
Pathways of Angiogenic and Inflammatory Cytokines in Multiple Myeloma: Role in Plasma Cell Clonal Expansion and Drug Resistance
 , ,
, ,  , and
, and
Abstract
:1. Introduction
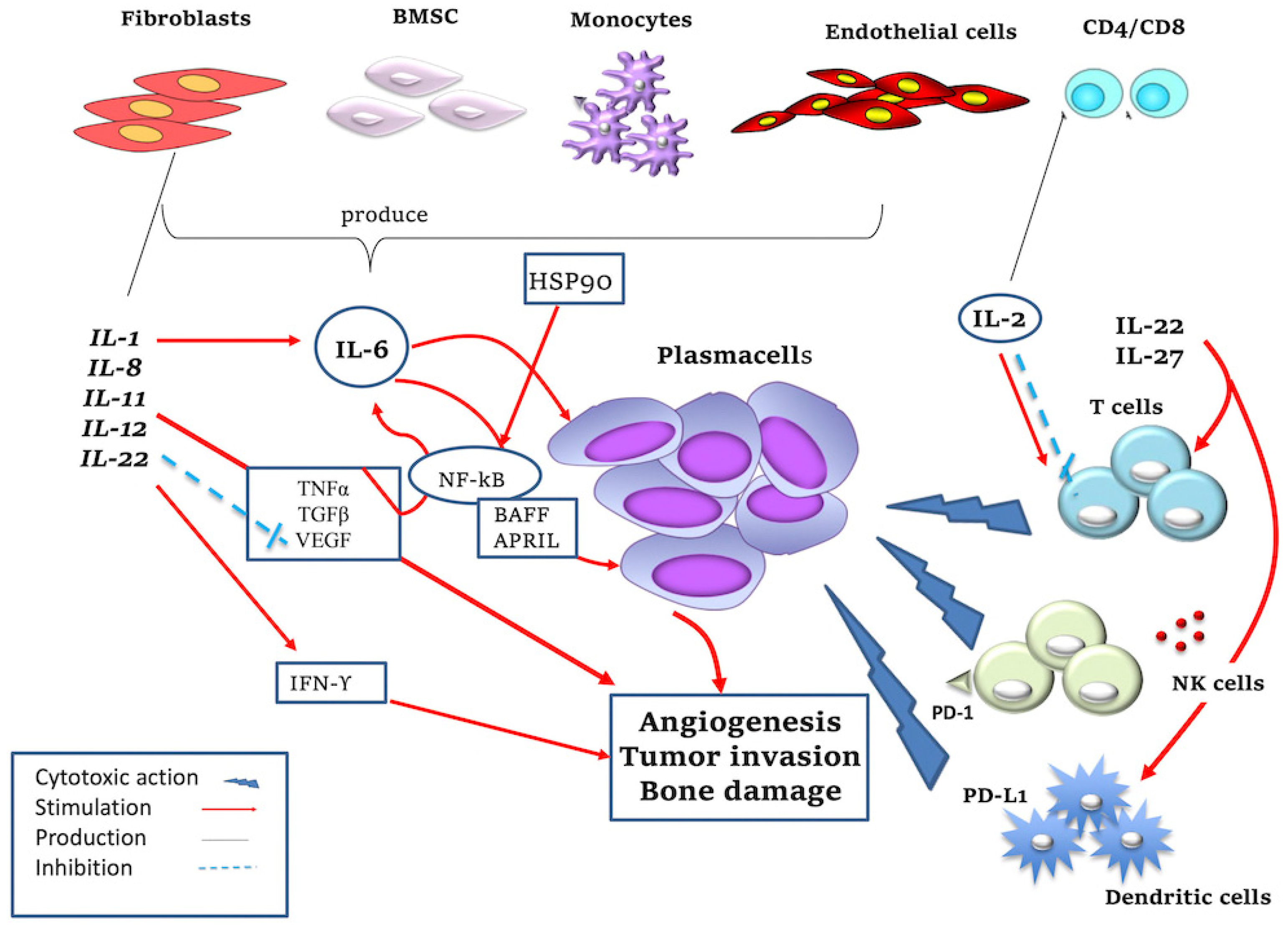
2. Pro- and Anti-Inflammatory Cytokines Induce Pro- and Anti-Tumor Effects
3. Angiogenic Cytokines and PCs Clonal Expansion
3.1. Vascular Endothelial Growth Factor A (VEGF-A)
3.2. Tumour Necrosis Factor α (TNFα)
3.3. Insulin-like Growth Factor-1 (IGF-1)
3.4. Matrix Metalloproteinases (MMP-2, MMP-9, uPA)
3.5. Fibroblast Growth Factor-2 (FGF-2)
3.6. Angiopoietin-1 and -2 (Ang-1 and -2)
3.7. Platelet-Derived Growth Factor-BB (PDGF-BB)
3.8. Hepatocyte Growth Factor (HGF)
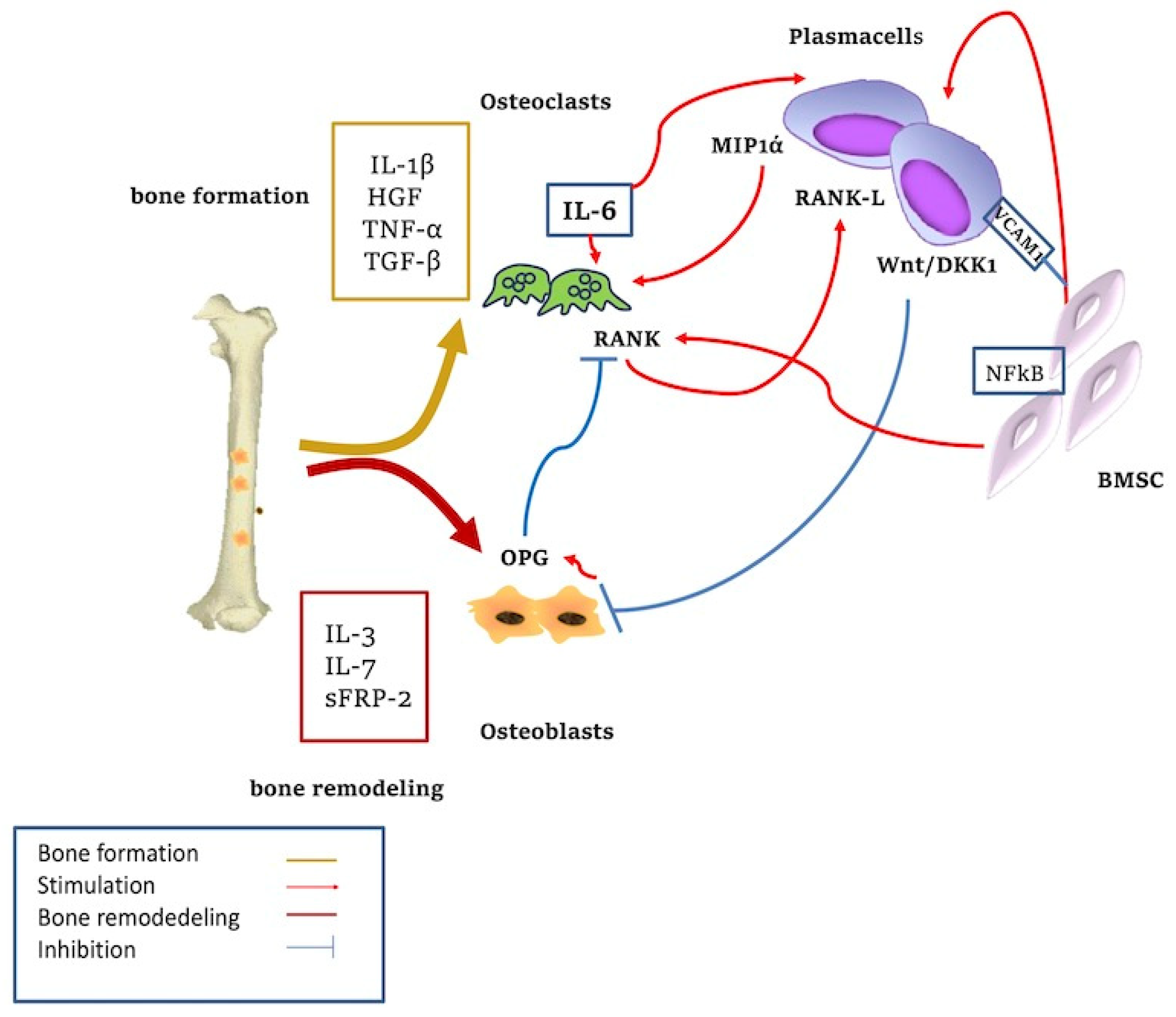
4. Cytokines and Bone Resorption
5. Roles of Cytokines in Drug Resistance in Multiple Myeloma
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| IMIDS | immunomodulatory agents |
| Anti-BCMA | anti-B cell maturation antigen |
| CRS | cytokine release syndrome |
| CRBN/DDB1 | complex-cereblon/DNA damage-binding protein1 |
| Treg | regulatory T cells |
References
- Akthar, S.; Tayyiba, A.; Faiyaz, A.; Khan, O.S.; Shadab Raza, S.; Kulinski, M.; El Omri, H.; Bhat, A.A.; Uddin, S. Cytokine-Mediated Dysregulation of Signaling Pathways in the Pathogenesis of Multiple Myeloma. Int. J. Mol. Sci. 2020, 21, 5002. [Google Scholar]
- Ria, R.; Reale, A.; Mangialardi, G.; Dammacco, F.; Ribatti, D.; Vacca, A. The Bone Marrow Microenvironment in Multiple Myeloma: Cellular and Molecular Basis of Disease Progression. In Cell Respiration and Cell Survival: Processes, Types and Effects; Nova Science Publishers: Hauppauge, NY, USA, 2009; Chapter 3; ISBN 978-1-60876-462-4. [Google Scholar]
- Ribatti, D.; Moschetta, M.; Vacca, A. Microenvironment and multiple myeloma spread. Thromb. Res. 2014, 133 (Suppl. S2), S102–S106. [Google Scholar] [CrossRef]
- Reale, A.; Carmichael, I.; Xu, R.; Mithraprabhu, S.; Khong, T.; Chen, M.; Fang, H.; Savvidou, I.; Ramachandran, M.; Bingham, N.; et al. Human myeloma cell- and plasma-derived extracellular vesicles contribute to functional regulation of stromal cells. Proteomics 2021, 21, e200011. [Google Scholar] [CrossRef]
- Dehghanifard, A.; Kaviani, S.; Abroun, S.; Mehdizadeh, M.; Saiedi, S.; Maali, A.; Ghaffari, S.; Azad, M. Various Signaling Pathways in Multiple Myeloma Cells and Effects of Treatment on These Pathways. Clin. Lymphoma Myeloma Leuk. 2018, 18, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Vacca, A. New Insights in Anti-Angiogenesis in Multiple Myeloma. Int. J. Mol. Sci. 2018, 19, 2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ria, R.; Melaccio, A.; Racanelli, V.; Vacca, A. Anti-VEGF drugs in the treatment of multiple myeloma patients. J. Clin. Med. 2020, 9, 1765. [Google Scholar] [CrossRef]
- Musolino, C.; Allegra, A.; Innao, V.; Allegra, A.G.; Pioggia, G.; Gangemi, S. Inflammatory and Anti-Inflammatory Equilibrium, Proliferative and Antiproliferative Balance: The Role of Cytokines in Multiple Myeloma. Mediat. Inflamm. 2017, 24, 1852517. [Google Scholar] [CrossRef] [Green Version]
- Ria, R.; Todoerti, K.; Berardi, S.; Coluccia, A.M.L.; De Luisi, A.; Mattioli, M.; Ronchetti, D.; Morabito, F.; Guarini, A.; Petrucci, M.T.; et al. Gene expression profiling of bone marrow endothelial cells in patients with multiple myeloma. Clin. Cancer Res. 2009, 15, 5369–5378. [Google Scholar] [CrossRef] [Green Version]
- Solimando, A.G.; Da Vià, M.C.; Cicco, S.; Leone, P.; Di Lernia, G.; Giannico, D.; DeSantis, V.; Frassanito, M.A.; Morizio, A.; Delgado Tascon, J.; et al. High-Risk Multiple Myeloma: Integrated Clinical and Omics Approach Dissects the Neoplastic Clone and the Tumor Microenvironment. J. Clin. Med. 2019, 8, 977. [Google Scholar] [CrossRef] [Green Version]
- De Smedt, E.; Lui, H.; Maes, K.; De Veirman, K.; Menu, E.; Vanderkerken, K.; De Bruyne, E. The epigenomic in Multiple Myeloma: Impact in tumor cell plasticity and drug response. Front. Oncol. 2018, 11, 566. [Google Scholar] [CrossRef]
- Allegra, A.; Innao, V.; Allegra, A.G.; Pugliese, M.; Di Salvo, E.; Ventura-Spagnolo, E.; Musolino, C.; Gangemi, S. Lymphocyte Subsets and Inflammatory Cytokines of Monoclonal Gammopathy of Undetermined Significance and Multiple Myeloma. Int. J. Mol. Sci. 2019, 20, 2822. [Google Scholar] [CrossRef]
- Leone, P.; Solimando, A.G.; Malerba, E.; Fasano, R.; Buonavoglia, A.; Pappagallo, F.; De Re, V.; Argentiero, A.; Silvestris, N.; Vacca, A.; et al. Actors on the Scene: Immune Cells in the Myeloma Niche. Front. Oncol. 2020, 10, 599098. [Google Scholar] [CrossRef]
- Gabay, C.; Lamacchia, C.; Palmer, G. IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 2010, 6, 232–241. [Google Scholar] [CrossRef]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [Green Version]
- Lust, J.A.; QLacy, M.; Zeldenrust, S.R.; Dispenzieri, A.; Gertz, M.A.; Witzig, T.E.; Kumar, S.; Hayman, S.R.; Russell, S.J.; Buadi, F.K.; et al. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1β-induced Mediators of in flammation interleukin 6 production and the myeloma proliferative component. Mayo Clin. Proc. 2009, 84, 114–122. [Google Scholar] [CrossRef]
