
Cardiac-Restricted Expression of VCP/TER94 RNAi or Disease Alleles Perturbs Drosophila Heart Structure and Impairs Function

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
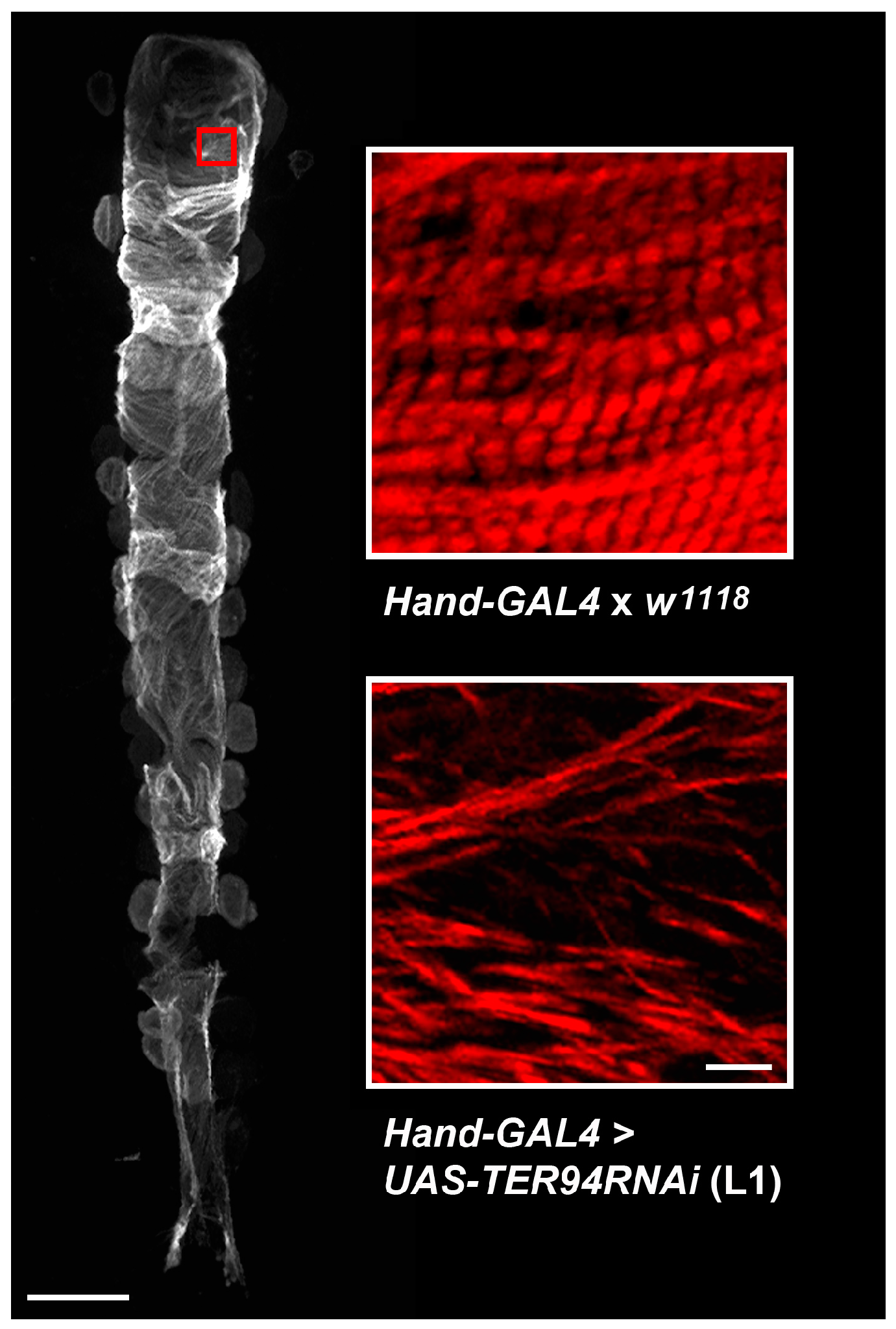
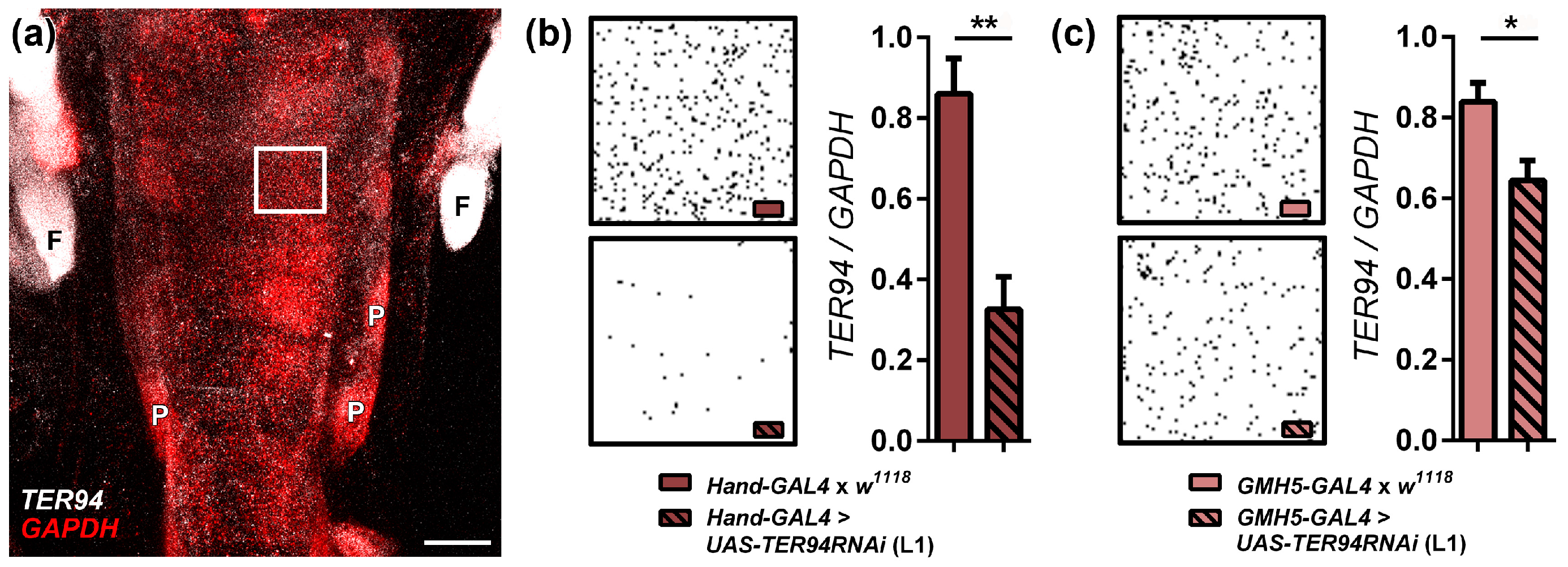
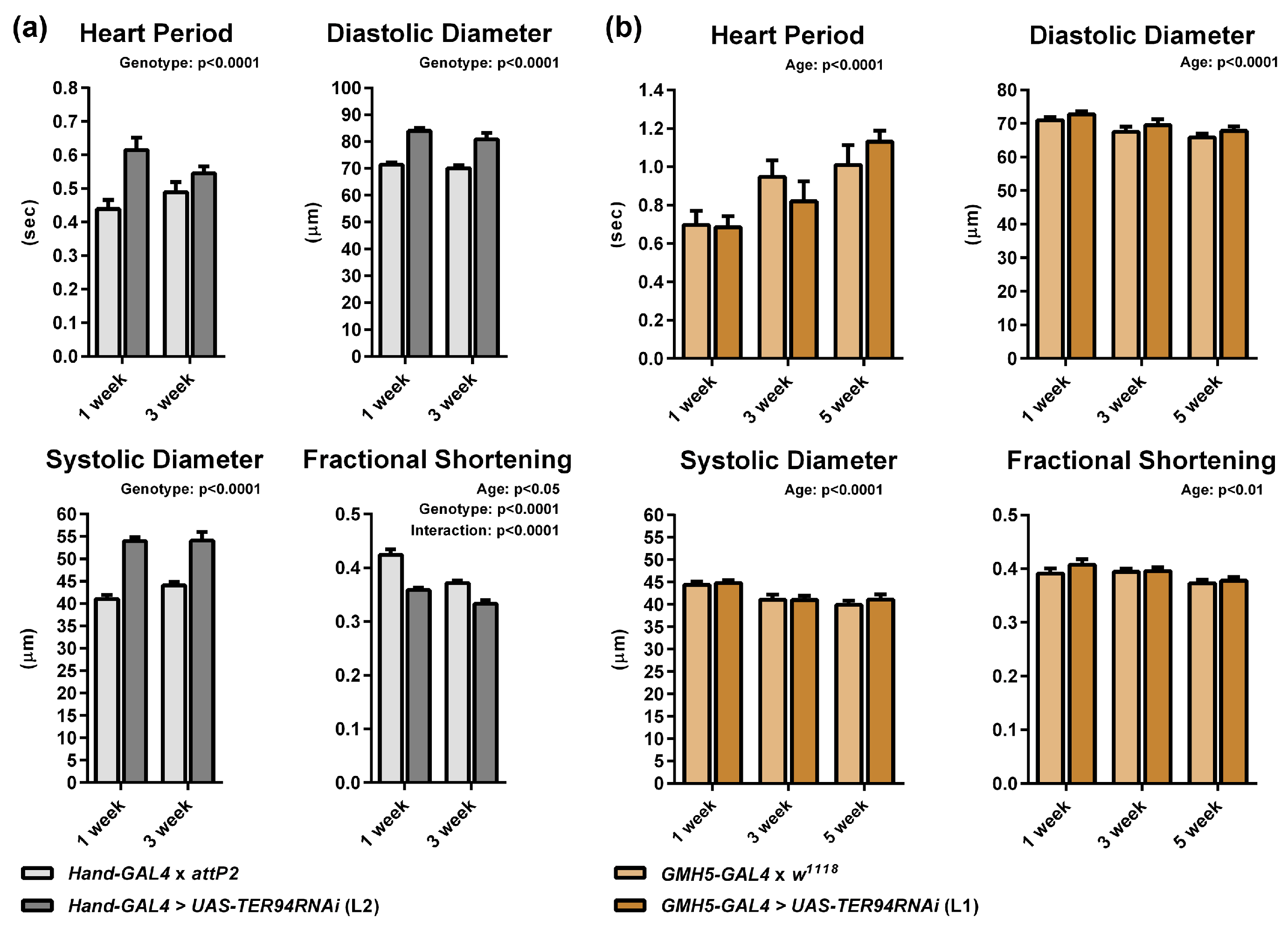
2.1. Cardiac-Specific Knockdown of TER94 Severely Affects Adult Drosophila Heart Structure and Function
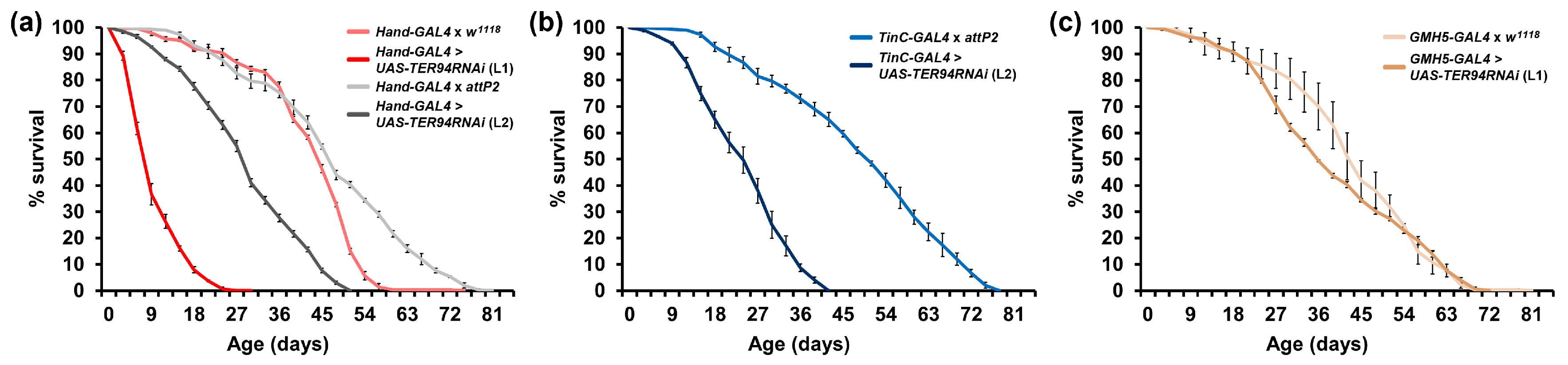
2.2. Modest Cardiac-Specific Knockdown of TER94 Has No Effect on Lifespan or Cardiac Function
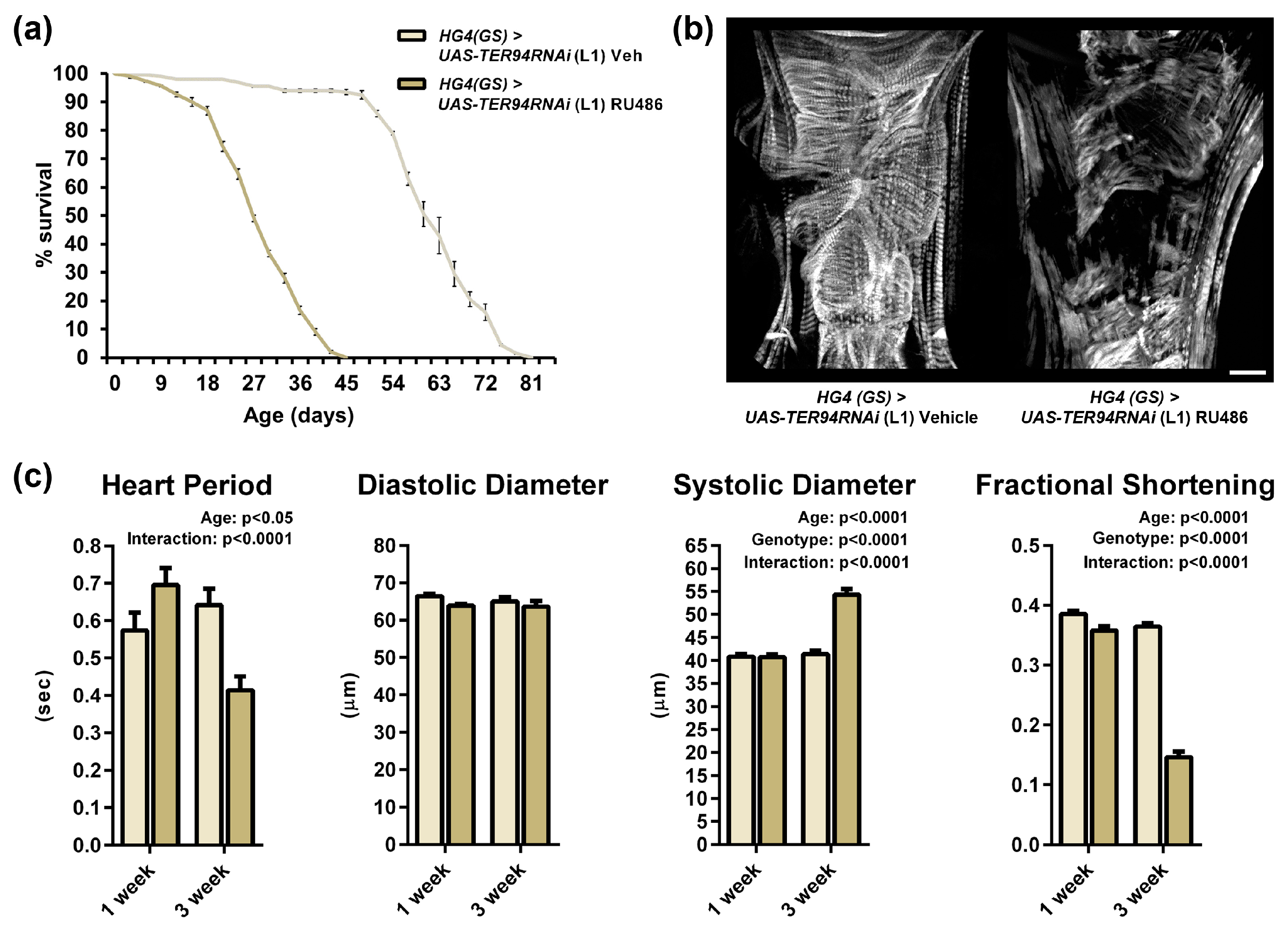
2.3. Heart-Specific Knockdown of TER94 Post-Development Affects Cardiac Structure and Function
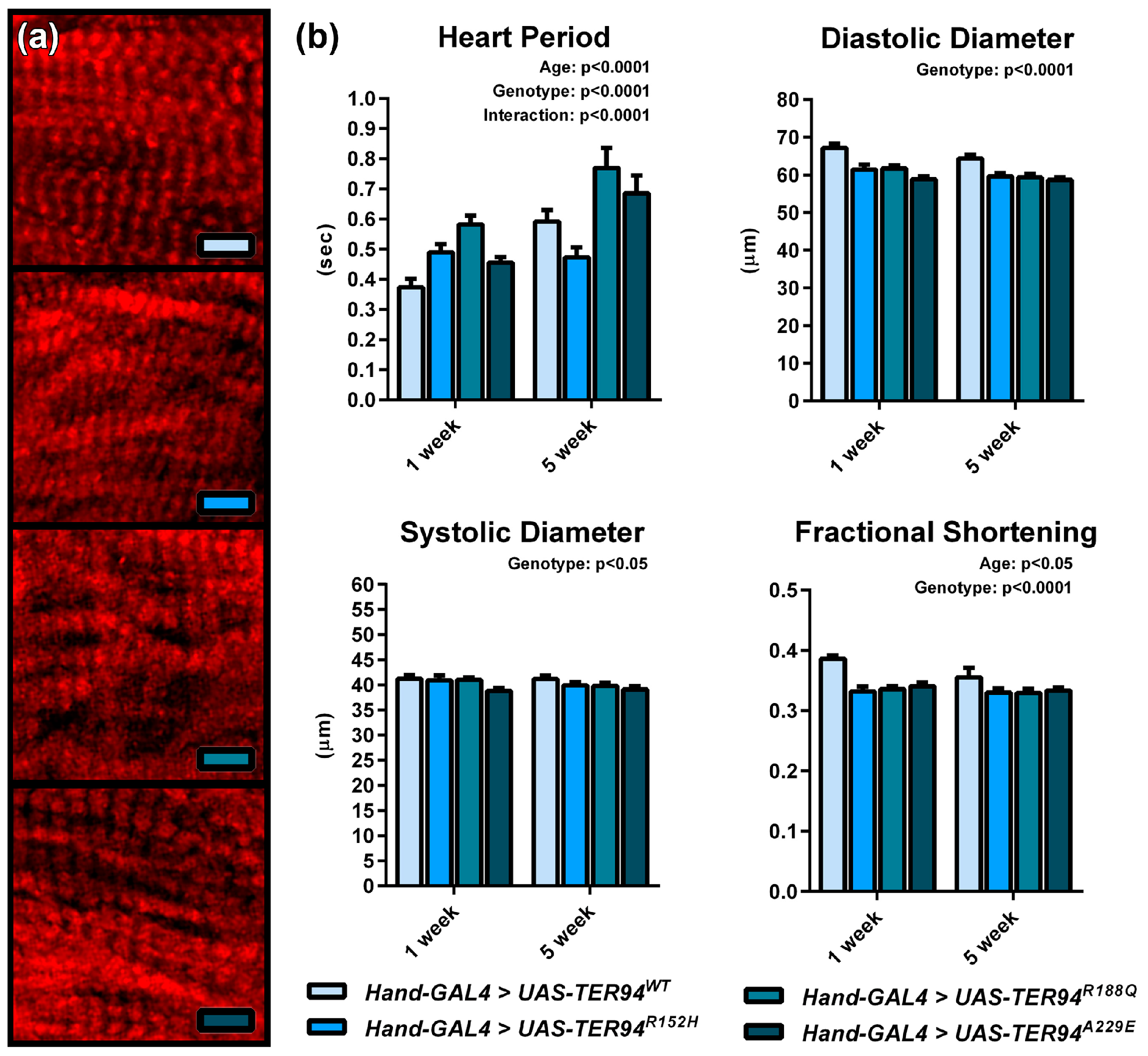
2.4. Expression of TER94 MSP Alleles Engenders a Restrictive Cardiac Phenotype in Adult Flies
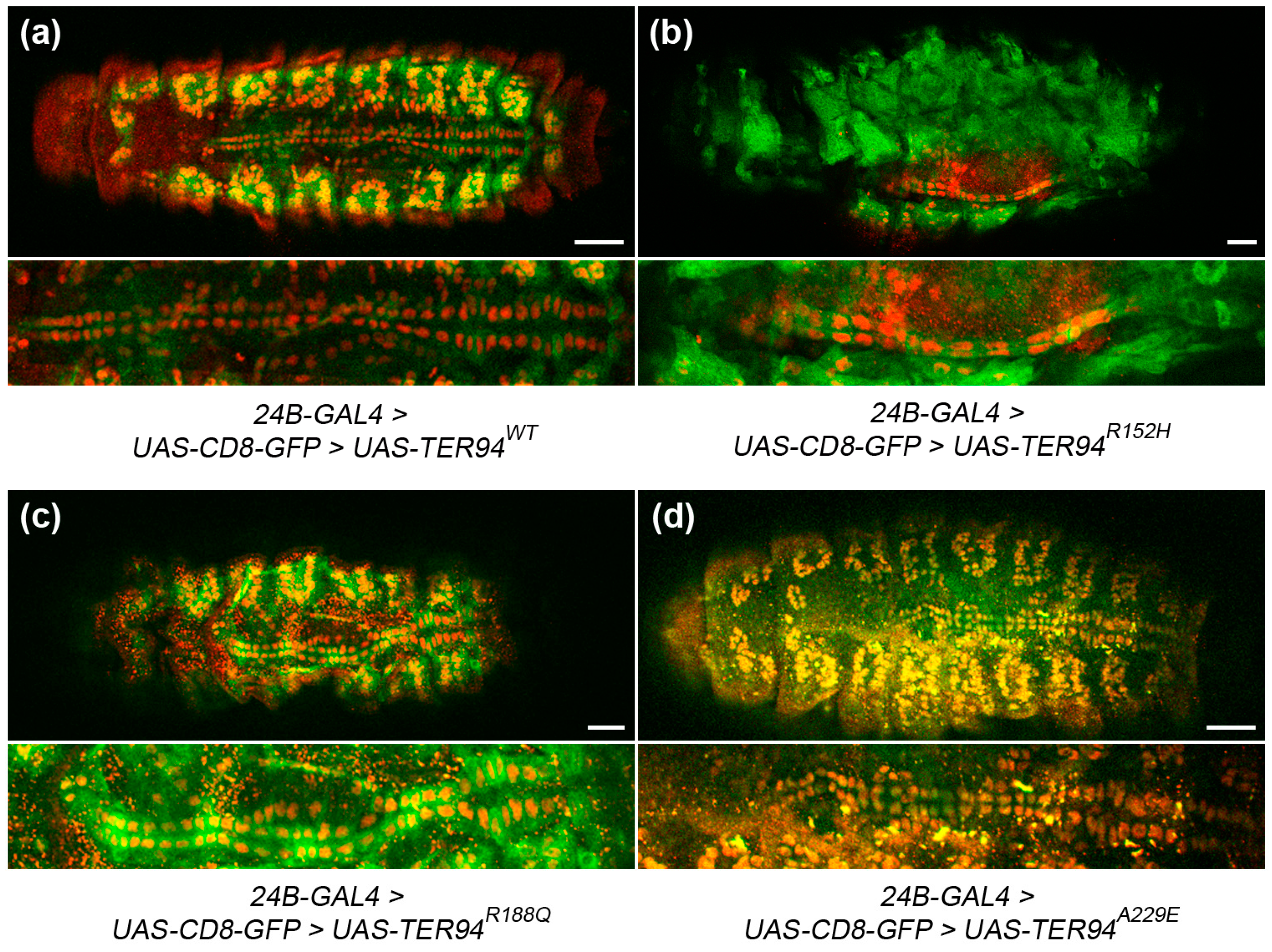
2.5. Expression of TER94 MSP Alleles in Muscle Progenitor Cells Causes Cardiac Defects in the Embryonic Heart
3. Discussion
4. Materials and Methods
4.1. Fly Stocks
4.2. Adult Drosophila Cardiac Tube Immunohistochemistry
4.3. Fluorescence in Situ Hybridization
4.4. Lifespan Analysis
4.5. Drosophila Cardiac Analysis
4.6. Drosophila Embryo Immunohistochemistry
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, Q.; Song, C.; Li, C.C. Molecular perspectives on p97-VCP: Progress in understanding its structure and diverse biological functions. J. Struct. Biol. 2004, 146, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Tang, W.K.; Ye, Y. Structure and function of the AAA+ ATPase p97/Cdc48p. Gene 2016, 583, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Weihl, C.C. Another VCP interactor: NF is enough. J. Clin. Investig. 