
Investigating Monophyly of Typhlocybini Based on Complete Mitochondrial Genomes with Characterization and Comparative Analysis of 19 Species (Hemiptera: Cicadellidae: Typhlocybinae)

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Mitogenome Sequence Analysis
2.3. Phylogenetic Analysis
3. Results
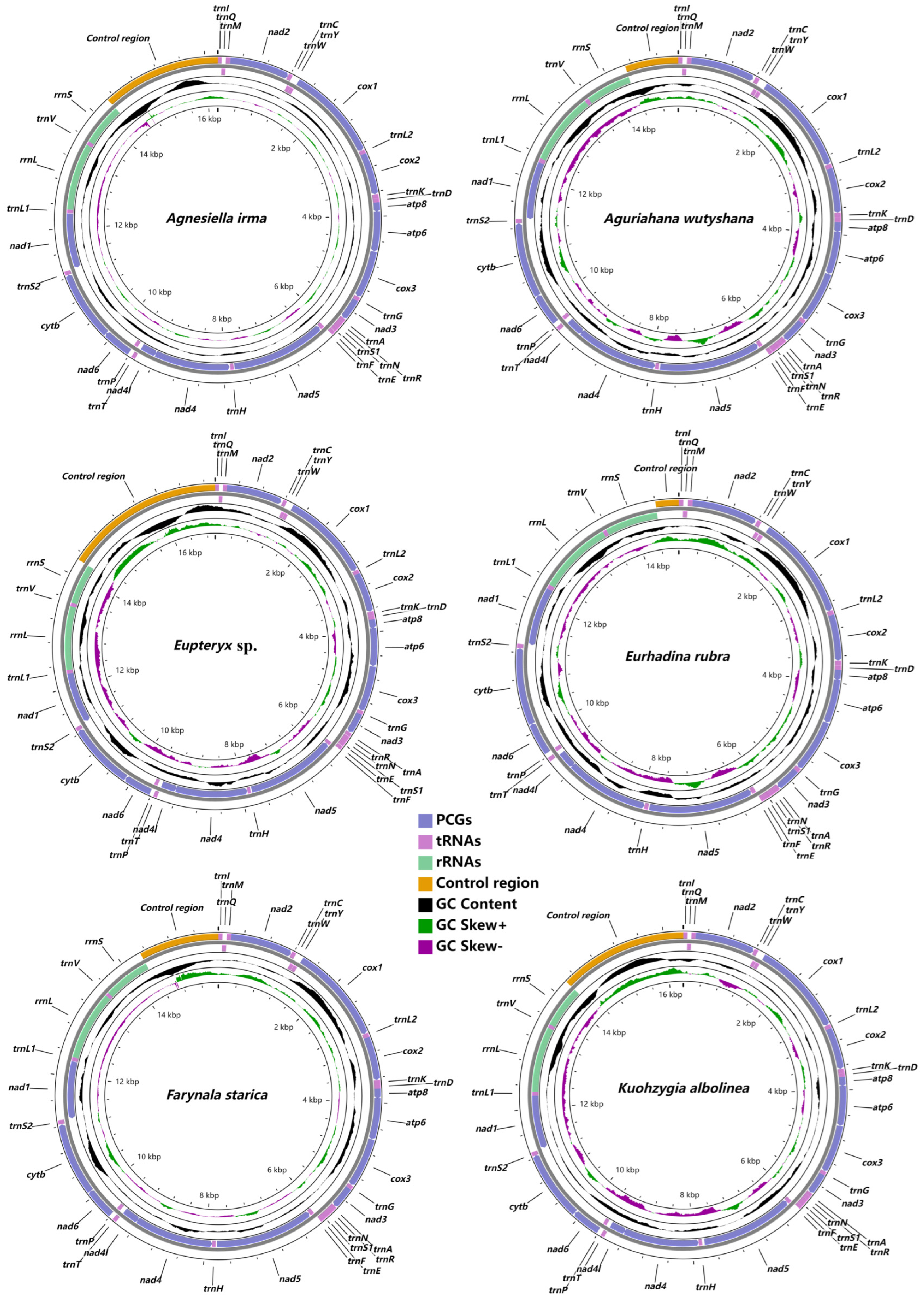
3.1. Mitogenome Organization and Gene Content
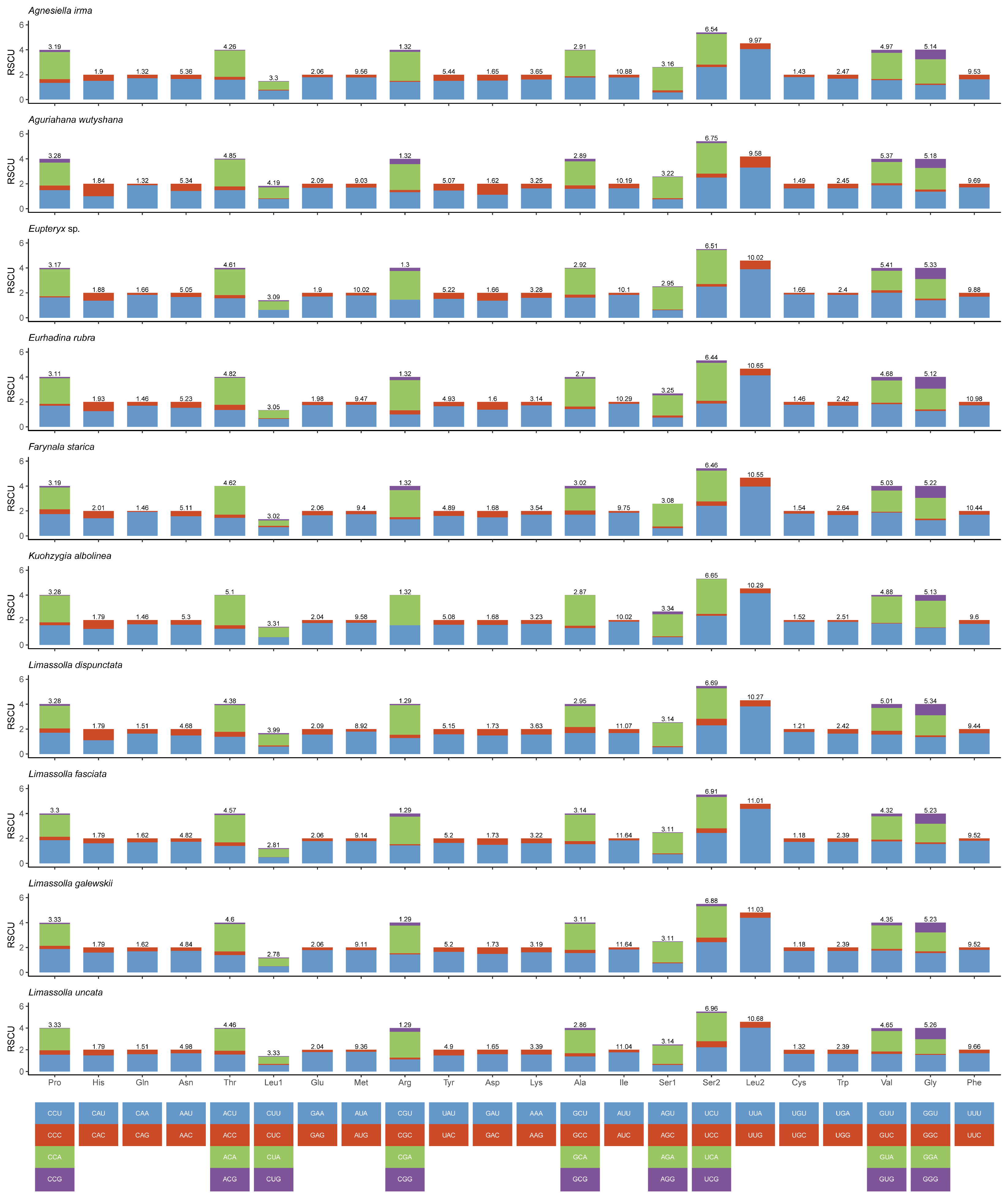
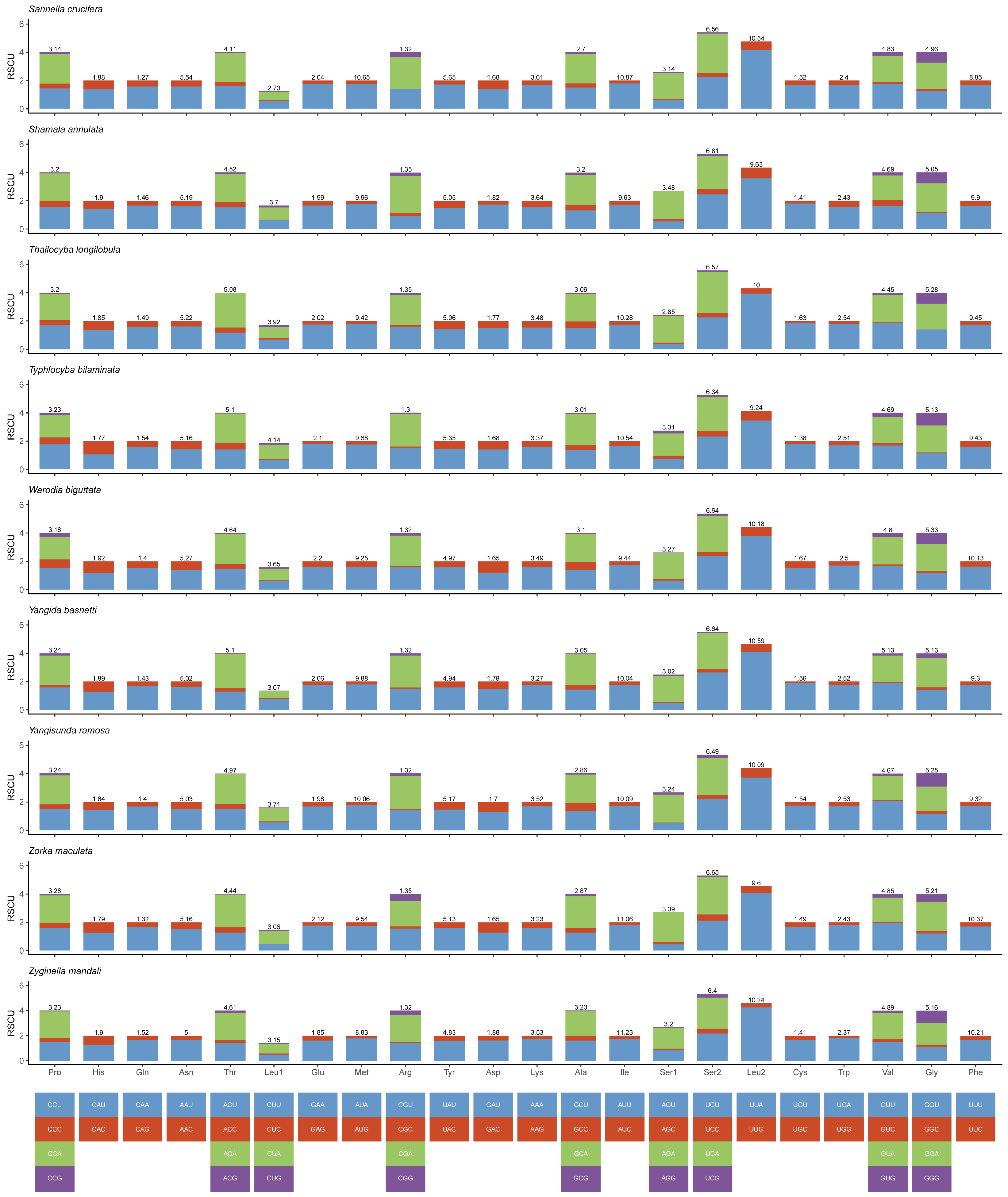
3.2. Protein-Coding Genes and Codon Usage
3.3. Transfer and Ribosomal RNA Genes
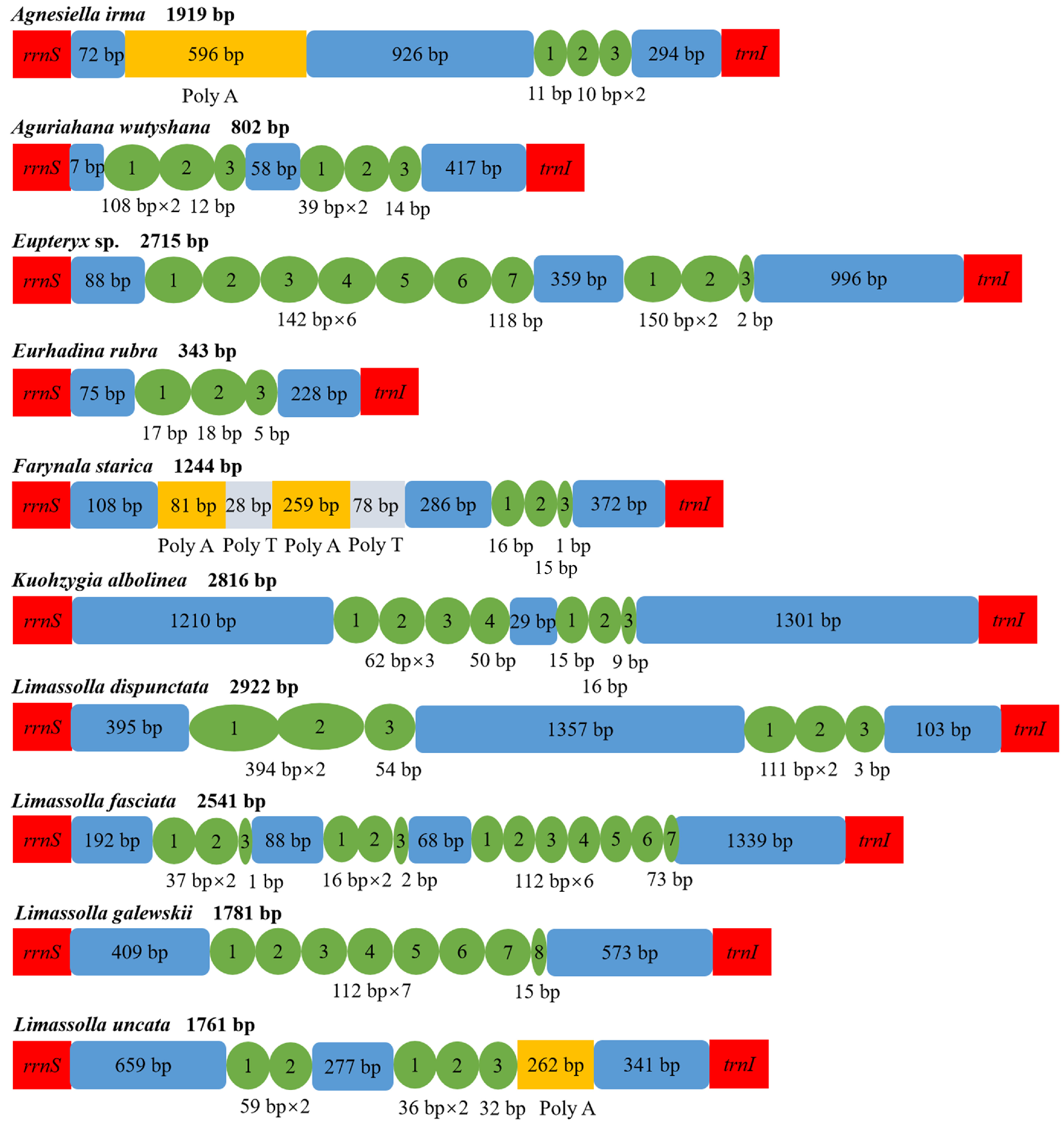
3.4. Control Region
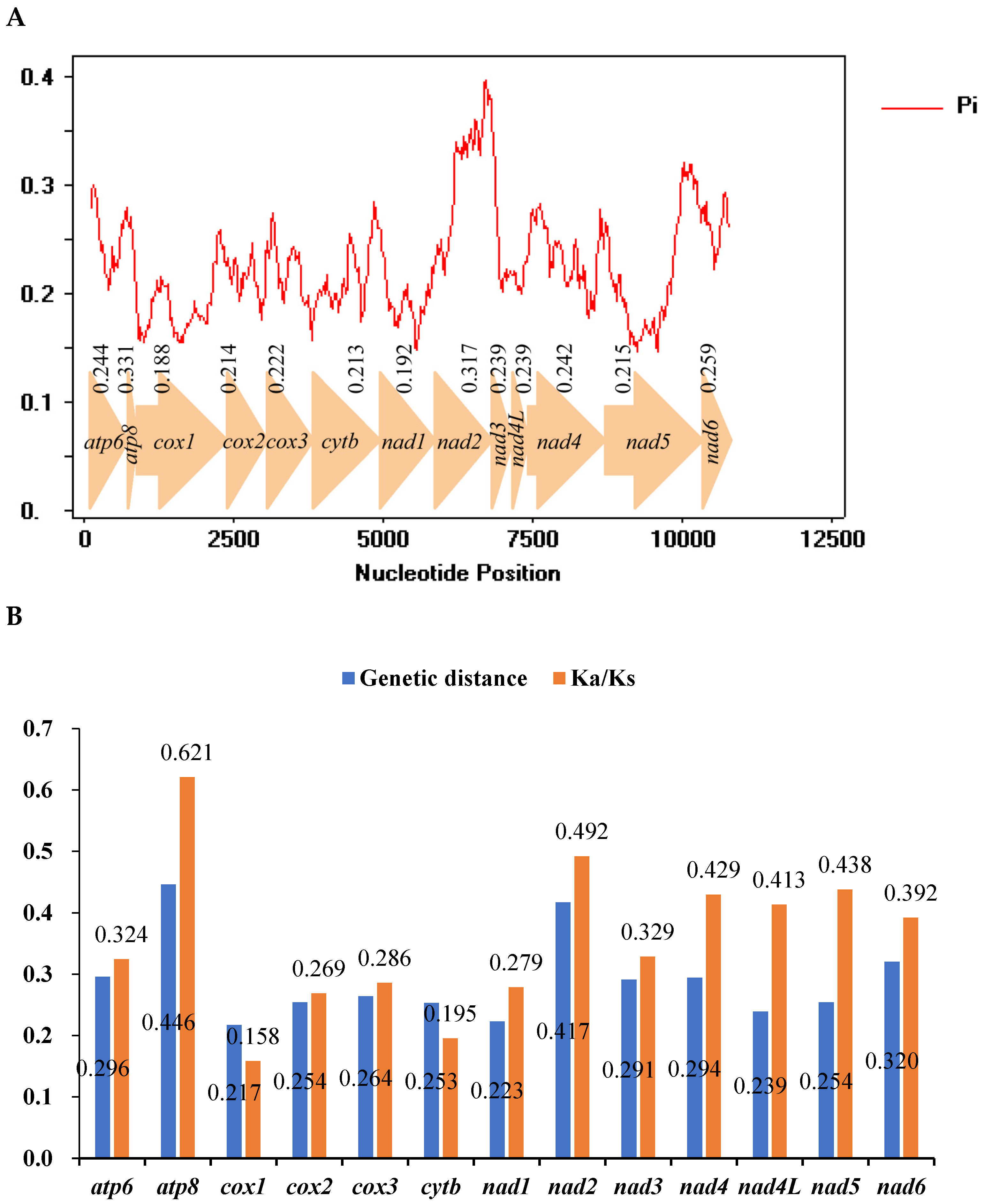
3.5. Nucleotide Diversity and Evolutionary Rate Analysis
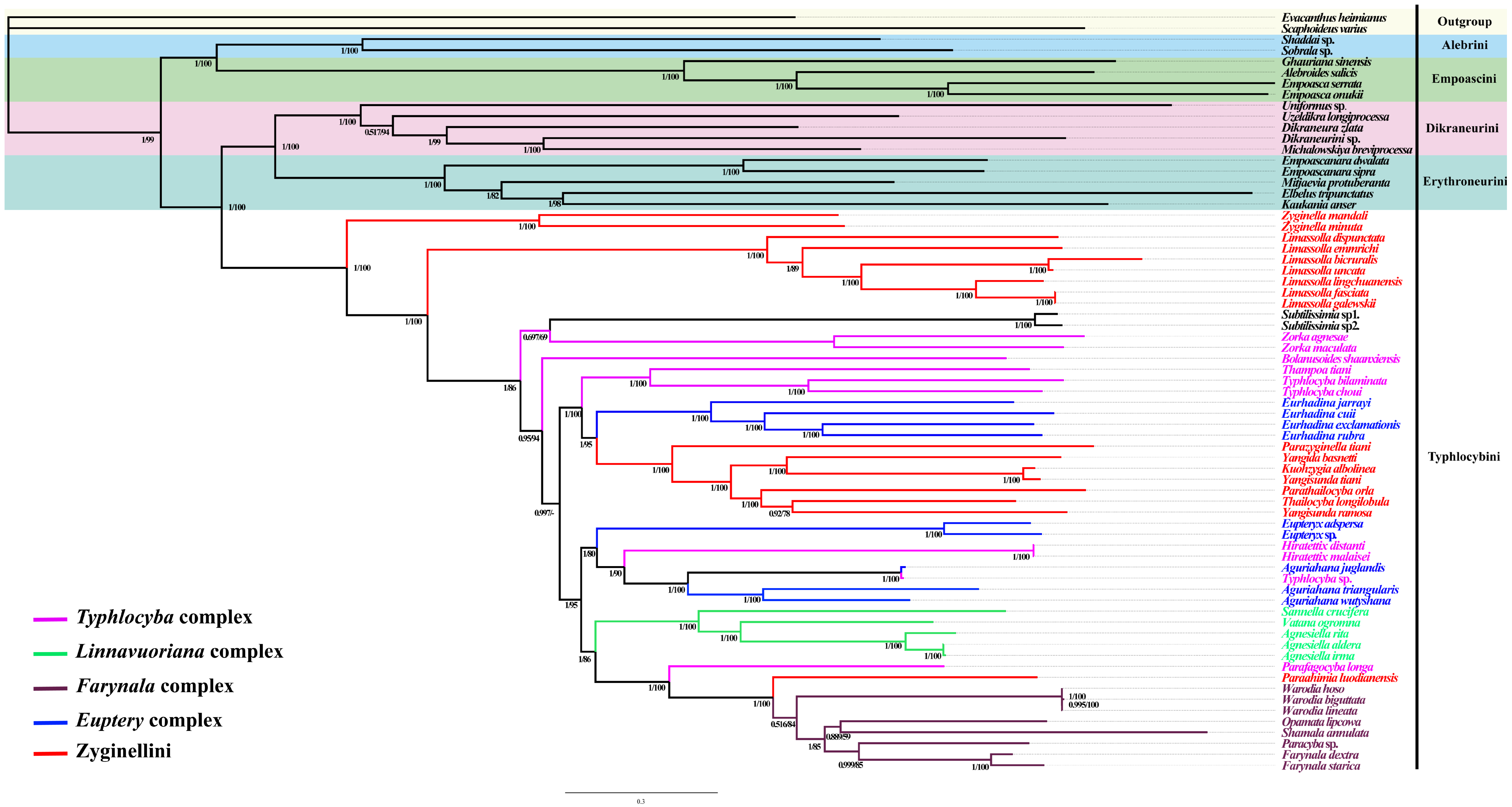
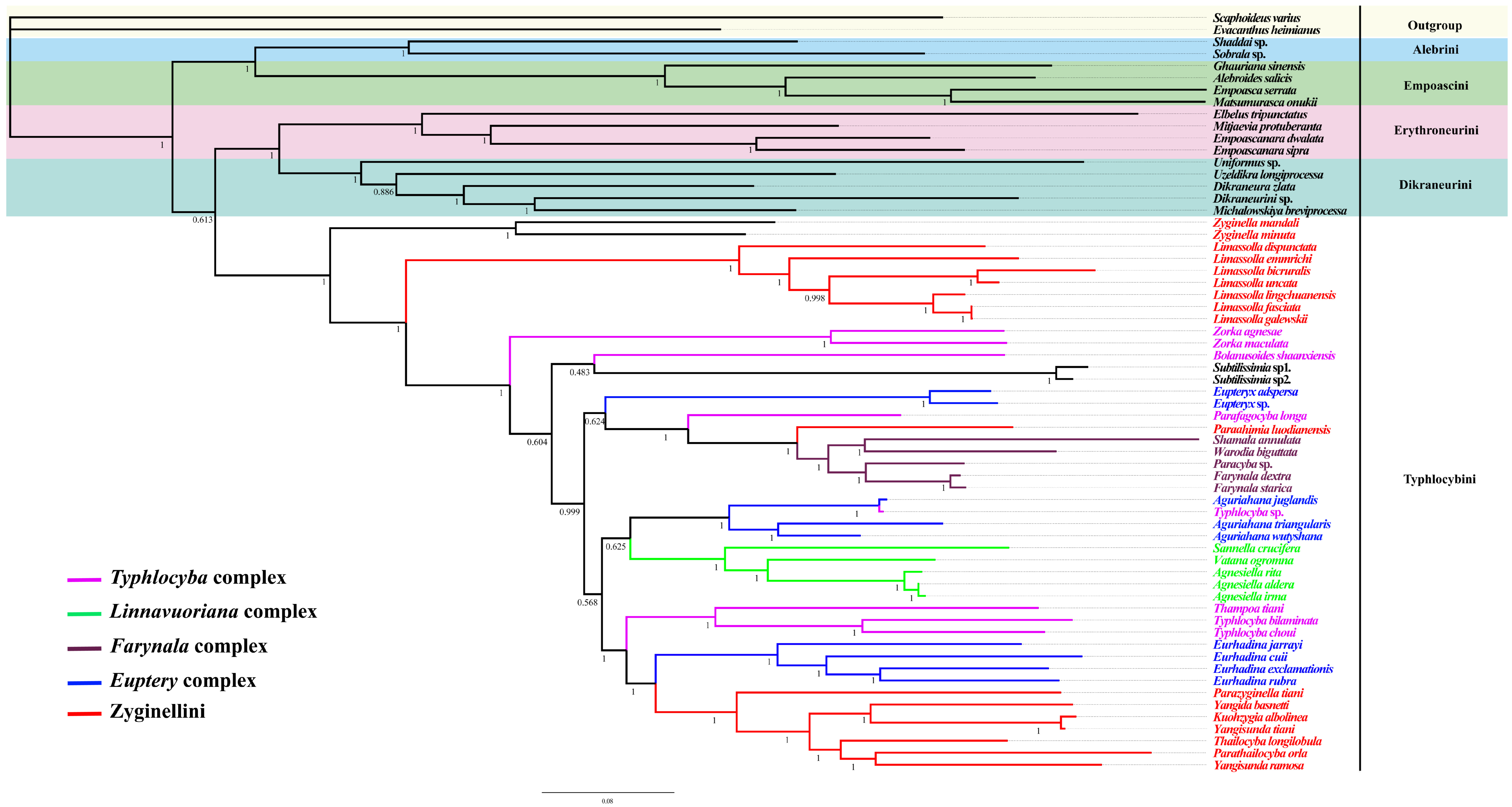
3.6. Phylogenetic Relationships
4. Discussion
4.1. Comparative Analysis of Typhlocybine Mitogenomes
4.2. The Monophyly of Typhlocybini
4.3. The Definition of Genera Complexes in Typhlocybini
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hamilton, K.G.A. New world species of Chlorita, Notus, and Forcipata (Rhynchota: Homoptera: Cicadellidae: Typhlocybinae) with a new tribe Forcipatini. Can. Entomol. 1998, 130, 491–507. [Google Scholar] [CrossRef]
- Balme, G.R. Phylogeny and Systematics of the Leafhopper Subfamily Typhlocybinae (Insecta: Hemiptera: Typhlocybinae). Ph.D. Thesis, North Carolina State University, Raleigh, NC, USA, 2007. [Google Scholar]
- Dietrich, C.H. South American leafhoppers of the tribe Typhlocybini (Hemiptera: Cicadellidae: Typhlocybinae). Zoologia 2013, 30, 519–568. [Google Scholar] [CrossRef]
- Dietrich, C.H.; Dmitriev, D.A. Review of the New World genera of the leafhopper tribe Erythroneurini (Hemiptera: Cicadellidae: Typhlocybinae). Bull. Ill. Nat. Hist. Surv. 2006, 37, 119–190. [Google Scholar] [CrossRef]
- Chasen, E.M.; Dietrich, C.H.; Backus, E.A.; Cullen, E.M. Potato leafhopper (Hemiptera: Cicadellidae) ecology and integrated pest management focused on alfalfa. J. Integr. Pest Manag. 2014, 5, A1–A8. [Google Scholar] [CrossRef]