- Lichtenstein, A.; Berenson, J.; Norman, D.; Chang, M.P.; Carlile, A. Production of cytokines by bone marrow cells obtained from patients with multiple myeloma. Blood 1989, 74, 1266–1273. [Google Scholar] [CrossRef] [Green Version]
- Klein, B.; Lu, Z.Y.; Gaillard, J.P.; Harousseau, J.L.; Bataille, R. Inhibiting IL-6 in human multiple myeloma. Curr. Top. Microbiol. Immunol. 1992, 182, 237–244. [Google Scholar]
- Cozzolino, F.; Torcia, M.; Aldinucci, D.; Rubartelli, A.; Miliani, A.; Shaw, A.R.; Lansdorp, P.M.; Di Guglielmo, R. Production of interleukin-1 by bone marrow myeloma cells. Blood 1989, 74, 380–387. [Google Scholar] [CrossRef] [Green Version]
- Donovan, K.; Lacy, M.; Kline, M.; Ahmann, G.; Heimbach, J.; Kyle, R.; Lust, J. Contrast in cytokine expression between patients with monoclonal gammopathy of undetermined significance or multiple myeloma. Leukemia 1998, 12, 593–600. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, A.; Wu, T.-C.; Hung, C.-F. Interleukin 2-Based Fusion Proteins for the Treatment of Cancer. J. Immunol. Res. 2021, 2021, 11. [Google Scholar] [CrossRef]
- De la Rosa, M.; Rutz, S.; Dorninger, H.; Scheffold, A. Interleukin-2 is essential for CD4+ CD25+ regulatory T cell function. Eur. J. Immunol. 2004, 34, 2480–2488. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.M., Jr.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.; et al. The PD-1/ PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef]
- Sarkar, S.; Germeraad, W.T.V.; Rouschop, K.M.A.; Steeghs, E.M.P.; Van Gelder, M.; Bos, G.M.J.; Wieten, L. Hypoxia-induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE 2013, 8, e64835. [Google Scholar] [CrossRef] [PubMed]
- Toshio, H. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2020, 33, 127–148. [Google Scholar]
- Hideshima, T.; Podar, K.; Chauhan, D.; Anderson, K.C. Cytokines and signal transduction. Best Pract. Res. Clin. Haematol. 2005, 18, 509–524. [Google Scholar] [CrossRef]
- Frassanito, M.A.; Cusmai, A.; Dammacco, F. Deregulated cytokine network and defective Th1 immune response in multiple myeloma. Clin. Exp. Immunol. 2001, 125, 190–197. [Google Scholar] [CrossRef]
- Matthes, T.; Manfroi, B.; Huard, B. Revisiting IL-6 Antagonism in Multiple Myeloma. Crit. Rev. Oncol. Hematol. 2016, 105, 1–4. [Google Scholar] [CrossRef]
- Jing, P.; Sun, Y.; Zhang, N.; Li, J.; Ta, F.; Wei, W.; Yu, S.; Ai, L. Characteristics of BAFF and APRIL factor expression in multiple myeloma and clinical significance. Oncol. Lett. 2017, 14, 2657–2662. [Google Scholar]
- Markham, A.; Patel, T. Siltuximab: First global approval. Drugs 2014, 74, 1147–1152. [Google Scholar] [CrossRef]
- Fousek, F.; Horn, L.A.; Palena, C. Interleukin-8: A Chemokine at the Intersection of Cancer Plasticity, Angiogenesis, and Immune Suppression. Pharmacol. Ther. 2021, 219, 107692. [Google Scholar] [CrossRef]
- Herrero, A.B.; García-Gómez, A.; Garayoa, M.; Corchete, L.A.; Hernández, J.M.; Miguel, J.S.; Gutierrez, N.C. Effects of IL-8 Up-Regulation on Cell Survival and Osteoclastogenesis in Multiple Myeloma. Am. J. Clin. Pathol. 2016, 186, 2171–2182. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of Interleukin (IL)-6-type Cytokine Signalling and Its Regulation. Biochem. J. 2003, 374 Pt 1, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Hongting, L.J.; Ng, B.; Lim, W. Interleukin-11: A Potential Biomarker and Molecular Therapeutic Target in Non-Small Cell Lung Cancer. Cells 2022, 11, 2257. [Google Scholar]
- Cook, S.A.; Schafer, S. Hiding in Plain Sight: Interleukin-11 Emerges as a Master Regulator of Fibrosis, Tissue Integrity, and Stromal Inflammation. Annu. Rev. Med. 2020, 71, 263–276. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Liu, Y.; Liu, R.; Qi, J.; Hou, Y.; Chang, J.; Ren, L. Upregulation of IL-11, an IL-6 Family Cytokine, Promotes Tumor Progression and Correlates with Poor Prognosis in Non-Small Cell Lung Cancer. Cell. Physiol. Biochem. 2018, 45, 2213–2224. [Google Scholar] [CrossRef]
- Giuliani, N.; Ferretti, M.; Bolzoni, M.; Storti, P.; Lazzaretti, M.; Dalla Palma, B.; Bonomini, S.; Martella, E.; Agnelli, L.; Neri, A.; et al. Increased Osteocyte Death in Multiple Myeloma Patients: Role in Myeloma-Induced Osteoclast Formation. Leukemia 2012, 26, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- De Raeve, H.; Vanderkerken, K. Immunomodulatory Drugs as a Therapy for Multiple Myeloma. Curr. Pharm. Biotechnol. 2006, 7, 415–421. [Google Scholar] [CrossRef]
- Dias, S.; Boyd, R.; Balkwill, F. IL-12 regulates VEGF and MMPs in a murine breast cancer model. Int. J. Cancer 1998, 78, 361–365. [Google Scholar] [CrossRef]
- Mirlekar, B.; Pylayeva-Gupta, Y. IL-12 Family Cytokines in Cancer and Immunotherapy. Cancers 2021, 13, 167. [Google Scholar] [CrossRef]
- Trinchieri, G.; Pflanz, S.; Kastelein, R.A. The IL-12 family of heterodimeric cytokines: New players in the regulation of T cell responses. Immunity 2003, 19, 641–644. [Google Scholar] [CrossRef] [Green Version]
