessary for mitochondrial growth, mitochondrial redistribution, and maintenance of mitochondrial length and shape [34]. In general, mitochondrial fusion positively impacts the maintenance of mitochondrial structure, whereas excessive mitochondrial fission is seen as deleterious in disease processes [35]. Dynamin family member proteins, which serve as GTPases, are primarily involved in the regulation of fission and fusion events [34]. Drp1 and Fis1 regulate mitochondrial fission, while fusion involving mitochondrial outer membranes is mediated by membrane-anchored dynamin family members Mfn1 and Mfn2, and fusion in the mitochondrial inner membrane is regulated by a single dynamin member Opa1 [35,36,37]. When mitochondria are partially damaged due to metabolic or oxidative stress, the mitochondria attempts a repair process wherein parts of the damaged mitochondria are combined by complementary fusion [35]. While mitochondrial fission is a necessary process for the maintenance of mitochondrial network, for mitochondrial biogenesis and the recycling of dysfunctional, damaged mitochondria, excessive fission can trigger mitochondrial fragmentation and apoptosis [35]. Both mitochondrial fission and fusion are indispensable events for proper cellular functions [38]. As such, disruption in these processes can have profound effects and contribute to diseases such as diabetes and diabetic retinopathy [6,12,18,21,22,23,39,40,41,42,43,44,45].
Increasing evidence points to abnormal mitochondrial structural changes as being one of the pathophysiological processes involving mitochondrial dysfunction in diabetic retinopathy. Current understanding suggests that alteration in fusion and fission gene expression may negatively impact mitochondrial structure. Taken together, these findings suggest that high glucose-induced mitochondrial structural damage involving excessive mitochondrial fragmentation plays a critical role in promoting apoptotic cell death associated with diabetic retinopathy.
5. HG-Induced Altered Mitochondrial Function Compromises Cellular Respiration
Oxygen consumption rate is a widely used index for studying mitochondrial metabolic activity [19,20,46]. In addition, extracellular acidification rate is useful in determining glycolytic flux [47]. Studies have shown that HG negatively impacts cellular respiration in retinal vascular cells. Reduced oxygen consumption is accompanied by altered mitochondrial membrane potential heterogeneity, cytochrome c release, and increased apoptosis [1]. Decreased oxygen consumption rate has been reported in isolated mitochondria of diabetic rat retinas concomitant with decreased CO2 production. In early stages of diabetes, retinal vascular cells exhibit an adaptive response whereas reduced cellular respiration occurs in late stages of diabetes [48].
Retinal endothelial cells grown in high glucose condition exhibit mitochondrial fragmentation concomitant with altered membrane potential heterogeneity [1]. Importantly, metabolic analysis revealed increased extracellular acidification, but reduced steady state/maximal oxygen consumption rate [1]. Increased extracellular acidification rate in these cells may be attributable to a compensatory mechanism counterbalancing high glucose-induced decreased mitochondrial oxygen consumption. Functionally, these respiratory changes are associated with increased cytochrome c release and apoptosis, suggesting that alteration in mitochondrial metabolism could be deleterious and promote cell loss seen in diabetic retinopathy.
In retinal pericytes, similar changes were observed under high glucose conditions. Excessive mitochondrial fragmentation and increased membrane potential heterogeneity were noted in retinal pericytes grown under high glucose medium [2]. While changes in membrane potential heterogeneity and steady state and maximum oxygen consumption rate were similar to those of retinal endothelial cells, extracellular acidification rate was decreased in pericytes under high glucose condition compared to those of endothelial cells [2]. This discrepancy may be due to the inability to compensate for high glucose-induced reduction in mitochondrial oxygen consumption in retinal pericytes. The observed difference in metabolic activity between endothelial cells and pericytes may be explained by differential transport of glucose between the two cell types. Specifically, GLUT1 activity was found to be reduced in pericytes, but not in endothelial cells under high glucose conditions [49]. Therefore, there may be a disparity in extracellular acidification and possibly even glycolytic levels in retinal pericytes and endothelial cells in response to high glucose. As expected, these metabolic changes in the mitochondria induced by high glucose led to increased apoptotic cell death in retinal pericytes. Further, this suggests that both retinal endothelial cells and pericytes may be susceptible to mitochondrial metabolic dysfunction induced by high glucose.