2011, 121, 4627–4630. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Weihl, C.C. The VCP/p97 system at a glance: Connecting cellular function to disease pathogenesis. J. Cell Sci. 2014, 127, 3877–3883. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Bug, M.; Bremer, S. Emerging functions of the VCP/p97 AAA-atpase in the ubiquitin system. Nat. Cell Biol. 2012, 14, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Halawani, D.; Latterich, M. P97: The cell’s molecular purgatory? Mol. Cell 2006, 22, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, R.; Goldberg, A.L. The p97/VCP Atpase is critical in muscle atrophy and the accelerated degradation of muscle proteins. EMBO J. 2012, 31, 3334–3350. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Sasagawa, Y.; Ogura, T. Recent advances in p97/VCP/Cdc48 cellular functions. Biochim. Biophys. Acta 2012, 1823, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Watts, G.D.; Wymer, J.; Kovach, M.J.; Mehta, S.G.; Mumm, S.; Darvish, D.; Pestronk, A.; Whyte, M.P.; Kimonis, V.E. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 2004, 36, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P. Multisystem proteinopathy: Intersecting genetics in muscle, bone, and brain degeneration. Neurology 2015, 85, 658–660. [Google Scholar] [CrossRef] [PubMed]
- Kimonis, V.E.; Kovach, M.J.; Waggoner, B.; Leal, S.; Salam, A.; Rimer, L.; Davis, K.; Khardori, R.; Gelber, D. Clinical and molecular studies in a unique family with autosomal dominant limb-girdle muscular dystrophy and Paget disease of bone. Genet. Med. 2000, 2, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Kimonis, V.E.; Fulchiero, E.; Vesa, J.; Watts, G. VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: Review of a unique disorder. Biochim. Biophys. Acta 2008, 1782, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Schroder, R.; Watts, G.D.; Mehta, S.G.; Evert, B.O.; Broich, P.; Fliessbach, K.; Pauls, K.; Hans, V.H.; Kimonis, V.; Thal, D.R. Mutant valosin-containing protein causes a novel type of frontotemporal dementia. Ann. Neurol. 2005, 57, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Hubbers, C.U.; Clemen, C.S.; Kesper, K.; Boddrich, A.; Hofmann, A.; Kamarainen, O.; Tolksdorf, K.; Stumpf, M.; Reichelt, J.; Roth, U.; et al. Pathological consequences of VCP mutations on human striated muscle. Brain 2007, 130, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Ponfick, M.; Ludolph, A.C.; Dekomien, G.; Uttner, I.; Kassubek, J.; Gdynia, H.J. Inclusion body myositis, Paget’s disease of the bone and frontotemporal dementia: Early involvement of the heart and respiratory muscles. Fortschr. Neurol. Psychiatr. 2012, 80, 344–347. [Google Scholar] [PubMed]
- Ju, J.S.; Weihl, C.C. Inclusion body myopathy, Paget’s disease of the bone and fronto-temporal dementia: A disorder of autophagy. Hum. Mol. Genet. 2010, 19, R38–R45. [Google Scholar] [CrossRef] [PubMed]