- Jarrell, K.R.; Rebek, E.J.; Wayadande, A.C.; Giles, K.L. Biology, ecology, and management of eastern grape leafhopper (Hemiptera: Cicadellidae), a key pest of vineyards in north America. J. Integr. Pest Manag. 2020, 11, 6. [Google Scholar] [CrossRef]
- Pinedo-Escatel, J.A.; Dmitriev, D. Redescription of the Dikraneurini leafhopper Dikrella mella Ruppel & DeLong, 1952 (Hemiptera, Cicadellidae) with a synoptic checklist of leafhoppers on avocado trees in Mexico. ZooKeys 2019, 857, 33910. [Google Scholar]
- Xu, Y.; Wang, Y.R.; Dietrich, C.H.; Fletcher, M.J.; Qin, D.Z. Review of Chinese species of the leafhopper genus Amrasca Ghauri (Hemiptera, Cicadellidae, Typhlocybinae), with description of a new species, species checklist and notes on the identity of the Indian cotton leafhopper. Zootaxa 2017, 4353, 360–370. [Google Scholar] [CrossRef]
- Dworakowska, I. The leafhopper tribe Zyginellini (Homoptera, Auchenorrhyncha, Cicadellidae, Typhlocybinae). Rev. Zool. Afr. 1979, 93, 299–331. [Google Scholar]
- Dworakowska, I. On some Typhlocybinae from Vietnam (Homoptera: Cicadellidae). Folia Entomol. Hungarica. 1977, 30, 9–47. [Google Scholar]
- Lin, S.H.; Huang, M.; Zhang, Y.L. Structural Features and Phylogenetic Implications of 11 New Mitogenomes of Typhlocybinae (Hemiptera: Cicadellidae). Insects 2021, 12, 678. [Google Scholar] [CrossRef]
- Cao, Y.H.; Dietrich, C.H.; Kits, J.H.; Richter, R.; Dmitriev, D.A.; Eyres, J.; Dettman, J.R.; Xu, Y.; Huang, M. Phylogenomics of microleafhoppers (Hemiptera: Cicadellidae: Typhlocybinae): Morphological evolution, divergence times, and biogeography. Insect Syst. Diver. 2023, 7. [Google Scholar] [CrossRef]
- Lu, L.; Dietrich, C.H.; Cao, Y.H.; Zhang, Y.L. A multi-gene phylogenetic analysis of the leafhopper subfamily Typhlocybinae (Hemiptera: Cicadellidae) challenges the traditional view of the evolution of wing venation. Mol. Phylogenet. Evol. 2021, 165, 107299. [Google Scholar] [CrossRef] [PubMed]
