
Estimation of the Whitefly Bemisia tabaci Genome Size Based on k-mer and Flow Cytometric Analyses

Abstract
:1. Introduction
2. Results

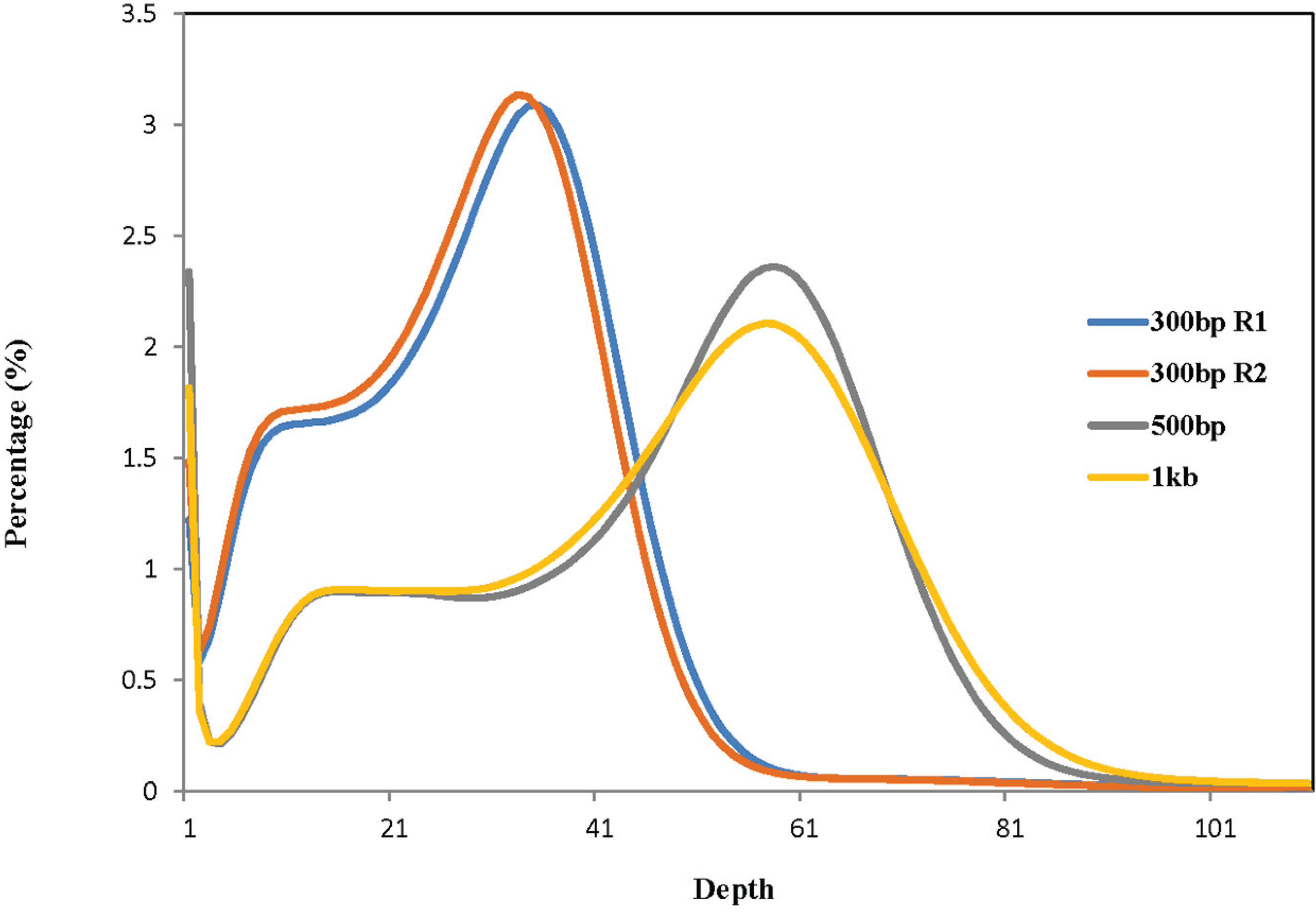{kind=link}
{kind=link}
| Library | Total High-Quality Cleaned Bases | Total Number of Corrected 27-mers | Peak Value of 27-mer Depth | Estimated Genome Size (bp) b |
|---|---|---|---|---|
| 300 bp R1 a | 28,576,003,381 | 23,334,888,827 | 35 | 666,711,109 |
| 300 bp R2 a | 27,654,048,868 | 22,412,934,314 | 33 | 679,179,828 |
| 500 bp | 48,793,881,921 | 39,521,025,705 | 58 | 681,396,995 |
| 1 Kb | 49,456,289,287 | 40,032,313,271 | 57 | 702,321,285 |

| Replicate | Sample | Standard a | DNA Content (pg) | |
|---|---|---|---|---|
| Male (haploid) | 1 | 100.80 | 343.51 | 0.73 |
| 2 | 103.20 | 366.74 | 0.70 | |
| 3 | 112.17 | 396.05 | 0.71 | |
| 4 | 116.46 | 419.92 | 0.69 | |
| Mean ± SD | 0.7075 ± 0.0171 | |||
| Coefficient of variation | 0.024 | |||
| Female (diploid) | 1 | 190.21 | 343.96 | 1.38 |
| 2 | 205.07 | 370.81 | 1.38 | |
| 3 | 230.07 | 399.54 | 1.44 | |
| 4 | 243.95 | 423.25 | 1.44 | |
| Mean ± SD | 1.41 ± 0.0346 | |||
| Coefficient of variation | 0.025 |

3. Discussion
4. Materials and Methods
4.1. Insect Rearing
4.2. DNA Extraction from Male Whiteflies
4.3. Genome Size Estimation by k-mer Analysis
4.4. Nuclear DNA Content Estimation by Flow Cytometry
5. Conclusions
Acknowledgements
Author Contributions
Conflict of Interests
References
- Martin, J.H.; Mound, L.A. An annotated check list of the world’s whiteflies (Insecta: Hemiptera: Aleyrodidae). Zootaxa 2007, 1492, 1–84. [Google Scholar]
- Abd-Rabou, S.; Simmons, A.M. Survey of reproductive host plants of Bemisia tabaci (Hemiptera: Aleyrodidae) in Egypt, including new host records. Entomol. News 2010, 121, 456–465. [Google Scholar] [CrossRef]
- Brown, J.K. Current status of Bemisia tabaci as a plant pest and virus vector in agroecosystems worldwide. FAO Plant Prot. Bull. 1994, 42, 3–32. [Google Scholar]
- Jones, D.R. Plant viruses transmitted by whiteflies. Eur. J. Plant Pathol. 2003, 109, 195–219. [Google Scholar] [CrossRef]
- Legg, J.P.; Owor, B.; Sseruwagi, P.; Ndunguru, J. Cassava mosaic virus disease in East and Central Africa: Epidemiology and management of a regional pandemic. Adv. Virus Res. 2006, 67, 355–418. [Google Scholar] [PubMed]
