
The Roles and Regulatory Mechanisms of Tight Junction Protein Cingulin and Transcription Factor Forkhead Box Protein O1 in Human Lung Adenocarcinoma A549 Cells and Normal Lung Epithelial Cells
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
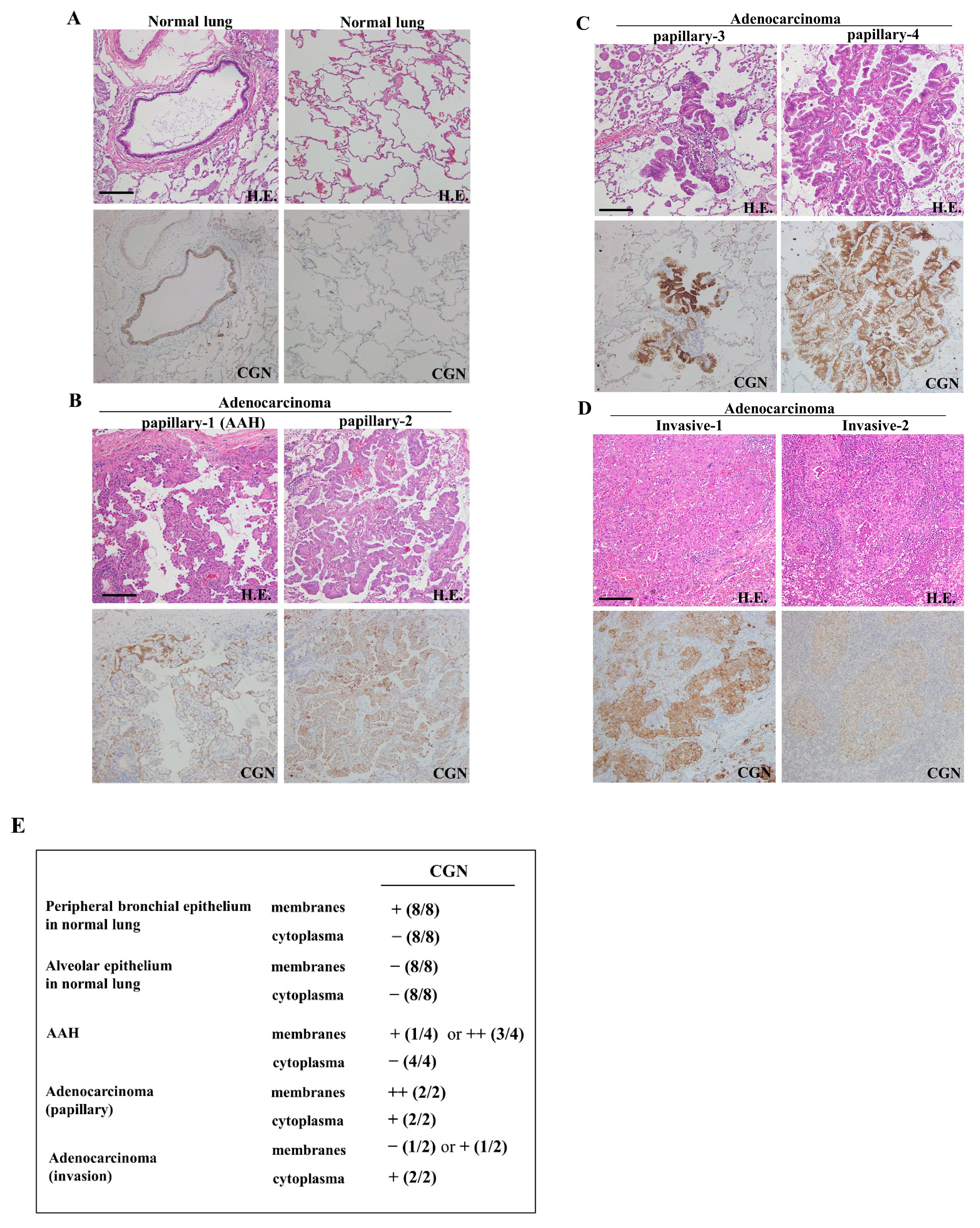
2.1. Expression and Localization of CGN in Lung Adenocarcinoma