- Huang, M. Systematic Study of Typhlocybini (Hemiptera: Cicadellidae: Typhlocybinae). Ph.D. Thesis, Northwest A&F University, Xianyang, China, 2003. (In Chinese). [Google Scholar]
- Yan, B. Study on Taxonomy and Molecular Systematics of Typhlocybini from China (Hemiptera: Cicadellidae: Typhlocybinae). Ph.D. Thesis, Guizhou University, Guiyang, China, 2019. (In Chinese). [Google Scholar]
- Kim, M.J.; Kang, A.R.; Jeong, H.C.; Kim, K.G.; Kim, I. Reconstructing intraordinal relationships in Lepidoptera using mitochondrial genome data with the description of two newly sequenced Lycaenids, Spindasis takanonis and Protantigius superans (Lepidoptera: Lycaenidae). Mol. Phylogenet. Evol. 2011, 61, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.N.; Penton, E.H.; Burns, J.M.; Janzen, D.H.; Hallwachs, W. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. USA 2004, 101, 14812–14817. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Song, W.; Shi, S.L.; Liu, Y.Q.; Pan, M.H.; Dai, F.Y.; Lu, C.; Xiang, Z.H. Mitochondrial genome nucleotide substitution pattern between domesticated silkmoth, Bombyx mori, and its wild ancestors, Chinese Bombyx mandarina and Japanese Bombyx mandarina. Genet. Mol. Biol. 2010, 33, 186–189. [Google Scholar] [CrossRef]
- Simon, C.; Hadrys, H. A comparative analysis of complete mitochondrial genomes among Hexapoda. Mol. Phylogenet. Evol. 2013, 69, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Buckley, T.R.; Frati, F.; Stewart, J.B.; Beckenbach, A.T. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 545–579. [Google Scholar] [CrossRef]
- Saccone, C.; Giorgi, C.D.; Gissi, C.; Pesole, G.; Reyes, A. Evolutionary genomics in Metazoa: The mitochondrial DNA as a model system. Gene 1999, 238, 195–209. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Yang, X.; Yuan, Z.W.; Li, C.; Song, Y.H. Complete mitochondrial genome of Eupteryx (Stacla) minusula (Hemiptera: Cicadellidae: Typhlocybinae) from China. Mitochondrial DNA Part B 2020, 5, 2375–2376. [Google Scholar] [CrossRef]