While mitochondrial dysfunction has been documented in retinal vascular cells, limited information is available with respect to retinal glial cells under high glucose conditions. Our recent study elucidated that retinal Müller cells grown in high glucose conditions also exhibit mitochondrial fragmentation with concomitant increase in membrane potential heterogeneity [3]. Comparable to retinal pericytes, retinal Müller cells showed decreased oxygen consumption rate. Though unlike retinal endothelial cells, retinal Müller cells showed decreased extracellular acidification rate, concomitant with cytochrome c release and apoptosis [3]. Taken together, these findings provide clear evidence that high glucose contributes to mitochondrial dysfunction in retinal vascular and glial cells by compromising mitochondrial function and cellular metabolism, thereby promoting apoptosis associated with the development of acellular capillaries, pericyte loss, and neuronal injury in diabetic retinopathy.
6. Mitochondrial Dysfunction-Mediated Apoptosis in Diabetic Retinopathy
Apoptosis or programmed cell death was initially believed to be regulated by the nucleus until it was discovered that enucleated cells also undergo apoptosis, and that the intrinsic apoptotic process is regulated by mitochondria [50]. A study showed that Bcl-2 is localized in the inner mitochondrial membrane [51], where it can perform either anti- or pro-apoptotic actions, thus confirming mitochondria’s ability to regulate apoptosis. Furthermore, opening and closure of mitochondria membrane transition pores [52] and subsequent release of cytochrome c [53] is known to modulate the intrinsic apoptotic pathway. While apoptosis can be triggered through various mechanisms, cytochrome c release is a pivotal culminating “point of no return” for the commitment towards apoptosis. Studies have shown that cytochrome c release can develop as a result of altered mitochondrial dynamics [1], activation of Drp1, a fission protein [54], and mitochondrial fragmentation. Our studies with retinal vascular cells have shown that HG-induced mitochondrial fragmentation (Figure 2) concomitant with altered membrane potential heterogeneity, reduced oxygen consumption rate, increased extracellular acidification, and that cytochrome c release promotes apoptosis, suggesting that mitochondrial dysfunction involving structural changes can mediate the retinal vascular cell death seen in diabetic retinopathy [1,2].
7. Mitochondrial Cx43 (mtCx43) Abnormalities and Mitochondrial Fragmentation
Presence of Cx43 in the mitochondria is a relatively recent discovery and has gained significant attention [55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71]. A study showed that inhibition of mtCx43 activity can promote cytochrome c, and thereby promote apoptosis [60]. In our lab using confocal microscopy, green fluorescent protein-tagged Cx43 was identified mostly in the inner mitochondrial membrane of retinal endothelial cells. Following Western blot analysis we also demonstrated that mtCx43 expression levels are significantly downregulated under high glucose condition, which in turn triggers mitochondrial fragmentation and subsequent release of cytochrome c in retinal endothelial cells [70]. These findings suggest that functional activity of mtCx43 channels is critical for maintaining mitochondrial morphology. Interestingly, inhibition of mtCx43 activity in isolated mitochondria promoted cytochrome c release [70], suggesting that mtCx43 changes may contribute to overall mitochondrial dysfunction evident in retinal vascular cells associated with diabetic retinopathy. While the exact role of mtCx43 in retinal vascular cells is not well understood, the scientific literature suggests that mtCx43 activity is mostly beneficial and that it confers cytoprotective effects in cardiac cells through diminishing reactive oxygen species (ROS) production in the cytosol and mitochondria, reducing mitochondrial calcium overload, mitochondrial membrane depolarization, and preventing the release of cytochrome c [65,67,68]. In addition, inhibition of mtCx43 has been shown to promote ROS release and induction of autophagy, indicating that proper mtCx43 activity is necessary to maintain mitochondrial integrity and metabolic activity of brown adipose tissue [61]. Collectively, these findings indicate that HG-induced downregulation of mtCx43 protein contributes to decreased Cx43 channel activity, altered mitochondrial morphology, and cytochrome c release. This could provide a framework for a novel mechanism underlying apoptotic retinal vascular cell death in diabetic retinopathy.