- Nalbandian, A.; Donkervoort, S.; Dec, E.; Badadani, M.; Katheria, V.; Rana, P.; Nguyen, C.; Mukherjee, J.; Caiozzo, V.; Martin, B.; et al. The multiple faces of valosin-containing protein-associated diseases: Inclusion body myopathy with Paget’s disease of bone, frontotemporal dementia, and amyotrophic lateral sclerosis. J. Mol. Neurosci. 2011, 45, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Weihl, C.C.; Temiz, P.; Miller, S.E.; Watts, G.; Smith, C.; Forman, M.; Hanson, P.I.; Kimonis, V.; Pestronk, A. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1186–1189. [Google Scholar] [CrossRef] [PubMed]
- Weihl, C.C.; Pestronk, A.; Kimonis, V.E. Valosin-containing protein disease: Inclusion body myopathy with Paget’s disease of the bone and fronto-temporal dementia. Neuromuscul. Disord. 2009, 19, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Lizano, P.; Rashed, E.; Kang, H.; Dai, H.; Sui, X.; Yan, L.; Qiu, H.; Depre, C. The valosin-containing protein promotes cardiac survival through the inducible isoform of nitric oxide synthase. Cardiovasc. Res. 2013, 99, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Hung, W.T.; Chang, H.C.; Wu, C.L.; Chiang, A.S.; Jackson, G.R.; Sang, T.K. Pathogenic VCP/TER94 alleles are dominant actives and contribute to neurodegeneration by altering cellular ATP level in a Drosophila IBMPFD model. PLoS Genet. 2011, 7, e1001288. [Google Scholar] [CrossRef] [PubMed]
- Ritson, G.P.; Custer, S.K.; Freibaum, B.D.; Guinto, J.B.; Geffel, D.; Moore, J.; Tang, W.; Winton, M.J.; Neumann, M.; Trojanowski, J.Q.; et al. TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J. Neurosci. 