- Legg, J.P. Epidemiology of a whitefly-transmitted cassava mosaic geminivirus pandemic in Africa. In Bemisia: Bionomics and Management of a Global Pest; Stansly, P.A., Naranjo, S.E., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 233–257. [Google Scholar]
- Diaz-Pendon, J.A.; Canizares, M.C.; Moriones, E.; Bejarano, E.R.; Czosnek, H.; Navas-Castillo, J. Tomato yellow leaf curl viruses: Menage a trois between the virus complex, the plant and the whitefly vector. Mol. Plant Pathol. 2010, 11, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, I.M.; Lapidot, M.; Thomma, B.P. Emerging viral diseases of tomato crops. Mol. Plant Microbe Interact. 2010, 23, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Navas-Castillo, J.; Fiallo-Olive, E.; Sanchez-Campos, S. Emerging virus diseases transmitted by whiteflies. Annu. Rev. Phytopathol. 2011, 49, 219–248. [Google Scholar] [CrossRef] [PubMed]
- Van der Vlugt, R.A.; Verbeek, M.; Dullemans, A.M.; Wintermantel, W.M.; Cuellar, W.J.; Fox, A.; Thompson, J.R. Torradoviruses. Annu. Rev. Phytopathol. 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Czosnek, H.; Ghanim, M. Back to basics: Are Begomoviruses whitefly pathogens? J. Integr. Agric. 2012, 11, 225–234. [Google Scholar] [CrossRef]
- Morin, S.; Ghanim, M.; Zeidan, M.; Czosnek, H.; Verbeek, M.; van den Heuvel, J.F.J.M. A GroEL homologue from endosymbiotic bacteria of the whitefly Bemisia tabaci is implicated in the circulative transmission of Tomato yellow leaf curl virus. Virology 1999, 256, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Morin, S.; Ghanim, M.; Sobol, I.; Czosnek, H. The GroEL protein of the whitefly Bemisia tabaci interacts with the coat protein of transmissible and nontransmissible begomoviruses in the yeast two-hybrid system. Virology 2000, 276, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, Y.; Zchori-Fein, E.; Mozes-Daube, N.; Kontsedalov, S.; Skaljac, M.; Brumin, M.; Sobol, I.; Czosnek, H.; Vavre, F.; Fleury, F.; et al. The transmission efficiency of Tomato yellow leaf curl virus by the whitefly Bemisia tabaci is correlated with the presence of a specific symbiotic bacterium species. J. Virol. 2010, 84, 9310–9317. [Google Scholar] [CrossRef] [PubMed]