- Giuliani, N.; Airoldi, I. Novel Insights into the Role of Interleukin-27 and Interleukin-23 in Human Malignant and Normal Plasma Cells. Clin. Cancer Res. 2011, 17, 6963–6970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbotti, G.; Barisione, G.; Airoldi, I.; Mezzanzanica, D.; Bagnoli, M.; Ferrero, S.; Petretto, A.; Fabbi, M.; Ferrini, S. IL-27 induces the expression of IDO and PD-L1 in human cancer cells. Oncotarget 2015, 6, 43267–43280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dondero, A.; Casu, B.; Bellora, F.; Vacca, A.; De Luisi, A.; Frassanito, M.A.; Cantoni, C.; Gaggero, S.; Olive, D.; Moretta, A.; et al. NK Cells and Multiple Myeloma-Associated Endothelial Cells: Molecular Interactions and Influence of IL-27. Oncotarget 2017, 8, 35088–35102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsirakis, G.; Pappa, C.A.; Kolovou, A.; Kokonozaki, M.; Neonakis, I.; Alexandrakis, M.G. Clinical significance of interleukin-22 in multiple myeloma. Hematology 2015, 20, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Berardi, S.; Caivano, A.; Ria, R.; Nico, B.; Savino, R.; Terracciano, R.; De Tullio, G.; Ferrucci, A.; De Luisi, A.; Moschetta, M.; et al. Four proteins governing overangiogenic endothelial cell phenotype in patients with multiple myeloma are plausible therapeutic targets. Oncogene 2012, 31, 2258–2269. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, M.; Turkbey, B.; Tan, E.; Kemp, T.J.; Pinto, L.A.; Berg, A.R.; Korde, N.; Minter, A.R.; Weiss, B.M.; Mena, E.; et al. Bone marrow angiogenesis in myeloma and its precursor disease: A prospective clinical trial. Leukemia 2014, 28, 413–416. [Google Scholar] [CrossRef]
- Solimando, A.G.; Da Vià, M.C.; Leone, P.; Borrelli, P.; Croci, G.A.; Tabares, P.; Brandl, A.; Di Lernia, G.; Bianchi, F.P.; Tafuri, S.; et al. Halting the vicious cycle within the multiple myeloma ecosystem: Blocking JAM-A on bone marrow endothelial cells restores angiogenic homeostasis and suppresses tumor progression. Haematologica 2021, 106, 1943–1956. [Google Scholar] [CrossRef]
- Ryan, V.L.; Flinchum, R.; Brown, N.; Ramsey, J.; Riccitelli, S.; Heuck, C.; Barlogie, B.; Shaughnessy, J.D., Jr. Translating a gene expression signature for multiple myeloma prognosis into a robust high-throughput assay for clinical use. BMC Med. Genom. 2014, 7, 25. [Google Scholar]
- Zhang, J.; Sattler, M.; Tonon, G.; Grabher, C.; Lababidi, S.; Zimmerhackl, A.; Raab, M.S.; Vallet, S.; Zhou, Y.; Cartron, M.-A.; et al. Targeting Angiogenesis via a c-Myc/Hypoxia-Inducible Factor-1α–Dependent Pathway in Multiple Myeloma. Cancer 2009, 69, 5082–5090. [Google Scholar] [CrossRef] [Green Version]
- Katoh, O.; Tauchi, H.; Kawaishi, K.; Kimura, A.; Satow, Y. Expression of the vascular endothelial growth factor (VEGF) receptor gene, KDR, in hematopoietic cells and inhibitory effect of VEGF on apoptotic cell death caused by ionizing radiation. Cancer Res. 1995, 55, 5687–5692. [Google Scholar]
- Braun, T.; Carvalho, G.; Fabre, C.; Grosjean, J.; Fenaux, P.; Kroemer, G. Targeting NF-kappaB in hematologic malignancies. Cell Death Differ. 2006, 13, 748–758. [Google Scholar] [CrossRef] [Green Version]
- Podar, K.; Tai, Y.-T.; Davies, F.; Lentzsch, S.; Sattler, M.; Hideshima, T.; Lin, B.K.; Gupta, D.; Shima, Y.; Chauhan, D.; et al. Vascular endothelial growth factor triggers signaling cascades mediating multiple myeloma cell growth and migration. Blood 2001, 98, 428–435. [Google Scholar] [CrossRef] [Green Version]
- Saltarella, I.; Morabito, F.; Giuliani, N.; Terragna, C.; Omedè, P.; Palumbo, A.; Bringhen, S.; De Paoli, L.; Martino, E.; LaRocca, A.; et al. Prognostic or predictive value of circulating cytokines and angiogenic factors for initial treatment of multiple myeloma in the GIMEMA MM0305 randomized controlled trial. J. Hematol. Oncol. 2019, 12, 4. [Google Scholar] [CrossRef]
- Mehta, A.K.; Gracias, D.T.; Croft, M. TNF activity and T cells. Cytokine 2018, 101, 14–18. [Google Scholar] [CrossRef]
- Bladè, J.; Filella, X.; Montoto, S.; Bosch, F.; Rosiñol, L.; Coca, F.; Giné, E.; Nadal, E.; Aymerich, M.; Rozman, M.; et al. Clinical relevance of interleukin 6 and tumor necrosis factor-alpha serum levels in monoclonal gammopathy of undetermined significance. Blood 1997, 90, 351. [Google Scholar]
- Roy, P.; Sarkar, U.A.; Basak, S. TheNF-κB Activating Pathways in Multiple Myeloma. Biomedicines 2018, 6, 59. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Pei, X.Y.; Rahmani, M.; Conrad, D.H.; Dent, P.; Grant, S. Interruption of the NF-kappaB pathway by Bay 11-7082 promotes UCN-01-mediated mitochondrial dysfunction and apoptosis in human multiple myeloma cells. Blood 2004, 103, 2761–2770. [Google Scholar] [CrossRef]
- Manier, S.; Sacco, A.; Leleu, X.; Ghobrial, I.M.; Roccaro, A.M. Bone Marrow Microenvironment in MultipleMyeloma Progression. BioMed Res. Int. 2012, 2012, 157496. [Google Scholar] [CrossRef]
- Greco, C. Reduction of serum IGF-1 levels in patients affected with monoclonal gammopathies of undetermined significance or multiple myeloma. Comparison with bFGF, VEGF and κ-ras gene mutation. J. Exp. Clin. Cancer Res. 2009, 28, 35. [Google Scholar] [CrossRef] [Green Version]
- Vacca, A.; Ribatti, D. Bone marrow angiogenesis in multiple myeloma. Leukemia 2006, 20, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Vacca, A. Angiogenesis in multiple myeloma. Chem. Immunol. Allergy 2014, 99, 180–196. [Google Scholar]