2010, 30, 7729–7739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.C.; Tresse, E.; Kolaitis, R.M.; Molliex, A.; Thomas, R.E.; Alami, N.H.; Wang, B.; Joshi, A.; Smith, R.B.; Ritson, G.P.; et al. VCP is essential for mitochondrial quality control by pink1/parkin and this function is impaired by VCP mutations. Neuron 2013, 78, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.J.; Chang, Y.C.; Chang, H.C.; Wang, C.K.; Hung, Y.C.; Lin, Y.E.; Chan, C.C.; Chen, C.H.; Chang, H.Y.; Sang, T.K. Derlin-1 regulates mutant VCP-linked pathogenesis and endoplasmic reticulum stress-induced apoptosis. PLoS Genet. 2014, 10, e1004675. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.T.; Lee, T.R.; Huang, S.H.; Lee, H.Y.; Sang, T.K.; Chan, H.L.; Lyu, P.C. Proteomic analysis of a Drosophila IBMPFD model reveals potential pathogenic mechanisms. Mol. Biosyst. 2012, 8, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Pinter, M.; Jekely, G.; Szepesi, R.J.; Farkas, A.; Theopold, U.; Meyer, H.E.; Lindholm, D.; Nassel, D.R.; Hultmark, D.; Friedrich, P. TER94, a Drosophila homolog of the membrane fusion protein CDC48/p97, is accumulated in nonproliferating cells: In the reproductive organs and in the brain of the imago. Insect Biochem. Mol. Biol. 1998, 28, 91–98. [Google Scholar] [CrossRef]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar] [PubMed]
- Finch, C.E.; Ruvkun, G. The genetics of aging. Annu. Rev. Genom. Hum. Genet. 2001, 2, 435–462. [Google Scholar] [CrossRef] [PubMed]
- Boulianne, G.L. Neuronal regulation of lifespan: Clues from flies and worms. Mech. Ageing Dev. 2001, 122, 883–894. [Google Scholar] [CrossRef]