8. Strategies Inhibiting Mitochondrial Fragmentation and Dysfunction
It is well established that mitochondrial fragmentation disrupts cellular homeostasis through destabilization of mitochondrial membranes, ultimately leading to cytochrome c release, apoptotic cell death, and contributing to mitochondria-related disorders. Excessive mitochondrial fission and altered mitochondrial dynamics have been documented to play a role in several disease processes including cardiovascular disease, neurodegenerative disorders [72], Huntington’s disease, diabetes, mitochondrial diseases, Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, and amyotrophic lateral sclerosis [73]. Involvement of increased levels of fission gene expression, in particular, Drp1, appears to play a central role in these disease processes. While studies are underway examining molecules capable of inhibiting excessive mitochondrial fragmentation for treatment of patients with mitochondria-related disorders, strategies to combat mitochondrial fragmentation using drugs promoting mitochondrial fusion are also underway. Recent research has revealed that abnormal mitochondrial dynamics is an early event in diabetes. Although mitochondrial fusion and fission are necessary events for maintenance of functional mitochondria and the dynamic mitochondrial network, the balance between fission and fusion is a delicate process. Excessive fission and reduced fusion can lead to accumulation of mitochondrial fragments, leading to mitochondrial dysfunction and development of mitochondria-related disease processes. In diabetes, strategies are being tested for preventing mitochondrial fragmentation that includes administration of mitochondrial division inhibitor-1 (mdivi-1), dynasore, P110, and 15-oxospiramilactone [42]. A clinical trial investigating mitochondrial dynamics has begun, looking into effects of dietary stearic acid on mitochondrial fusion [74]. Another clinical trial currently ongoing entitled “Hyperglycemia and Mitochondrial Function in The Endothelium of Humans” (ClinicalTrials.gov Identifier #NCT02682342) is examining the effects of hyperglycemia on mitochondrial network, specifically fragmentation and fission in diabetic patients. The future prospect of targeting excessive mitochondrial fragmentation as a therapeutic strategy for diabetic retinopathy is promising.
9. Conclusions
Recent strategies for preventing excessive mitochondrial fragmentation in diseases have gained attention. It is evident that mitochondrial morphology changes play an important role in contributing to biochemical and molecular alterations underlying disease processes, including those of diabetic retinopathy. While mitochondrial fragmentation has been implicated in pathophysiological changes involving kidneys, liver, heart, pancreas, and skeletal muscles in diabetes, only limited information is available with respect to diabetic retinopathy. The consequences of mitochondrial fragmentation are significant, affecting various functions including cellular metabolism, cellular respiration, and triggering apoptotic death. Each of these cellular events is closely associated with the development and progression of retinal lesions in diabetic retinopathy. As such, an understanding of molecular mechanisms underlying mitochondrial structural changes is necessary. The challenge would be to identify novel targets for maintaining intact mitochondrial morphology in diabetic retinopathy.
Funding
This work was supported in part by a grant NIH R01 EY027082 (SR).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Trudeau, K.; Molina, A.J.; Guo, W.; Roy, S. High glucose disrupts mitochondrial morphology in retinal endothelial cells: Implications for diabetic retinopathy. Am. J. Pathol. 2010, 177, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Trudeau, K.; Molina, A.J.; Roy, S. High glucose induces mitochondrial morphology and metabolic changes in retinal pericytes. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8657–8664. [Google Scholar] [CrossRef] [PubMed]