- Ria, R.; Piccoli, C.; Cirulli, T.; Falzetti, F.; Mangialardi, G.; Guidolin, D.; Tabilio, A.; Di Renzo, N.; Guarini, A.; Ribatti, D.; et al. Endothelial differentiation of hematopoietic stem and progenitor cells from patients with multiple myeloma. Clin. Cancer Res. 2008, 14, 1678–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribatti, D.; Nico, B.; Vacca, A. Importance of the bone marrow microenvironment in inducing the angiogenic response in multiple myeloma. Oncogene 2006, 25, 4257–4266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vacca, A. Bone marrow angiogenesis in patients with active multiple myeloma. Semin. Oncol. 2001, 28, 543–550. [Google Scholar] [CrossRef]
- Ting, K.R.; Henry, M.; Meiller, J.; Larkin, A.; Clynes, M.; Meleady, P.; Bazou, D.; Dowling, P.; O’Gorman, P. Novel panel of protein biomarkers to predict response to bortezomib-containing induction regimens in multiple myeloma patients. BBA Clin. 2017, 8, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Belloni, D.; Marcatti, M.; Ponzoni, M.; Ciceri, F.; Veschini, L.; Corti, A.; Cappio, F.C.; Ferrarini, M.; Ferrero, E. Angiopoietin-2 in bone marrow milieu promotes multiple myeloma-associated angiogenesis. Exp. Cell Res. 2015, 330, 1–12. [Google Scholar] [CrossRef]
- Giuliani, N.; Colla, S.; Lazzaretti, M.; Sala, R.; Roti, G.; Mancini, C.; Bonomini, S.; Lunghi, P.; Hojden, M.; Genestreti, G.; et al. Proangiogenic properties of human myeloma cells: Production of angiopoietin-1 and its potential relationship to myeloma-induced angiogenesis. Blood 2003, 102, 638–645. [Google Scholar] [CrossRef] [Green Version]
- Pappa, C.A.; Tsirakis, G.; Samiotakis, P.; Tsigaridaki, M.; Alegakis, A.; Goulidaki, N.; Alexandrakis, M.G. Serum levels of angiopoietin-2 are associated with the growth of multiple myeloma. Cancer Investig. 2013, 31, 385–389. [Google Scholar] [CrossRef]
- Podar, K.; Anderson, K.C. The pathophysiologic role of VEGF in hematologic malignancies: Therapeutic implications. Blood 2005, 105, 13831395. [Google Scholar] [CrossRef]
- Bilalis, A.; Pouliou, E.; Roussou, M.; Papanikolaou, A.; Tassidou, A.; Economopoulos, T.; Terpos, E. Increased expression of platelet derived growth factor receptor β on trephine biopsies correlates with advanced myeloma. J. Balk. Union Oncol. 2017, 22, 1032–1037. [Google Scholar]
- Terpos, E.; Anargyrou, E.; Katodritou, E.; Kastritis, E.; Papatheodorou, A.; Christoulas, D.; Pouli, A.; Michalis, E.; Delimpasi, S.; Gkotzamanidou, M.; et al. Greek myeloma study group, Greece. Circulating angiopoietin-1 to angiopoietin-2 ratio is an independent prognostic factor for survival in newly diagnosed patients with multiple myeloma who received therapy with novel antimyeloma agents. Int. J. Cancer 2012, 130, 735–742. [Google Scholar] [CrossRef]
- Coluccia, A.M.L.; Cirulli, T.; Neri, P.; Mangieri, D.; Colanardi, M.C.; Gnoni, A.; Di Renzo, N.; Dammacco, F.; Tassone, P.; Ribatti, D.; et al. Validation of PDGFRβ and c-Src tyrosine kinases as tumor/vessel targets in patients with multiple myeloma: Preclinical efficacy of the novel, orally available inhibitor dasatinib. Blood 2008, 12, 1346–1356. [Google Scholar] [CrossRef]
- Tsirakis, G.; Pappa, C.A.; Kanellou, P.; Stratinaki, M.A.; Xekalou, A.; Psarakis, F.E.; Sakellaris, G.; Alegakis, A.; Stathopoulos, E.N.; Alexandrakis, M.G. Role of platelet-derived growth factor-AB in tumour growth and angiogenesis in relation with other angiogenic cytokines in multiple myeloma. Hematol. Oncol. 2012, 30, 131–136. [Google Scholar] [CrossRef]
- Ferrucci, A.; Moschetta, M.; Frassanito, M.A.; Berardi, S.; Catacchio, I.; Ria, R.; Racanelli, V.; Caivano, A.; Solimando, A.G.; Vergara, D.; et al. A HGF/cMET Autocrine Loop Is Operative in Multiple Myeloma Bone Marrow Endothelial Cellsand May Represent a Novel Therapeutic Target. Clin. Cancer Res. 2014, 20, 5796–5807. [Google Scholar] [CrossRef] [Green Version]
- Moschetta, M.; Kawano, Y.; Podar, K. Targeting the Bone Marrow Microenvironment. Cancer Treat. Res. 2016, 169, 63–102. [Google Scholar]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [Green Version]
- Balakumaran, A.; Robey, P.G.; Fedarko, N.; Landgren, O. Bone marrow microenvironment in myelomagenesis: Its potential role in early diagnosis. Expert Rev. Mol. Diagn. 2010, 10, 465–480. [Google Scholar] [CrossRef]
- Gnoni, A.; Brunetti, O. Immune system and bone microenvironment: Rationale for targeted cancer therapies. Oncotarget 2020, 11, 480–487. [Google Scholar]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Dimopoulos, M.A. Pathogenesis of bone disease in multiple myeloma: From bench to bedside. Blood Cancer J. 2018, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Jurczyszyn, A.; Zebzda, A.; Czepiel, J.; Gdula-Argasińska, J.; Perucki, W.; Skotnicki, A.B.; Majka, M. The analysis of the relationship between multiple myeloma cells and their microenvironment. J. Cancer 2015, 6, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Giuliani, N.; Colla, S.; Morandi, F.; Lazzaretti, M.; Sala, R.; Bonomini, S.; Grano, M.; Colucci, S.; Svaldi, M.; Rizzoli, V. Myeloma cells block RUNX2/CBFA1 activity in human bone marrow osteoblast progenitors and inhibit osteoblast formation and differentiation. Blood 2005, 106, 2472–2483. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, G.B.; Gupta, V.A.; Vertino, P.M.; Boise, L.H. Cell of Origin and Genetic Alterations in the Pathogenesis of Multiple Myeloma. Front. Immunol. 2019, 10, 1121. [Google Scholar]