- Kliot, A.; Ghanim, M. The role of bacterial chaperones in the circulative transmission of plant viruses by insect vectors. Viruses 2013, 5, 1516–1535. [Google Scholar] [CrossRef] [PubMed]
- Ghanim, M. A review of the mechanisms and components that determine the transmission efficiency of Tomato yellow leaf curl virus (Geminiviridae; Begomovirus) by its whitefly vector. Virus Res. 2014, 186, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Avidov, Z. Bionomics of the tobacco whitefly (Bemisia tabaci Gennad.) in Israel. Ktavim 1956, 7, 25–41. [Google Scholar]
- Azab, A.K.; Megahed, M.M.; El-Mirsawi, H.D. On the biology of Bemisia tabaci (Genn.) (Hemiptera-Homoptera: Aleyrodidae). Bull. Entomol. Soc. Egypte 1971, 55, 305–315. [Google Scholar]
- Bethke, J.A.; Paine, T.D.; Nuessly, G.S. Comparative biology, morphometrics, and development of two populations of Bemisia tabaci (Homoptera: Aleyrodidae) on cotton and poinsettia. Ann. Entomol. Soc. Am. 1991, 84, 407–411. [Google Scholar] [CrossRef]
- Schrader, F. Sex determination in the white-fly (Trialeurodes vaporariorum). J. Morphol. 1920, 34, 266–305. [Google Scholar] [CrossRef]
- Blackman, R.L.; Cahill, M. The karyotype of Bemisia tabaci (Hemiptera: Aleyrodidae). Bull. Entomol. Res. 1998, 88, 213–215. [Google Scholar] [CrossRef]
- Costa, H.S.; Westcot, D.M.; Ullman, D.E.; Johnson, M.W. Ultrastructure of the endosymbionts of the whitefly, Bemisia tabaci and Trialeurodes vaporariorum. Protoplasma 1993, 176, 106–115. [Google Scholar] [CrossRef]
- Costa, H.S.; Westcot, D.M.; Ullman, D.E.; Rosell, R.; Brown, J.B.; Johnson, M.W. Morphological variation in Bemisia endosymbionts. Protoplasma 1995, 189, 194–202. [Google Scholar] [CrossRef]
- Szklarzewicz, T.; Moskal, A. Ultrastructure, distribution, and transmission of endosymbionts in the whitefly Aleurochiton aceris Modeer (Insecta, Hemiptera, Aleyrodinea). Protoplasma 2001, 218, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P. Biology of bacteriocyte-associated endosymbionts of plant sap-sucking insects. Ann. Rev. Microbiol. 2005, 59, 155–189. [Google Scholar] [CrossRef] [PubMed]