- Tien, T.; Zhang, J.; Muto, T.; Kim, D.; Sarthy, V.P.; Roy, S. High Glucose Induces Mitochondrial Dysfunction in Retinal Muller Cells: Implications for Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2915–2921. [Google Scholar] [CrossRef] [PubMed]
- Ayanga, B.A.; Badal, S.S.; Wang, Y.; Galvan, D.L.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Dynamin-Related Protein 1 Deficiency Improves Mitochondrial Fitness and Protects against Progression of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2733–2747. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef]
- Edwards, J.L.; Quattrini, A.; Lentz, S.I.; Figueroa-Romero, C.; Cerri, F.; Backus, C.; Hong, Y.; Feldman, E.L. Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia 2010, 53, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xiang, H.; Chen, R.; Yang, J.; Yang, X.; Zhou, J.; Liu, H.; Zhao, S.; Xiao, J.; Chen, P.; et al. S1PR2 antagonist ameliorate high glucose-induced fission and dysfunction of mitochondria in HRGECs via regulating ROCK1. BMC Nephrol. 2019, 20, 135. [Google Scholar] [CrossRef]
- Choo, H.J.; Kim, J.H.; Kwon, O.B.; Lee, C.S.; Mun, J.Y.; Han, S.S.; Yoon, Y.S.; Yoon, G.; Choi, K.M.; Ko, Y.G. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia 2006, 49, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Coughlan, M.T.; Nguyen, T.V.; Penfold, S.A.; Higgins, G.C.; Thallas-Bonke, V.; Tan, S.M.; Van Bergen, N.J.; Sourris, K.C.; Harcourt, B.E.; Thorburn, D.R.; et al. Mapping time-course mitochondrial adaptations in the kidney in experimental diabetes. Clin. Sci. (Lond.) 2016, 130, 711–720. [Google Scholar] [CrossRef]
- Dlaskova, A.; Spacek, T.; Santorova, J.; Plecita-Hlavata, L.; Berkova, Z.; Saudek, F.; Lessard, M.; Bewersdorf, J.; Jezek, P. 4Pi microscopy reveals an impaired three-dimensional mitochondrial network of pancreatic islet beta-cells, an experimental model of type-2 diabetes. Biochim. Biophys. Acta 2010, 1797, 1327–1341. [Google Scholar] [CrossRef]
- Harano, Y.; DePalma, R.G.; Miller, M. Fatty acid oxidation, citric acid cycle activity, and morphology of mitochondria in diabetic rat liver. Proc. Soc. Exp. Biol. Med. 1969, 131, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Jheng, H.F.; Tsai, P.J.; Guo, S.M.; Kuo, L.H.; Chang, C.S.; Su, I.J.; Chang, C.R.; Tsai, Y.S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [PubMed]
- Makino, A.; Scott, B.T.; Dillmann, W.H. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia 2010, 53, 1783–1794. [Google Scholar] [CrossRef] [Green Version]
- Molina, A.J.; Wikstrom, J.D.; Stiles, L.; Las, G.; Mohamed, H.; Elorza, A.; Walzer, G.; Twig, G.; Katz, S.; Corkey, B.E.; et al. Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes 2009, 58, 2303–2315. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Tao, L.; Xiaohui, X.; Zelin, Z.; Jiangang, L.; Zhao, S.; Weikang, H.; Hongchao, X.; Qiujing, W.; Xin, L. Polydatin impairs mitochondria fitness and ameliorates podocyte injury by suppressing Drp1 expression. J. Cell Physiol. 2017, 232, 2776–2787. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, K.; Matsuura, T.; Narama, I. Histochemical and morphometrical analysis of skeletal muscle in spontaneous diabetic WBN/Kob rat. Acta Neuropathol. 2001, 102, 264–270. [Google Scholar]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Sheu, S.S.; Robotham, J.L.; Yoon, Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 2008, 79, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Devi, T.S.; Somayajulu, M.; Kowluru, R.A.; Singh, L.P. TXNIP regulates mitophagy in retinal Muller cells under high-glucose conditions: Implications for diabetic retinopathy. Cell Death Dis. 2017, 8, e2777. [Google Scholar] [CrossRef] [PubMed]