- Barille-Nion, S.; Bataille, R. New insights in myeloma-induced osteolysis. Leuk. Lymphoma 2003, 44, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Lentzsch, S.; Gries, M.; Janz, M.; Bargou, R.; Dörken, B.; Mapara, M.Y. Macrophage inflammatory protein 1-α (MIP-1 α) triggers migration and signaling cascades mediating survival and proliferation in multiple myeloma (MM) cells. Blood 2003, 101, 3568–3573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, R.E.; Brown, J.E.; Lawson, M.A.; Chantry, A.D. Myeloma Bone Disease: The Osteoblast in the Spotlight. J. Clin. Med. 2021, 10, 3973. [Google Scholar] [CrossRef]
- Calvani, N.; Cafforio, P.; Silvestris, F.; Dammacco, F. Functional osteoclast-like transformation of cultured human myeloma cell lines. Br. J. Haematol. 2005, 130, 926–938. [Google Scholar] [CrossRef]
- Sun, L.; Iqbal, J.; Dolgilevich, S.; Yuen, T.; Wu, X.; Moonga, B.S.; Adebanjo, O.A.; Bevis, P.J.R.; Lund, F.; Huang, C.L.; et al. Disordered osteoclast formation and function in a CD38 (ADP-ribosyl cyclase)-deficient mouse establishes an essential role for CD38 in bone resorption. FASEB J. 2003, 17, 369–375. [Google Scholar] [CrossRef] [Green Version]
- An, G.; Acharya, C.; Feng, X.; Wen, K.; Zhong, M.; Zhang, L.; Munshi, N.C.; Qiu, L.; Tai, Y.-T.; Anderson, K.C. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: Therapeutic implication. Blood 2016, 128, 1590–1603. [Google Scholar] [CrossRef] [Green Version]
- Horenstein, A.L.; Quarona, V.; Toscani, D.; Costa, F.; Chillemi, A.; Pistoia, V.; Giuliani, N.; Malavasi, F. Adenosine generated in the bone marrow niche through a CD38-mediated pathway correlates with progression of human myeloma. Mol. Med. 2016, 22, 694–704. [Google Scholar] [CrossRef] [Green Version]
- Saltarella, I.; Desantis, V.; Melaccio, A.; Solimando, A.G.; Lamanuzzi, A.; Ria, R.; Storlazzi, C.T.; Mariggiò, M.A.; Vacca, A.; Frassanito, M.A. Mechanisms of Resistance to Anti-CD38 Daratumumab in Multiple Myeloma. Cells 2020, 9, 167. [Google Scholar] [CrossRef] [Green Version]
- Hagen, P.; Zhang, J.; Barton, K. High-risk disease in newly diagnosed multiple myeloma: Beyond the R-ISS and IMWG definitions. Blood Cancer J. 2022, 12, 83. [Google Scholar] [CrossRef]
- De la Puente, P.; Muz, B.; Azab, F.; Luderer, M.; Azab, A.K. Molecularly Targeted Therapies in Multiple Myeloma. Leuk. Res. Treat. 2014, 976567, 8. [Google Scholar] [CrossRef]
- Zheng, Z.; Liu, T.; Zheng, J.; Hu, J. Clarifying the molecular mechanism associated with carfilzomib resistance in human multiple myeloma using microarray gene expression profile and genetic interaction network. Oncol. Targets Ther. 2017, 10, 1327–1334. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kikuchi, J. Epigenetic mechanisms of cell adhesion-mediated drug resistance in multiple myeloma. Int. J. Hematol. 2016, 104, 281–292. [Google Scholar] [CrossRef]
- Kortüm, K.M.; Mai, E.K.; Hanafiah, N.H.; Shi, C.-X.; Zhu, Y.-X.; Bruins, L.; Barrio, S.; Jedlowski, P.; Merz, M.; Xu, J.; et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood 2016, 128, 1226–1233. [Google Scholar] [CrossRef] [Green Version]
- Bellone, S.; Roque, D.; Cocco, E.; Gasparrini, S.; Bortolomai, I.; Buza, N.; Abu-Khalaf, M.; Silasi, D.-A.; Ratner, E.; Azodi, M.; et al. Downregulation of membrane complement inhibitors CD55 and CD59 by siRNA sensitizes uterine serous carcinoma overexpressing Her2/neu to complement and antibody-dependent cell cytotoxicity in vitro: Implications for trastuzumab-based immunotherapy. Br. J. Cancer 2012, 106, 1543–1550. [Google Scholar] [CrossRef]
- Kankeu, L.; Sirpilla, O.; Sakemura, R.; Siegler, R.L.; Kenderian, S.S. CAR T cell therapy and the tumor microenvironment: Current challenges and opportunities. Mol. Ther. Oncolytics 2022, 25, 69–77. [Google Scholar] [CrossRef]
- Thakurta, A.; Pierceall, W.E.; Amatangelo, M.D.; Flynt, E.; Agarwal, A. Developing next generation immunomodulatory drugs and their combinations in multiple myeloma. Oncotarget 2021, 12, 1555–1563. [Google Scholar] [CrossRef]
- Robak, P.; Drozdz, I.; Szemraj, J. Robak Drug resistance in multiple myeloma. Cancer Treat. Rev. 2018, 70, 199–208. [Google Scholar] [CrossRef]
- Ria, R.; Vacca, A. Bone Marrow Stromal T. Cells-Induced Drug Resistance in Multiple Myeloma. Int. J. Mol. Sci. 2020, 21, 613. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K. Treatment Strategies Considering Micro-Environment and Clonal Evolution in Multiple Myeloma. Cancers 2021, 13, 215. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Santis, V.; Saltarella, I.; Lamanuzzi, A.; Melaccio, A.; Solimando, A.G.; Mariggiò, M.A.; Racanelli, V.; Paradiso, A.; Vacca, A.; Frassanito, M.A. MicroRNAs-Based Nano-Strategies as New Therapeutic Approach in Multiple Myeloma to Overcome Disease Progression and Drug Resistance. Int. J. Mol. Sci. 2020, 21, 3084. [Google Scholar] [CrossRef] [PubMed]
- Damiano, J.S.; Cress, A.E.; Hazlehurst, L.A.; Shtil, A.; Dalton, W.S. Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999, 93, 1658–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J. Cx43 expressed on bone marrow stromal cells plays an essential role in multiple myeloma cell survival and drug resistance. Arch. Med. Sci. 2017, 13, 236–245. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Rodriguez, F.; Teixido, J. VLA-4-dependent myeloma cell adhesion. Leuk. Lymphoma 2001, 41, 239–245. [Google Scholar] [CrossRef]