- Chiel, E.; Gottlieb, Y.; Zchori-Fein, E.; Mozes-Daube, N.; Katzir, N.; Inbar, M.; Ghanim, M. Biotype-dependent secondary symbiont communities in sympatric populations of Bemisia tabaci. Bull. Entomol. Res. 2007, 97, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Bing, X.L.; Ruan, Y.M.; Rao, Q.; Wang, X.W.; Liu, S.S. Diversity of secondary endosymbionts among different putative species of the whitefly Bemisia tabaci. Insect Sci. 2013, 20, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Zchori-Fein, E.; Lahav, T.; Freilich, S. Variations in the identity and complexity of endosymbiont combinations in whitefly hosts. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Zang, L.S.; Chen, W.Q.; Liu, S.S. Comparison of performance on different host plants between the B biotype and a non-B biotype of Bemisia tabaci from Zhejiang, China. Entomol. Exp. Appl. 2006, 121, 221–227. [Google Scholar] [CrossRef]
- Houndété, T.A.; Kétoh, G.K.; Hema, O.S.; Brévault, T.; Glitho, I.A.; Martin, T. Insecticide resistance in field populations of Bemisia tabaci (Hemiptera: Aleyrodidae) in West Africa. Pest Manag. Sci. 2010, 66, 1181–1185. [Google Scholar]
- Bedford, I.D.; Briddon, R.W.; Brown, J.K.; Roswell, R.C.; Markham, P.G. Geminivirus transmission and biological characterisation of Bemisia tabaci (Gennadius) biotypes from different geographic regions. Ann. Appl. Biol. 1994, 125, 311–325. [Google Scholar] [CrossRef]
- Dinsdale, A.; Cook, L.; Riginos, C.; Buckley, Y.M.; de Barro, P.J. Refined global analysis of Bemisia tabaci (Hemiptera: Sternorrhyncha: Aleyrodoidea: Aleyrodidae) mitochondrial cytochrome oxidase 1 to identify species level genetic boundaries. Ann. Entomol. Soc. Am. 2010, 103, 196–208. [Google Scholar] [CrossRef]
- Arumuganathan, K.; Earle, E.D. Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 1991, 9, 208–218. [Google Scholar] [CrossRef]
- Dolezel, J.; Greilhuber, J. Nuclear genome size: Are we getting closer? Cytometry A 2010, 77, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Shi, Y.; Yuan, J.; Hu, X.; Zhang, H.; Li, N.; Li, Z.; Chen, Y.; Mu, D.; Fan, W. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Available online: http://arxiv.org/ftp/arxiv/papers/1308/1308.2012.pdf (accessed on 5 March 2015).