- Yuan, X.W.; Xiong, K.N.; Li, C.; Song, Y.H. The complete mitochondrial genome of Limassolla lingchuanensis (Hemiptera: Cicadellidae: Typhlocybinae). Mitochondrial DNA Part B 2020, 5, 229–230. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Cai, W.Z.; Li, H. Deep-level phylogeny of Cicadomorpha inferred from mitochondrial genomes sequenced by NGS. Sci. Rep. 2017, 7, 10429. [Google Scholar] [CrossRef]
- Han, C.; Yan, B.; Yu, X.F.; Yang, M.F. Complete mitochondrial genome of Zyginella minuta (Cicadellidae: Typhlocybinae: Zyginellini) from China, with its phylogenetic analysis. Mitochondrial DNA Part B 2020, 5, 2795–2796. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Stothard, P. The CG View Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, 181–184. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.L.; Li, W.X.; Jakovlić, I.; Zou, H.; Zhang, J.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. bioRxiv 2018, 20, 489088. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870. [Google Scholar] [CrossRef]
- Shi, R.; Yu, X.F.; Yang, M.F. Complete mitochondrial genome of Ghauriana sinensis (Hemiptera: Cicadellidae: Typhlocybinae). Mitochondrial DNA Part B 2020, 5, 1367–1368. [Google Scholar] [CrossRef]
- Liu, J.H.; Sun, C.Y.; Long, J.; Guo, J.J. Complete mitogenome of tea green leafhopper, Empoasca onukii (Hemiptera: Cicadellidae) from Anshun, Guizhou Province in China. Mitochondrial DNA Part B 2017, 2, 808–809. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.W.; Li, C.; Song, Y.H. Characterization of the complete mitochondrial genome of Mitjaevia protuberanta (Hemiptera:Cicadellidae: Typhlocybinae). Mitochondrial DNA Part B 2020, 5, 601–602. [Google Scholar] [CrossRef]
- Chen, X.X.; Yuan, Z.W.; Li, C.; Song, Y.H. Complete mitochondrial genome sequence of Empoascanara dwalata (Hemiptera: Cicadellidae: Typhlocybinae). Mitochondrial DNA Part B 2020, 5, 2260–2261. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Chen, X.X.; Li, C.; Song, Y.H. The complete mitochondrial genome of Empoascanara sipra (Hemiptera: Cicadellidae: Typhlocybinae) with phylogenetic consideration. Mitochondrial DNA Part B 2020, 5, 260–261. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.H.; Yuan, X.W.; Li, C. The mitochondrial genome of Paraahimia luodianensis (Hemiptera: Cicadellidae: Typhlocybinae), a new genus and species from China. Mitochondrial DNA Part B 2020, 5, 1351–1352. [Google Scholar] [CrossRef]
- Jiang, J.; Yuan, X.W.; Yuan, Z.W.; Song, Y.H. The complete mitochondrial genome of Parathailocyba orla (Hemiptera: Cicadellidae: Typhlocybinae). Mitochondrial DNA Part B 2020, 5, 1981–1982. [Google Scholar] [CrossRef]
- Wang, J.J.; Yang, M.F.; Dai, R.H.; Li, H. Complete mitochondrial genome of Evacanthus heimianus (Hemiptera: Cicadellidae: Evacanthinae) from China. Mitochondrial DNA Part B 2019, 4, 284–285. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. Partition Finder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2016, 34, 772–773. [Google Scholar]
- Lam-Tung, N.; Schmidt, H.A.; Arndt, V.H.; Quang, M.B. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Ann. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470. [Google Scholar] [CrossRef] [PubMed]
- Garey, J.R.; Wolstenholme, D.R. Platyhelminth mitochondrial DNA: Evidence for early evolutionary origin of a tRNAserAGN that contains a dihydrouridine arm replacement loop, and of serine-specifying AGA and AGG codons. J. Mol. Evol. 1989, 28, 374–387. [Google Scholar] [CrossRef]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486–487. [Google Scholar] [CrossRef]
- Brabec, J.; Kostadinova, A.; Scholz, T.; Littlewood, D.T.J. Complete mitochondrial genomes and nuclear ribosomal RNA operons of two species of Diplostomum (Platyhelminthes: Trematoda): A molecular resource for taxonomy and molecular epidemiology of important fish pathogens. Parasit. Vectors 2015, 8, 336. [Google Scholar] [CrossRef]