- Cheung, W.C.; Van, N.B. Distinct IL-6 signal transduction leads to growth arrest and death in B cells or growth promotion and cell survival in myeloma cells. Leukemia 2002, 16, 1182–1188. [Google Scholar] [CrossRef] [Green Version]
- Abdi, J.; Chen, G.; Chang, H. Drug resistance in multiple myeloma: Latest findings and new concepts on molecular mechanisms. Oncotarget 2013, 4, 2186–2207. [Google Scholar] [CrossRef] [Green Version]
- Ria, R.; Catacchio, I.; Berardi, S.; De Luisi, A.; Caivano, A.; Piccoli, C.; Ruggieri, V.; Frassanito, M.A.; Ribatti, D.; Nico, B.; et al. HIF-1α of bone marrow endothelial cells implies relapse and drug resistance in patients with multiple myeloma and may act as a therapeutic target. Clin. Cancer Res. 2014, 20, 847–858. [Google Scholar] [CrossRef] [Green Version]
- Katz, B.Z. Adhesion molecules-The lifelines of multiple myeloma cells. Semin. Cancer Biol. 2010, 20, 186–195. [Google Scholar] [CrossRef]
- Vacca, A.; Di Loreto, M.; Ribatti, D.; Di Stefano, R.; Gadaleta-Caldarola, G.; Iodice, G.; Caloro, D.; Dammacco, F. Bone marrow of patients with active multiple myeloma: Angiogenesis and plasma cell adhesion molecules LFA-1, VLA-4, LAM-1, and CD44. Am. J. Hematol. 1995, 50, 9–14. [Google Scholar] [CrossRef]
- Barker, H.F.; Ball, J.; Drew, M.; Hamilton, M.S.; Franklin, I.M. The role of adhesion molecules in multiple myeloma. Leuk. Lymphoma 1992, 8, 189–196. [Google Scholar] [CrossRef]
- Hideshima, T.; Bergsagel, P.L.; Kuehl, W.M.; Anderson, K.C. Advances in biology of multiple myeloma: Clinical applications. Blood 2004, 104, 607–618. [Google Scholar] [CrossRef]
- Noborio-Hatano, K.; Kikuchi, J.; Takatoku, M.; Shimizu, R.; Wada, T.; Ueda, M.; Nobuyoshi, M.; Oh, I.; Sato, K.; Suzuki, T.; et al. Bortezomib overcomes cell-adhesion-mediated drug resistance through downregulation of VLA-4 expression in multiple myeloma. Oncogene 2009, 28, 231–242. [Google Scholar] [CrossRef] [Green Version]
- Pulido, R.; Elices, M.J.; Campanero, M.R.; Osborn, L.; Schiffer, S.; García-Pardo, A.; Lobb, R.; E Hemler, M.; Sánchez-Madrid, F. Functional evidence for three distinct and independently inhibitable adhesion activities mediated by the human integrin VLA-4. Correlation with distinct alpha 4 epitopes. J. Biol. Chem. 1991, 266, 10241–10245. [Google Scholar] [CrossRef]
- Roccaro, A.; Hideshima, T.; Richardson, P.; Russo, D.; Ribatti, D.; Vacca, A.; Dammacco, F.; Anderson, K. Bortezomib as an antitumor agent. Curr. Pharm. Biothechnol. 2006, 7, 441–448. [Google Scholar] [CrossRef]
- Hideshima, T.; Chauhan, D.; Richardson, P.; Mitsiades, C.; Mitsiades, N.; Hayashi, T.; Munshi, N.; Dang, L.; Castro, A.; Palombella, V. NF-kappa B as a therapeutic target in multiple myeloma. J. Biol. Chem. 2002, 277, 16639–16647. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D.; Vacca, A. Novel Therapeutic Approaches Targeting Vascular Endothelial Growth Factor and its receptors in hematological malignancies. Curr. Cancer Drug Targets 2005, 5, 573–578. [Google Scholar] [CrossRef]
- Segeren, M.C.; Sonneveld, P.; van der Holt, B.; Vellenga, E.; Croockewit, A.J.; Verhoef, G.E.G.; Cornelissen, J.J.; Schaafsma, M.R.; Marivan Oers, M.H.J.; Wijermans, P.W. Overall and event-free survival are not improved by the use of myeloablative therapy following intensified chemotherapy in previously untreated patients with multiple myeloma: A prospective randomized phase 3 study. Blood 2003, 101, 2144–2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amato, R.J. Thalidomide is an inhibitor of angiogemesis. Proc. Natl. Acad. Sci. USA 1994, 91, 4082–4085. [Google Scholar] [CrossRef] [Green Version]
- Bhutani, M.; Foureau, D.M.; Atrash, S.; Voorhees, P.M.; Usmani, S.Z. Extramedullary multiple myeloma. Leukemia 2020, 34, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Da Vià, M.C.; Solimando, A.G.; Garitano-Trojaola, A.; Barrio, S.; Munawar, U.; Strifler, S.; Haertle, L.; Rhodes, N.; Teufel, E.; Vogt, C.; et al. CIC Mutation as a Molecular Mechanism of Acquired Resistance to Combined BRAF-MEK Inhibition in Extramedullary Multiple Myeloma with Central Nervous System Involvement. Oncologist 2019, 25, 112–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neri, P.; Bahlis, N.J.; Lonial, S. New Strategies in Multiple Myeloma: Immunotherapy as a Novel Approach to Treat Patients With Multiple Myeloma. Clin. Cancer Res. 2016, 22, 5959–5965. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E. Isatuximab: A Review of Its Use in Multiple Myeloma. Target Oncol. 2021, 16, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Bardenstein, D.S.; Cheyer, C.; Okada, N.; Morgan, B.P.; Medof, M.E. Cell surface regulators of complement, 5I2 antigen, and CD59, in the rat eye and adnexal tissues. Investig. Ophthalmol. Vis. Sci. 1999, 40, 519–524. [Google Scholar]
- Gavriatopoulu, M.; Ntanasis-Stathopoulos, J.; Dimopoulos, M.A.; Terpos, E. Anti-BCMA Antibodies in the Future Management of Multiple Myeloma. Expert Rev. Anticancer Ther. 2019, 19, 319–326. [Google Scholar] [CrossRef]
- Larysa, S.; Dardac, A.; Madduri, D.; Richard, S.; Richter, J. B-cell maturation antigen (BCMA) in multiple myeloma: The new frontier of targeted therapies. Ther. Adv. Hematol. 2021, 12, 1–16. [Google Scholar]