- Brown, J.K.; Lambert, G.M.; Ghanim, M.; Czosnek, H.; Galbraith, D.W. Nuclear DNA content of the whitefly Bemisia tabaci (Aleyrodidae: Hemiptera) estimated by flow cytometry. Bull. Entomol. Res. 2005, 95, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wang, S.; Wu, Q.; Zhou, X.; Xie, W.; Zhang, Y. Flow cytometry and K-mer analysis estimates of the genome sizes of Bemisia tabaci B and Q (Hemiptera: Aleyrodidae). Front. Physiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.D.; Celniker, S.E.; Holt, R.A.; Evans, C.A.; Gocayne, J.D.; Amanatides, P.G.; Scherer, S.E.; Li, P.W.; Hoskins, R.A.; Galle, R.F.; et al. The genome sequence of Drosophila melanogaster. Science 2000, 287, 2185–2195. [Google Scholar] [CrossRef] [PubMed]
- Holt, R.A.; Subramanian, G.M.; Halpern, A.; Sutton, G.G.; Charlab, R.; Nusskern, D.R.; Wincker, P.; Clark, A.G.; Ribeiro, J.M.; Wides, R.; et al. The genome sequence of the malaria mosquito Anopheles gambiae. Science 2002, 298, 129–149. [Google Scholar] [CrossRef] [PubMed]
- The International Aphid Genomics Consortium. Genome sequence of the pea aphid Acyrthosiphon pisum. PLoS Biol 2010, 8, e1000313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mita, K.; Kasahara, M.; Sasaki, S.; Nagayasu, Y.; Yamada, T.; Kanamori, H.; Namiki, N.; Kitagawa, M.; Yamashita, H.; Yasukochi, Y.; et al. The genome sequence of silkworm, Bombyx mori. DNA Res. 2004, 11, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Kelley, J.L.; Peyton, J.T.; Fiston-Lavier, A.S.; Teets, N.M.; Yee, M.C.; Bustamante, C.D.; Lee, R.E.; Denlinger, D.L. Compact genome of the Antarctic midge is likely an adaptation to an extreme environment. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fang, X.; Yang, P.; Jiang, X.; Jiang, F.; Zhao, D.; Li, B.; Cui, F.; Wei, J.; Ma, C.; et al. The locust genome provides insight into swarm formation and long-distance flight. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Shatters, R.G., Jr.; Powell, C.A.; Boykin, L.M.; Liansheng, H.; McKenzie, C.L. Improved DNA barcoding method for Bemisia tabaci and related Aleyrodidae: Development of universal and Bemisia tabaci biotype-specific mitochondrial cytochrome c oxidase I polymerase chain reaction primers. J. Econ. Entomol. 2009, 102, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.; Anders, S.; Lawrence, M.; Aboyoun, P.; Pagès, H.; Gentleman, R. ShortRead: A Bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics 2009, 25, 2607–2608. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.R.; Schatz, M.C.; Salzberg, S.L. Quake: Quality-aware detection and correction of sequencing errors. Genome Biol. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Waterman, M.S. Estimating the repeat structure and length of DNA sequences using ℓ-tuples. Genome Res. 2003, 13, 1916–1922. [Google Scholar] [PubMed]
- Simmons, A.M.; Elsey, K.D. Overwintering and cold tolerance of Bemisia argentifolii (Homoptera: Aleyrodidae) in coastal South Carolina. J. Entomol. Sci. 1995, 30, 497–506. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.; Hasegawa, D.K.; Arumuganathan, K.; Simmons, A.M.; Wintermantel, W.M.; Fei, Z.; Ling, K.-S. Estimation of the Whitefly Bemisia tabaci Genome Size Based on k-mer and Flow Cytometric Analyses. Insects 2015, 6, 704-715. https://doi.org/10.3390/insects6030704
Chen W, Hasegawa DK, Arumuganathan K, Simmons AM, Wintermantel WM, Fei Z, Ling K-S. Estimation of the Whitefly Bemisia tabaci Genome Size Based on k-mer and Flow Cytometric Analyses. Insects. 2015; 6(3):704-715. https://doi.org/10.3390/insects6030704
Chicago/Turabian StyleChen, Wenbo, Daniel K. Hasegawa, Kathiravetpillai Arumuganathan, Alvin M. Simmons, William M. Wintermantel, Zhangjun Fei, and Kai-Shu Ling. 2015. "Estimation of the Whitefly Bemisia tabaci Genome Size Based on k-mer and Flow Cytometric Analyses" Insects 6, no. 3: 704-715. https://doi.org/10.3390/insects6030704