- Cammarato, A.; Ahrens, C.H.; Alayari, N.N.; Qeli, E.; Rucker, J.; Reedy, M.C.; Zmasek, C.M.; Gucek, M.; Cole, R.N.; Van Eyk, J.E.; et al. A mighty small heart: The cardiac proteome of adult Drosophila melanogaster. PLoS ONE 2011, 6, e18497. [Google Scholar] [CrossRef] [PubMed]
- Neely, G.G.; Kuba, K.; Cammarato, A.; Isobe, K.; Amann, S.; Zhang, L.; Murata, M.; Elmen, L.; Gupta, V.; Arora, S.; et al. A global in vivo Drosophila RNAi screen identifies NOT3 as a conserved regulator of heart function. Cell 2010, 141, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Tricoire, H.; Palandri, A.; Bourdais, A.; Camadro, J.-M.; Monnier, V. Methylene blue rescues heart defects in a Drosophila model of Friedreich’s ataxia. Hum. Mol. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- Landsverk, M.L.; Li, S.; Hutagalung, A.H.; Najafov, A.; Hoppe, T.; Barral, J.M.; Epstein, H.F. The UNC-45 chaperone mediates sarcomere assembly through myosin degradation in Caenorhabditis elegans. J. Cell Biol. 2007, 177, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Ritz, D.; Vuk, M.; Kirchner, P.; Bug, M.; Schutz, S.; Hayer, A.; Bremer, S.; Lusk, C.; Baloh, R.H.; Lee, H.; et al. Endolysosomal sorting of ubiquitylated caveolin-1 is regulated by VCP and Ubxd1 and impaired by VCP disease mutations. Nat. Cell Biol. 2011, 13, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Janiesch, P.C.; Kim, J.; Mouysset, J.; Barikbin, R.; Lochmuller, H.; Cassata, G.; Krause, S.; Hoppe, T. The ubiquitin-selective chaperone CDC-48/p97 links myosin assembly to human myopathy. Nat. Cell Biol. 2007, 9, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.