- Gomes-Silva, D.; Ramos, C.A. Cancer Immunotherapy Using CAR-T Cells: From the Research Bench to the Assembly Line. Biotechnol. J. 2018, 13, 1700097. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, Z.; Dang, Q.; Xu, H.; Lv, J.; Li, H.; Han, X. Immunosuppression in tumor immune microenvironment and its optimization from CAR-T cell therapy. Theranostics 2022, 12, 6273–6290. [Google Scholar] [CrossRef]
- Borja, P.; María-Victoria, M.; González-Calle, V. Anti-BCMA CAR T-cell Therapy: Changing the Natural History of Multiple Myeloma. Hemasphere 2022, 6, e691. [Google Scholar]
- Kleber, M. BCMA in Multiple Myeloma—A Promising Key to Therapy. J. Clin. Med. 2021, 10, 4088. [Google Scholar] [CrossRef]
- Neumeister, P.; Schulz, E.; Pansy, K.; Szmyra, M.; Deutsch, A.J. Targeting the Microenvironment for Treating Multiple Myeloma. Int. J. Mol. Sci. 2022, 23, 7627. [Google Scholar] [CrossRef]
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D.C. Differential Targeting of Prosurvival Bcl-2 Proteins by Their BH3-Only Ligands Allows Complementary Apoptotic Function. Mol. Cell 2005, 17, 393–403. [Google Scholar] [CrossRef]
- Shehata, M.; Schnabl, S.; Demirtas, D.; Hilgarth, M.; Hubmann, R.; Ponath, E.; Badrnya, S.; Lehner, C.; Hoelbl, A.; Duechler, M.; et al. Reconstitution of PTEN Activity by CK2 Inhibitors and Interference with the PI3-K/Akt Cascade Counteract the Antiapoptotic Effect of Human Stromal Cells in Chronic Lymphocytic Leukemia. Blood 2010, 116, 2513–2521. [Google Scholar] [CrossRef]
- Di Marzo, L.; Desantis, V.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; Fumarulo, R.; Vacca, A.; Frassanito, M.A. Microenvironment Drug Resistance in Multiple Myeloma: Emerging New Players. Oncotarget 2016, 7, 60698–60711. [Google Scholar] [CrossRef] [Green Version]
- Sutton, V.R.; Davis, J.E.; Cancilla, M.; Johnstone, R.; Ruefli, A.A.; Sedelies, K.; Browne, K.A.; Trapani, J. Initiation of Apoptosis by Granzyme B Requires Direct Cleavage of Bid, but Not Direct Granzyme B–Mediated Caspase Activation. J. Exp. Med. 2000, 192, 1403–1414. [Google Scholar] [CrossRef]
- Catalán, E.; Jaime-Sánchez, P.; Aguilo, N.; Simon, M.M.; Froelich, C.J.; Pardo, J. Mouse Cytotoxic T Cell-Derived Granzyme B Activates the Mitochondrial Cell Death Pathway in a Bim-Dependent Fashion. J. Biol. Chem. 2015, 290, 6868–6877. [Google Scholar] [CrossRef] [Green Version]
- Cullen, S.P.; Brunet, M.; Martin, S.J. Granzymes in Cancer and Immunity. Cell Death Differ. 2010, 17, 616–623. [Google Scholar] [CrossRef] [Green Version]
- Ewen, C.L.; Kane, K.P.; Bleackley, R.C. A Quarter Century of Granzymes. Cell Death Differ. 2011, 19, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Holthof, L.C.; Mutis, T. Challenges for Immunotherapy in Multiple Myeloma: Bone Marrow Microenvironment-Mediated Immune Suppression and Immune Resistance. Cancers 2020, 12, 988. [Google Scholar] [CrossRef]
- Giallongo, C.; Tibullo, D.; Camiolo, G.; Parrinello, N.L.; Romano, A.; Puglisi, F.; Barbato, A.; Conticello, C.; Lupo, G.; Anfuso, C.D. TLR4 Signaling Drives Mesenchymal Stromal Cells Commitment to Promote Tumor Microenvironment Transformation in Multiple Myeloma. Cell Death Dis. 2019, 10, 704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- André, T.; Najar, M.; Stamatopoulos, B.; Pieters, K.; Pradier, O.; Bron, D.; Meuleman, N.; Lagneaux, L. Immune Impairments in Multiple Myeloma Bone Marrow Mesenchymal Stromal Cells. Cancer Immunol. Immunother. 2015, 64, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Franssen, L.E.; Mutis, T.; Lokhorst, H.M.; van de Donk, N.W.J.C. Immunotherapy in Myeloma: How Far Have We Come? Ther. Adv. Hematol. 2019, 10, 2040620718822660. [Google Scholar] [CrossRef] [PubMed]
- Görgün, G.T.; Whitehill, G.; Anderson, J.L.; Hideshima, T.; Maguire, C.; Laubach, J.; Raje, N.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Tumor-Promoting Immune-Suppressive Myeloid-Derived Suppressor Cells in the Multiple Myeloma Microenvironment in Humans. Blood 2013, 121, 2975–2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Nicola, M.; Carlo-Stella, C.; Magni, M.; Milanesi, M.; Longoni, P.D.; Matteucci, P.; Grisanti, P.; Gianni, A.M. Human Bone Marrow Stromal Cells Suppress T-Lymphocyte Proliferation Induced by Cellular or Nonspecific Mitogenic Stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef]
- Hwang, W.; Jung, K.; Jeon, Y.; Yun, S.; Kim, T.W.; Choi, I. Knockdown of the Interleukin-6 Receptor Alpha Chain of Dendritic Cell Vaccines Enhances the Therapeutic Potential against IL-6 Producing Tumors. Vaccine 2010, 29, 34–44. [Google Scholar] [CrossRef]
- Ohno, Y.; Kitamura, H.; Takahashi, N.; Ohtake, J.; Kaneumi, S.; Sumida, K.; Homma, S.; Kawamura, H.; Minagawa, N.; Shibasaki, S.; et al. IL-6 down-Regulates HLA Class II Expression and IL-12 Production of Human Dendritic Cells to Impair Activation of Antigen-Specific CD4+ T Cells. Cancer Immunol. Immunother. 2016, 65, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Sumida, K.; Wakita, D.; Narita, Y.; Masuko, K.; Terada, S.; Watanabe, K.; Satoh, T.; Kitamura, H.; Nishimura, T. Anti-IL-6 Receptor MAb Eliminates Myeloid-Derived Suppressor Cells and Inhibits Tumor Growth by Enhancing T-Cell Responses. Eur. J. Immunol. 2012, 42, 2060–2072. [Google Scholar] [CrossRef]