- Devi, T.S.; Yumnamcha, T.; Yao, F.; Somayajulu, M.; Kowluru, R.A.; Singh, L.P. TXNIP mediates high glucose-induced mitophagic flux and lysosome enlargement in human retinal pigment epithelial cells. Biol. Open 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, H.Y.; Kang, J.M.; Jun, H.H.; Kim, D.J.; Park, S.H.; Sung, M.J.; Heo, J.H.; Yang, D.H.; Lee, S.H.; Lee, S.Y. Chloroquine and amodiaquine enhance AMPK phosphorylation and improve mitochondrial fragmentation in diabetic tubulopathy. Sci. Rep. 2018, 8, 8774. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef]
- Parone, P.A.; Da Cruz, S.; Tondera, D.; Mattenberger, Y.; James, D.I.; Maechler, P.; Barja, F.; Martinou, J.C. Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS ONE 2008, 3, e3257. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.C.; Coughlan, M.T. Mitochondrial dysfunction and mitophagy: The beginning and end to diabetic nephropathy? Br. J. Pharmacol. 2014, 171, 1917–1942. [Google Scholar] [CrossRef] [PubMed]
- Soleimanpour, S.A.; Ferrari, A.M.; Raum, J.C.; Groff, D.N.; Yang, J.; Kaufman, B.A.; Stoffers, D.A. Diabetes Susceptibility Genes Pdx1 and Clec16a Function in a Pathway Regulating Mitophagy in beta-Cells. Diabetes 2015, 64, 3475–3484. [Google Scholar] [CrossRef] [PubMed]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef] [PubMed]
- Mitra, K.; Wunder, C.; Roysam, B.; Lin, G.; Lippincott-Schwartz, J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc. Natl. Acad. Sci. USA 2009, 106, 11960–11965. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, T.; Yamasaki, Y. Ultra-high-resolution scanning electron microscopy of mitochondria and sarcoplasmic reticulum arrangement in human red, white, and intermediate muscle fibers. Anat. Rec. 1997, 248, 214–223. [Google Scholar] [CrossRef]
- Vanhorebeek, I.; De Vos, R.; Mesotten, D.; Wouters, P.J.; De Wolf-Peeters, C.; Van den Berghe, G. Protection of hepatocyte mitochondrial ultrastructure and function by strict blood glucose control with insulin in critically ill patients. Lancet 2005, 365, 53–59. [Google Scholar] [CrossRef]
- Van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Elgass, K.; Pakay, J.; Ryan, M.T.; Palmer, C.S. Recent advances into the understanding of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 150–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppins, S.; Lackner, L.; Nunnari, J. The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 2007, 76, 751–780. [Google Scholar] [CrossRef]
- Lee, S.E.; Ma, W.; Rattigan, E.M.; Aleshin, A.; Chen, L.; Johnson, L.L.; D’Agati, V.D.; Schmidt, A.M.; Barile, G.R. Ultrastructural features of retinal capillary basement membrane thickening in diabetic swine. Ultrastruct. Pathol. 2010, 34, 35–41. [Google Scholar] [CrossRef]
- Chang, J.Y.; Yu, F.; Shi, L.; Ko, M.L.; Ko, G.Y. Melatonin Affects Mitochondrial Fission/Fusion Dynamics in the Diabetic Retina. J. Diabetes Res. 2019, 2019, 8463125. [Google Scholar] [CrossRef]
- Civitarese, A.E.; Ravussin, E. Mitochondrial energetics and insulin resistance. Endocrinology 2008, 149, 950–954. [Google Scholar] [CrossRef]
- Duraisamy, A.J.; Mohammad, G.; Kowluru, R.A. Mitochondrial fusion and maintenance of mitochondrial homeostasis in diabetic retinopathy. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Rovira-Llopis, S.; Banuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Caino, M.C. Mitochondrial Dynamics in Type 2 Diabetes and Cancer. Front. Endocrinol. (Lausanne) 2018, 9, 211. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, L.; Li, X.; Jiang, Z.; Sun, L.; Zhao, G.; Zhou, G.; Zhang, H.; Shang, J.; Wang, T. Mitochondrial fusion/fission process involved in the improvement of catalpol on high glucose-induced hepatic mitochondrial dysfunction. Acta Biochim. Biophys. Sin. (Shanghai) 2015, 47, 730–740. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Q.; Kowluru, R.A. Diabetic retinopathy and damage to mitochondrial structure and transport machinery. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8739–8746. [Google Scholar] [CrossRef]