- Demari-Silva, B.; Foster, P.G.; Oliveira-de, T.M.P.; Bergo, E.S.; Sanabani, S.S.; Pessôa, R.; Sallum, M.A.M. Mitochondrial genomes and comparative analyses of Culex camposi, Culex coronator, Culex usquatus and Culex usquatissimus (Diptera: Culicidae), members of the Coronator group. BMC Genom. 2015, 16, 831. [Google Scholar] [CrossRef]
- Kirschbaum, B. Die Cicadinen der gegend von Wiesbaden und Frankfurt, A.M. Nebsteiner anzahl neuer oder Schwer zu unterscheidender Arten aus anderen Gegenden Europa’s Tabellarisch Beschrieben. Nassau Ver. f. Naturk. Jahrb. 1868, 21, 1–202. [Google Scholar]
- Distant, W.L.; Rhynchota, I.V. Homoptera and Appendix (Pt.). In The Fauna of British India, Including Ceylon and Burma; Taylor & Francis: London, UK, 1908; p. XV+501. [Google Scholar]
- McAtee, W.L. Genera and subgenera of Eupteryginae (Homoptera; Jassidae). Proc. Zool. Soc. Lond. 1934, 104, 93–117. [Google Scholar] [CrossRef]
- Young, D.A.J. A reclassification of western Hemisphere Typhlocybinae (Homoptera: Cicadellidae). Univ. Kans. Sci. Bull. 1952, 35, 3–217. [Google Scholar] [CrossRef]
- Mahmood, S.H.; Ahmed, M. Problems of higher classification of Typhlocybinae (Cicadellidae: Homoptera). Univ. Stud. Karachi 1968, 5, 72–79. [Google Scholar]
- Anufriev, G.A. Les cicadellidaes de le Territoire Maritime. Horae Soc. Entomol. Unionis Sov. 1978, 60, 1–215. [Google Scholar]
- Hamilton, K.G.A. Classification, Morphology and Phylogeny of the family Cicadellidae (Rhynchota: Homoptera). In Proceedings of the the 1st International Workshop on Biotaxonomy Classification and Biology of Leafhoppers and Planthoppers (Auchenorrhyncha) of Economic Importance, London, UK, 4–7 October 1983; pp. 15–37. [Google Scholar]
- Huang, M.; Zhang, Y.L. Taxonomic study of the genus Vatana Dworakowska (Auchenorrhyncha: Cicadellidae: Typhlocybinae: Typhlocybini). Proc. Entomol. Soc. Wash. 2006, 108, 945–952. [Google Scholar]
- Dworakowska, I. Contribution to the taxonomy of genera related to Eupteryx Curt. with description of two new genera (Homoptera, Typhlocybinae). Bull. L’academie Pol. Sci. (Ser. Sci. Biol.) 1969, 17, 381–385. [Google Scholar]