- Holthof, L.C.; Van der Horst, H.J.; Poels, R.; van der Schans, J.T.; Gelderloos, A.T.; Li, F.; Lokhorst, H.; Zweegman, S.; Themeli, M.; Van de Donk, N.W.J.C.; et al. The Impact and Modulation of Microenvironment-Induced Immune Resistance Against CAR T Cell and Antibody Treatments in Multiple Myeloma. Blood 2019, 134, 137. [Google Scholar] [CrossRef]
- Frerichs, K.A.; Nagy, N.A.; Lindenbergh, P.L.; Bosman, P.; Soto, J.M.; Broekmans, M.; Groen, R.W.J.; Themeli, M.; Nieuwenhuis, L.; Stege, C.; et al. CD38-Targeting Antibodies in Multiple Myeloma: Mechanisms of Action and Clinical Experience. Expert Rev. Clin. Immunol. 2018, 14, 197–206. [Google Scholar] [CrossRef]
- Lee, A.H.S.; E Gillett, C.; Ryder, K.; Fentiman, I.S.; Miles, D.W.; Millis, R.R. Different patterns of inflammation and prognosis in invasive carcinoma of the breast. Histopathology 2006, 48, 692–701. [Google Scholar] [CrossRef]
- Boissinot, M.; Vilaine, M.; Hermouet, S. The hepatocyte growth factor (HGF)/ met axis: A neglected target in the treatment of chronic myeloproliferative neoplasms? Cancers 2014, 6, 1631–1669. [Google Scholar] [CrossRef]
- Lust, J.A.; Lacy, M.Q.; Zeldenrust, S.R.; Witzig, T.E.; Moon-Tasson, L.L.; Dinarello, C.A.; Donovan, K.A. Reduction in C-reactive protein indicates successful targeting of the IL-1/IL-6 axis resulting in improved survival in early stage multiple myeloma. Am. J. Hematol. 2016, 91, 571–574. [Google Scholar] [CrossRef]
- Scavelli, C.; Di Pietro, G.; Cirulli, T.; Coluccia, M.; Boccarelli, A.; Giannini, T.; Mangialardi, G.; Bertieri, R.; Coluccia, A.M.L.; Ribatti, D.; et al. Zoledronic acid affects ocer-angiogenic phenotype of endothelial cells in patients with multiple myeloma. Mol. Cancer Ther. 2007, 6 Pt 1, 3256–3262. [Google Scholar] [CrossRef] [Green Version]
- Ria, R.; Reale, A.; Moschetta, M.; Mangialardi, G.; Dammacco, F.; Vacca, A. A retrospective study of skeletal and disease-free survival benefits of zoledronic acid therapy in patients with multiple myeloma treated with novel agents. Int. J. Clin. Exp. Med. 2013, 6, 30–38. [Google Scholar]
- Ribatti, D.; Nico, B.; Mangieri, D.; Maruotti, N.; Longo, V.; Vacca, A.; Cantatore, F.P. Neridronate inhibits angiogenesis in vitro and in vivo. Clin. Rheumatol. 2007, 26, 1094–1098. [Google Scholar] [CrossRef]
- Allegra, A. Nanoparticles in oncology: The new theragnostic molecules. Anti-Cancer Agents Med. Chem. 2011, 11, 669–686. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drugs | Mechanism of Action | Cytokines Involved | Resistance | References |
|---|---|---|---|---|
| Proteasome inhibitors (Bortezomib, carfilzomib ixazomib) | Blockade of NF-kB; interferes with growth arrest, apoptosis, cell cycle progression, inflammation and immune surveillance | Inhibits adhesion of MM cells to BMSCs; reduced production of VEGF-2, FGF-2,IL-6, IGF-I, Ang-1 and Ang-2 | Interaction between cytokine-cytokine receptors; autophagy | Zheng et al., 2017 [94] Furukawa Y et al., 2016 [95] |
| IMIDs(Thalidomide, Lenalidomide pomalidomide) | Selectively enhance degradation of the transcription factors IKZF1 and IKFZ3; anti-angiogenic properties | Modulate TNF-α;reduction of FGF-2,VEGF, IL-6 by MM cells and BMSCs;reduce adhesion between MM PCs and BMSC | Downregulation of cerebron expression | Kortum et al., 2016 [96] |
| Monoclonal antibodies (elotuzumab, daratumumab, isatuximab) | Antibody-dependent cellular cytotoxicity (ADCC), complement activation, antibody-dependent phagocytosis, direct effect on target cells | Effects mediated by INFγ and TNFα | CDC mechanism mediated by the overexpression of CD55 and CD59 | Bellone et al., 2012 [97] |
| Anti-BCMA BITE CAR | ADC inducing cytotoxic cell death Simultaneous binding to T cell and tumor antigens Chimeric antigen receptors against specific tumor-associated antigen | IL-6, IL-10, TNFα inducing a (CRS) | immunosuppressive TME, production IL-2 by Treg MSC protection | Kankeu Fonkoua et al., 2022 [98] |
| CELmoDs | rapid degradation of distinct cell types mediated by physical interactions with the CRBN/DDB1 complex | IL-2 up-regulation | Thakurta et al., 2021 [99] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melaccio, A.; Reale, A.; Saltarella, I.; Desantis, V.; Lamanuzzi, A.; Cicco, S.; Frassanito, M.A.; Vacca, A.; Ria, R. Pathways of Angiogenic and Inflammatory Cytokines in Multiple Myeloma: Role in Plasma Cell Clonal Expansion and Drug Resistance. J. Clin. Med. 2022, 11, 6491. https://doi.org/10.3390/jcm11216491
Melaccio A, Reale A, Saltarella I, Desantis V, Lamanuzzi A, Cicco S, Frassanito MA, Vacca A, Ria R. Pathways of Angiogenic and Inflammatory Cytokines in Multiple Myeloma: Role in Plasma Cell Clonal Expansion and Drug Resistance. Journal of Clinical Medicine. 2022; 11(21):6491. https://doi.org/10.3390/jcm11216491
Chicago/Turabian StyleMelaccio, Assunta, Antonia Reale, Ilaria Saltarella, Vanessa Desantis, Aurelia Lamanuzzi, Sebastiano Cicco, Maria Antonia Frassanito, Angelo Vacca, and Roberto Ria. 2022. "Pathways of Angiogenic and Inflammatory Cytokines in Multiple Myeloma: Role in Plasma Cell Clonal Expansion and Drug Resistance" Journal of Clinical Medicine 11, no. 21: 6491. https://doi.org/10.3390/jcm11216491