- Paltauf-Doburzynska, J.; Malli, R.; Graier, W.F. Hyperglycemic conditions affect shape and Ca2+ homeostasis of mitochondria in endothelial cells. J. Cardiovasc. Pharmacol. 2004, 44, 423–436. [Google Scholar] [CrossRef]
- TeSlaa, T.; Teitell, M.A. Techniques to monitor glycolysis. Methods Enzymol. 2014, 542, 91–114. [Google Scholar] [CrossRef]
- Osorio-Paz, I.; Uribe-Carvajal, S.; Salceda, R. In the Early Stages of Diabetes, Rat Retinal Mitochondria Undergo Mild Uncoupling due to UCP2 Activity. PLoS ONE 2015, 10, e0122727. [Google Scholar] [CrossRef]
- Mandarino, L.J.; Finlayson, J.; Hassell, J.R. High glucose downregulates glucose transport activity in retinal capillary pericytes but not endothelial cells. Investig. Ophthalmol. Vis. Sci. 1994, 35, 964–972. [Google Scholar]
- Jacobson, M.D.; Burne, J.F.; Raff, M.C. Programmed cell death and Bcl-2 protection in the absence of a nucleus. EMBO J. 1994, 13, 1899–1910. [Google Scholar] [CrossRef]
- Hockenbery, D.; Nunez, G.; Milliman, C.; Schreiber, R.D.; Korsmeyer, S.J. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 1990, 348, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Petit, P.; Zamzami, N.; Vayssiere, J.L.; Mignotte, B. The biochemistry of programmed cell death. FASEB J. 1995, 9, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef]
- Estaquier, J.; Arnoult, D. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 2007, 14, 1086–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boengler, K.; Dodoni, G.; Rodriguez-Sinovas, A.; Cabestrero, A.; Ruiz-Meana, M.; Gres, P.; Konietzka, I.; Lopez-Iglesias, C.; Garcia-Dorado, D.; Di Lisa, F.; et al. Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc. Res. 2005, 67, 234–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boengler, K.; Schulz, R. Connexin 43 and Mitochondria in Cardiovascular Health and Disease. Adv. Exp. Med. Biol. 2017, 982, 227–246. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Stahlhofen, S.; van de Sand, A.; Gres, P.; Ruiz-Meana, M.; Garcia-Dorado, D.; Heusch, G.; Schulz, R. Presence of connexin 43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res. Cardiol. 2009, 104, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Gadicherla, A.K.; Wang, N.; Bulic, M.; Agullo-Pascual, E.; Lissoni, A.; De Smet, M.; Delmar, M.; Bultynck, G.; Krysko, D.V.; Camara, A.; et al. Mitochondrial Cx43 hemichannels contribute to mitochondrial calcium entry and cell death in the heart. Basic Res. Cardiol. 2017, 112, 27. [Google Scholar] [CrossRef]
- Garcia-Dorado, D.; Ruiz-Meana, M.; Rodriguez-Sinovas, A. Connexin 43 phosphorylation in subsarcolemmal mitochondria: A general cardioprotective signal targeted by fibroblast growth factor-2? Cardiovasc. Res. 2014, 103, 1–2. [Google Scholar] [CrossRef]
- Goubaeva, F.; Mikami, M.; Giardina, S.; Ding, B.; Abe, J.; Yang, J. Cardiac mitochondrial connexin 43 regulates apoptosis. Biochem. Biophys. Res. Commun. 2007, 352, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.N.; Kwon, H.J.; Im, S.W.; Son, Y.H.; Akindehin, S.; Jung, Y.S.; Lee, S.J.; Rhyu, I.J.; Kim, I.Y.; Seong, J.K.; et al. Connexin 43 is required for the maintenance of mitochondrial integrity in brown adipose tissue. Sci. Rep. 2017, 7, 7159. [Google Scholar] [CrossRef] [PubMed]
- Kirca, M.; Kleinbongard, P.; Soetkamp, D.; Heger, J.; Csonka, C.; Ferdinandy, P.; Schulz, R. Interaction between connexin 43 and nitric oxide synthase in mice heart mitochondria. J. Cell Mol. Med. 2015, 19, 815–825. [Google Scholar] [CrossRef]