- Huang, M.; Zhang, Y.L. A new leafhopper genus Comahadina Huang and Zhang (Hemiptera: Cicadellidae: Typhlocybinae) and a key to genera of Eupteryx-complex. Zootaxa 2010, 2353, 65–68. [Google Scholar] [CrossRef]
- Dworakowska, I. Typhlocybini of Asia (Homoptera, Auchenorrhyncha, Cicadellidae). Entomol. Abh. Berichte Staatl. Mus. Tierkd. Dresd. 1982, 45, 99–181. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subfamily | Tribe | Species | Accession Number |
|---|---|---|---|
| Evacanthinae | Evacanthus heimianus | MG813486 | |
| Deltocephalinae | Scaphoideus varius | KY817245 | |
| Typhlocybinae | Alebrini | Shaddai sp. | MZ014457 |
| Sobrala sp. | MZ014458 | ||
| Empoascini | Ghauriana sinensis | MN699874 | |
| Alebroides salicis | MZ014449 | ||
| Empoasca serrata | MZ014453 | ||
| Empoasca onukii | NC_037210 | ||
| Dikraneurini | Dikraneura zlata | MZ014450 | |
| Dikraneurini sp. | MZ014451 | ||
| Uniformus sp. | MW272457 | ||
| Uzeldikra longiprocessa | MW284821 | ||
| Michalowskiya breviprocessa | MW264489 | ||
| Erythroneurini | Elbelus tripunctatus | MZ014452 | |
| Kaukania anser | MZ014456 | ||
| Mitjaevia protuberanta | NC_047465 | ||
| Empoascanara dwalata | MT350235 | ||
| Empoascanara sipra | MN604278 | ||
| Typhlocybini | Agnesiella aldera | MW284835 | |
| Agnesiella irma | ON022031 | ||
| Agnesiella rita | MW284822 | ||
| Aguriahana wutyshana | ON000924 | ||
| Aguriahana juglandis | MW284823 | ||
| Aguriahana triangularis | MW284824 | ||
| Bolanusoides shaanxiensis | MN661136 | ||
| Eupteryx adspersa | MZ014454 | ||
| Eupteryx sp. | ON022032 | ||
| Eurhadina cuii | MW284836 | ||
| Eurhadina exclamationis | MW284837 | ||
| Eurhadina rubra | ON022033 | ||
| Eurhadina jarrary | MZ014455 | ||
| Farynala dextra | MW284838 | ||
| Farynala starica | ON022034 | ||
| Hiratettix distanti | MW284839 | ||
| Hiratettix malaisei | MW284840 | ||
| Opamata lipcowa | MW284842 | ||
| Paracyba sp. | MW284842 | ||
| Parafagocyba longa | MW284825 | ||
| Sannella crucifera | OL960659 | ||
| Shamala annulata | ON022035 | ||