-S.; Miller, S.E.; Hanson, P.I.; Weihl, C.C. Impaired protein aggregate handling and clearance underlie the pathogenesis of p97/VCP-associated disease. J. Biol. Chem. 2008, 283, 30289–30299. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.-S.; Fuentealba, R.A.; Miller, S.E.; Jackson, E.; Piwnica-Worms, D.; Baloh, R.H.; Weihl, C.C. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 2009, 187, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Cammarato, A.; Dambacher, C.M.; Knowles, A.F.; Kronert, W.A.; Bodmer, R.; Ocorr, K.; Bernstein, S.I. Myosin transducer mutations differentially affect motor function, myofibril structure, and the performance of skeletal and cardiac muscles. Mol Biol Cell 2008, 19, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, M.C.; Kaushik, G.; Engler, A.J.; Lehman, W.; Cammarato, A. A Drosophila melanogaster model of diastolic dysfunction and cardiomyopathy based on impaired troponin-T function. Circ. Res. 2014, 114, e6–e17. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Saiz, V.; Buchberger, A. Imbalances in p97 co-factor interactions in human proteinopathy. EMBO Rep. 2010, 11, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Alayari, N.N.; Vogler, G.; Taghli-Lamallem, O.; Ocorr, K.; Bodmer, R.; Cammarato, A. Fluorescent labeling of Drosophila heart structures. J. Vis. Exp. 2009. [Google Scholar] [CrossRef] [PubMed]
- Vogler, G.; Ocorr, K. Visualizing the beating heart in Drosophila. J. Vis. Exp. 2009, 31, e1425. [Google Scholar]