- Lu, G.; Haider, H.; Porollo, A.; Ashraf, M. Mitochondria-specific transgenic overexpression of connexin-43 simulates preconditioning-induced cytoprotection of stem cells. Cardiovasc. Res. 2010, 88, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecoraro, M.; Pinto, A.; Popolo, A. Inhibition of Connexin 43 translocation on mitochondria accelerates CoCl2-induced apoptotic response in a chemical model of hypoxia. Toxicol. In Vitro 2018, 47, 120–128. [Google Scholar] [CrossRef]
- Pecoraro, M.; Sorrentino, R.; Franceschelli, S.; Del Pizzo, M.; Pinto, A.; Popolo, A. Doxorubicin-Mediated Cardiotoxicity: Role of Mitochondrial Connexin 43. Cardiovasc. Toxicol. 2015, 15, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Sinovas, A.; Boengler, K.; Cabestrero, A.; Gres, P.; Morente, M.; Ruiz-Meana, M.; Konietzka, I.; Miro, E.; Totzeck, A.; Heusch, G.; et al. Translocation of connexin 43 to the inner mitochondrial membrane of cardiomyocytes through the heat shock protein 90-dependent TOM pathway and its importance for cardioprotection. Circ. Res. 2006, 99, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Sinovas, A.; Ruiz-Meana, M.; Denuc, A.; Garcia-Dorado, D. Mitochondrial Cx43, an important component of cardiac preconditioning. Biochim. Biophys. Acta Biomembr. 2018, 1860, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Meana, M.; Rodriguez-Sinovas, A.; Cabestrero, A.; Boengler, K.; Heusch, G.; Garcia-Dorado, D. Mitochondrial connexin43 as a new player in the pathophysiology of myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2008, 77, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Srisakuldee, W.; Makazan, Z.; Nickel, B.E.; Zhang, F.; Thliveris, J.A.; Pasumarthi, K.B.; Kardami, E. The FGF-2-triggered protection of cardiac subsarcolemmal mitochondria from calcium overload is mitochondrial connexin 43-dependent. Cardiovasc. Res. 2014, 103, 72–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trudeau, K.; Muto, T.; Roy, S. Downregulation of mitochondrial connexin 43 by high glucose triggers mitochondrial shape change and cytochrome C release in retinal endothelial cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6675–6681. [Google Scholar] [CrossRef] [PubMed]
- Wasilewski, M.; Scorrano, L. The changing shape of mitochondrial apoptosis. Trends Endocrinol. Met. 2009, 20, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Sharma, S. Dynamin-related protein-1 as potential therapeutic target in various diseases. Inflammopharmacology 2017, 25, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Inhibitors of mitochondrial fission as a therapeutic strategy for diseases with oxidative stress and mitochondrial dysfunction. J. Alzheimers Dis. 2014, 40, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Senyilmaz-Tiebe, D.; Pfaff, D.H.; Virtue, S.; Schwarz, K.V.; Fleming, T.; Altamura, S.; Muckenthaler, M.U.; Okun, J.G.; Vidal-Puig, A.; Nawroth, P.; et al. Dietary stearic acid regulates mitochondria in vivo in humans. Nat. Commun. 2018, 9, 3129. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Changes in mitochondrial morphology and its implications in diabetic retinopathy. Diagram illustrates the effects of diabetes/hyperglycemia on mitochondrial morphology changes leading to mitochondrial dysfunction, contributing to the development and progression of diabetic retinopathy. Dashed arrows suggest possible mechanisms that are still under investigation.
Figure 1.
Changes in mitochondrial morphology and its implications in diabetic retinopathy. Diagram illustrates the effects of diabetes/hyperglycemia on mitochondrial morphology changes leading to mitochondrial dysfunction, contributing to the development and progression of diabetic retinopathy. Dashed arrows suggest possible mechanisms that are still under investigation.

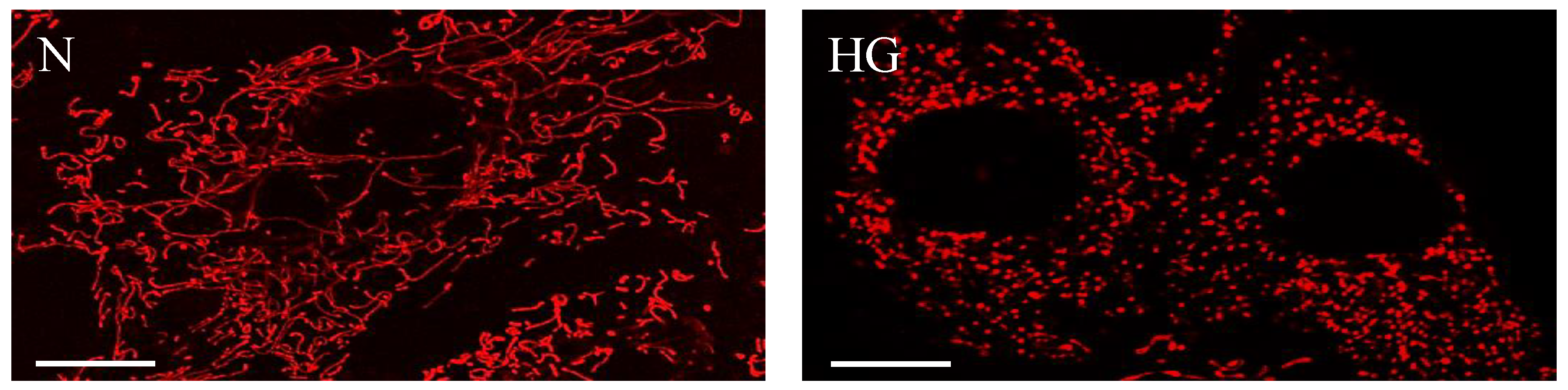
Figure 2.