| Subtilissimia sp1. | MW284826 | ||
| Subtilissimia sp2. | MW284827 | ||
| Thampoa tiani | MW284828 | ||
| Typhlocyba bilaminata | ON022036 | ||
| Typhlocyba choui | MW284829 | ||
| Typhlocyba sp. | KY039138 | ||
| Vatana ogromna | MW284830 | ||
| Warodia biguttata | ON022037 | ||
| Warodia hoso | MW284831 | ||
| Warodia lineata | MW284832 | ||
| Zorka agnesae | MW284834 | ||
| Zorka maculata | ON022038 | ||
| Zyginellini | Kuohzygia albolinea | OL960654 | |
| Limassolla lingchuanensis | NC_046037 | ||
| Limassolla bicruralis | MT683892 | ||
| Limassolla dispunctata | OL960655 | ||
| Limassolla emmrichi | MW272458 | ||
| Limassolla fasciata | OL960656 | ||
| Limassolla galewskii | OL960657 | ||
| Limassolla uncata | OL960654 | ||
| Paraahimia luodianensis | NC_047464 | ||
| Parathailocyba orla | MN894531 | ||
| Parazyginella tiani | MT683891 | ||
| Thailocyba longilobula | OL960660 | ||
| Yangida basnetti | ON000925 | ||
| Yangisunda ramosa | OL960661 | ||
| Yangisunda tiani | MZ014459 | ||
| Zyginella mandali | ON055365 | ||
| Zyginella minuta | MT488436 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Lei, Y.; Dietrich, C.H.; Huang, M. Investigating Monophyly of Typhlocybini Based on Complete Mitochondrial Genomes with Characterization and Comparative Analysis of 19 Species (Hemiptera: Cicadellidae: Typhlocybinae). Insects 2023, 14, 842. https://doi.org/10.3390/insects14110842
Zhou X, Lei Y, Dietrich CH, Huang M. Investigating Monophyly of Typhlocybini Based on Complete Mitochondrial Genomes with Characterization and Comparative Analysis of 19 Species (Hemiptera: Cicadellidae: Typhlocybinae). Insects. 2023; 14(11):842. https://doi.org/10.3390/insects14110842
Chicago/Turabian StyleZhou, Xian, Yuejie Lei, Christopher H. Dietrich, and Min Huang. 2023. "Investigating Monophyly of Typhlocybini Based on Complete Mitochondrial Genomes with Characterization and Comparative Analysis of 19 Species (Hemiptera: Cicadellidae: Typhlocybinae)" Insects 14, no. 11: 842. https://doi.org/10.3390/insects14110842