- Fink, M.; Callol-Massot, C.; Chu, A.; Ruiz-Lozano, P.; Izpisua Belmonte, J.C.; Giles, W.; Bodmer, R.; Ocorr, K. A new method for detection and quantification of heartbeat parameters in Drosophila, zebrafish, and embryonic mouse hearts. Biotechniques 2009, 46, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Cammarato, A.; Ocorr, S.; Ocorr, K. Enhanced assessment of contractile dynamics in Drosophila hearts. Biotechniques 2015, 58, 77–80. [Google Scholar] [PubMed]
- Harlow, E.; Lane, D. Preparing early whole-mount Drosophila embryos for immunostaining. CSH Protoc. 2006, 2006. [Google Scholar] [CrossRef] [PubMed]







© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viswanathan, M.C.; Blice-Baum, A.C.; Sang, T.-K.; Cammarato, A. Cardiac-Restricted Expression of VCP/TER94 RNAi or Disease Alleles Perturbs Drosophila Heart Structure and Impairs Function. J. Cardiovasc. Dev. Dis. 2016, 3, 19. https://doi.org/10.3390/jcdd3020019
Viswanathan MC, Blice-Baum AC, Sang T-K, Cammarato A. Cardiac-Restricted Expression of VCP/TER94 RNAi or Disease Alleles Perturbs Drosophila Heart Structure and Impairs Function. Journal of Cardiovascular Development and Disease. 2016; 3(2):19. https://doi.org/10.3390/jcdd3020019
Chicago/Turabian StyleViswanathan, Meera C., Anna C. Blice-Baum, Tzu-Kang Sang, and Anthony Cammarato. 2016. "Cardiac-Restricted Expression of VCP/TER94 RNAi or Disease Alleles Perturbs Drosophila Heart Structure and Impairs Function" Journal of Cardiovascular Development and Disease 3, no. 2: 19. https://doi.org/10.3390/jcdd3020019