High glucose compromises mitochondrial morphology and promotes mitochondrial fragmentation. Representative confocal images of RRECs grown in normal (N) medium (left) stained with mitotracker red show long, tubular networks of mitochondria. In parallel, cells grown in high glucose (HG) medium (right) exhibit significant mitochondrial fragmentation. Scale bars: 10 µm.
Figure 2.
High glucose compromises mitochondrial morphology and promotes mitochondrial fragmentation. Representative confocal images of RRECs grown in normal (N) medium (left) stained with mitotracker red show long, tubular networks of mitochondria. In parallel, cells grown in high glucose (HG) medium (right) exhibit significant mitochondrial fragmentation. Scale bars: 10 µm.


{kind=link}
{kind=link}
Table 1.
Mitochondrial morphology changes in diabetic tissues.
| Tissue Type | Cell Type | Species | Changes in Mitochondrial Morphology | References |
|---|---|---|---|---|
| Retina | Retinal endothelial cells | Rat | Fragmentation | [1] |
| Retinal pericytes | Bovine | Fragmentation | [2] | |
| Retinal Müller cells | Rat | Fragmentation | [3] | |
| Retinal Müller cells | Human | Fragmentation | [21] | |
| Retinal pigmented epithelial cells | Human | Fragmentation | [22] | |
| Kidney | Renal glomerular cells | Human | Fragmentation | [7] |
| Proximal tubule epithelial cells | Rat | Fragmentation | [9] | |
| Proximal tubule epithelial cells | Human | Fragmentation | [23] | |
| Podocytes | Mouse | Fragmentation | [16] | |
| Liver | Hepatocytes | Rat | Increased mitochondria size and density | [11] |
| Epithelial cells | Rat | Fragmentation | [19,20] | |
| Heart | Coronary endothelial cells | Mouse | Fragmentation | [14] |
| Myoblasts | Rat | Fragmentation | [19] | |
| Ventricular myocytes | Rat | Fragmentation | [20] | |
| Aortic endothelial cells | Bovine | Fragmentation | [20] | |
| Aortic smooth muscle cells | Mouse | Fragmentation | [20] | |
| Aortic endothelial cells | Human | Fragmentation | [18] | |
| Venous endothelial cells | Human | Fragmentation | [18] | |
| Pancreas | β-islet cells | Rat | Fragmentation | [10] |
| β-islet cells | Mouse | Fragmentation | [15] | |
| Fat | Adipocytes | Mouse | Decreased mitochondria size | [8] |
| Skeletal Muscle | Myoblast | Mouse | Fragmentation | [12] |
| Skeletal muscle cells | Human | Decreased mitochondria size and larger vacuoles | [13] | |
| Skeletal muscle cells | Mouse | Swelling and lysis of mitochondrial cristae | [17] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Roy, S.; Kim, D.; Sankaramoorthy, A. Mitochondrial Structural Changes in the Pathogenesis of Diabetic Retinopathy. J. Clin. Med. 2019, 8, 1363. https://doi.org/10.3390/jcm8091363
AMA Style
Roy S, Kim D, Sankaramoorthy A. Mitochondrial Structural Changes in the Pathogenesis of Diabetic Retinopathy. Journal of Clinical Medicine. 2019; 8(9):1363. https://doi.org/10.3390/jcm8091363
Chicago/Turabian StyleRoy, Sayon, Dongjoon Kim, and Aravind Sankaramoorthy. 2019. "Mitochondrial Structural Changes in the Pathogenesis of Diabetic Retinopathy" Journal of Clinical Medicine 8, no. 9: 1363. https://doi.org/10.3390/jcm8091363
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.