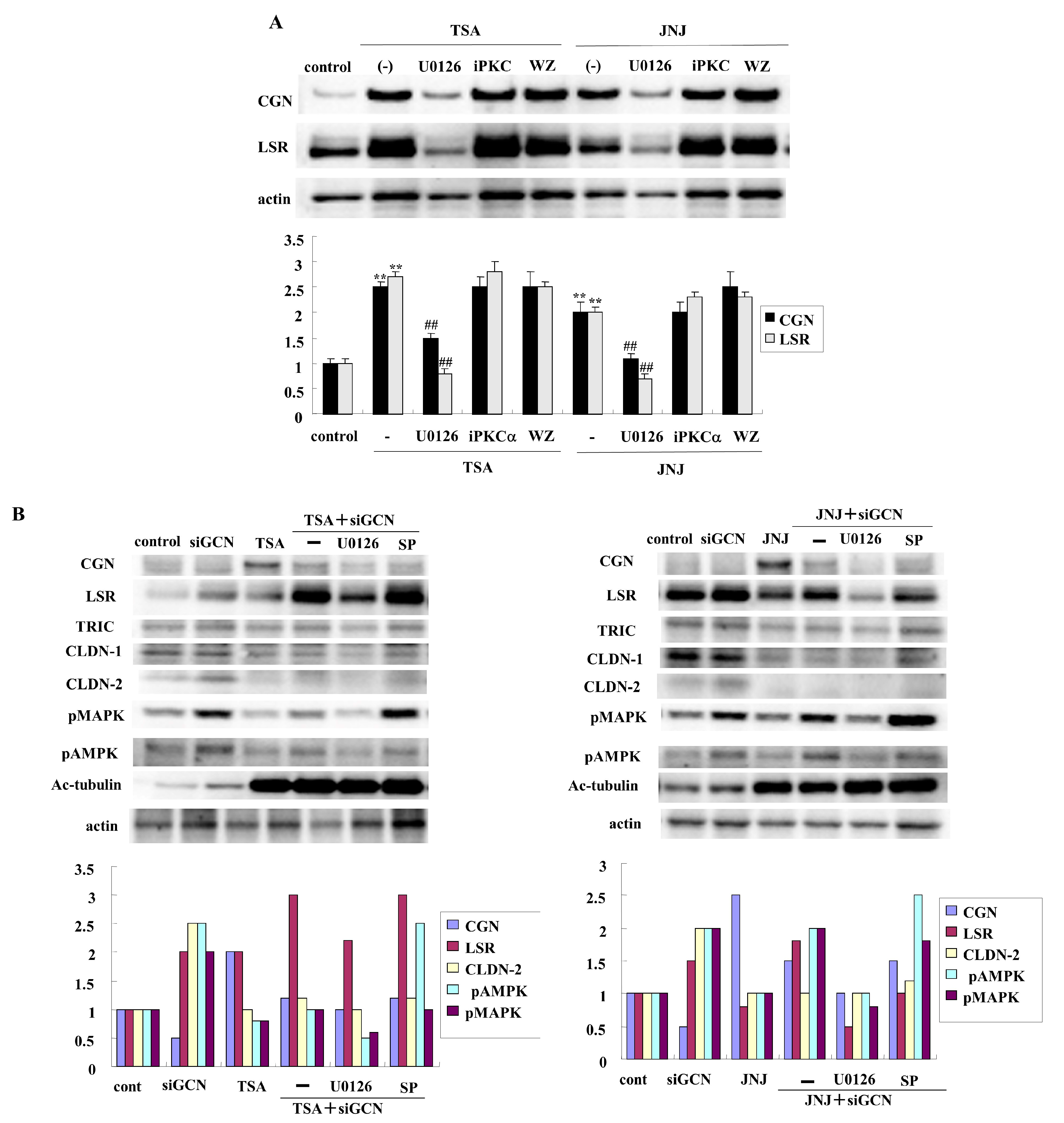
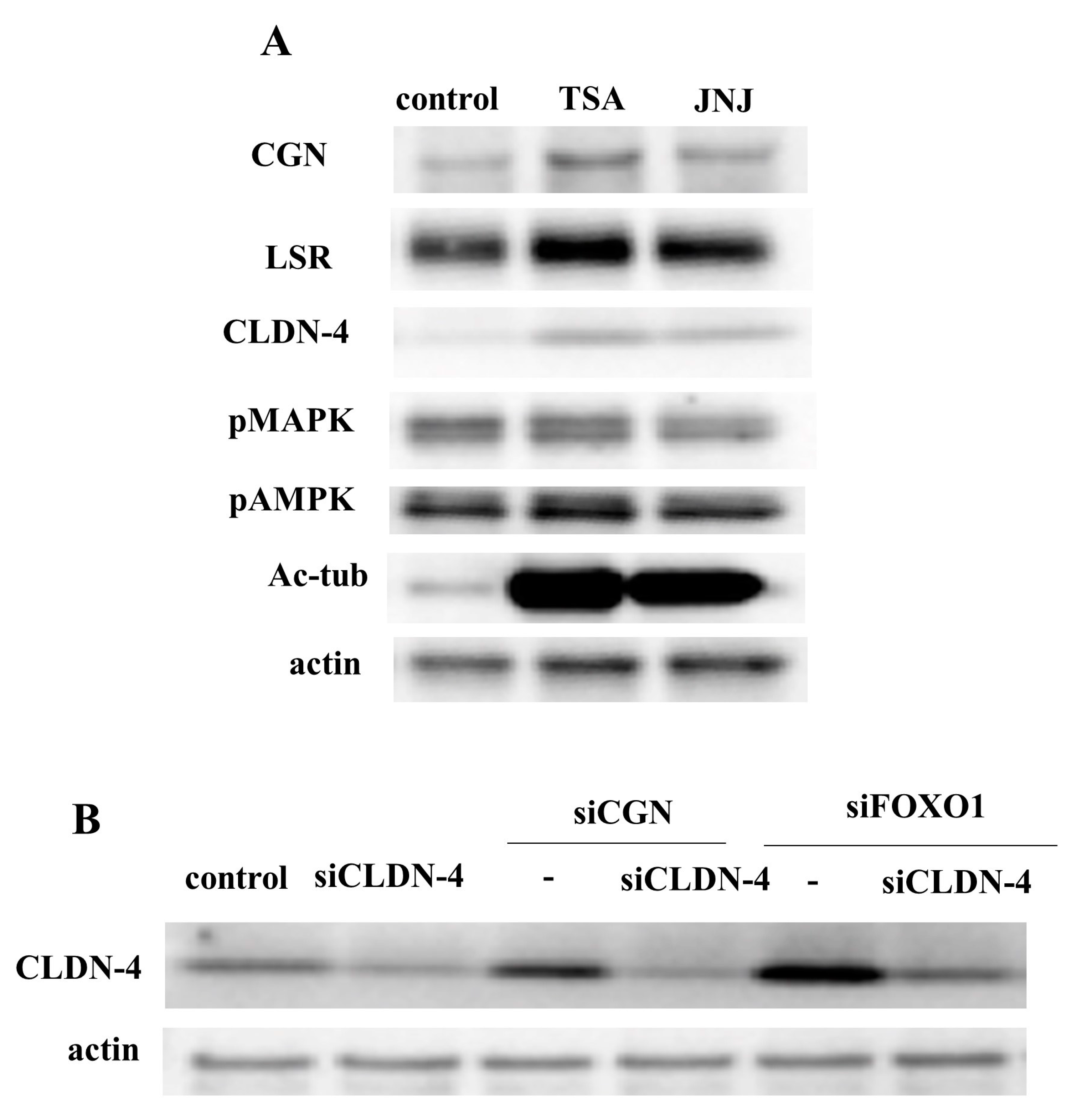
2.2. HDAC Inhibitors TSA and Quisinostat Increased CGN and Angulin-1/LSR in Lung Adenocarcinoma Cell Line A549
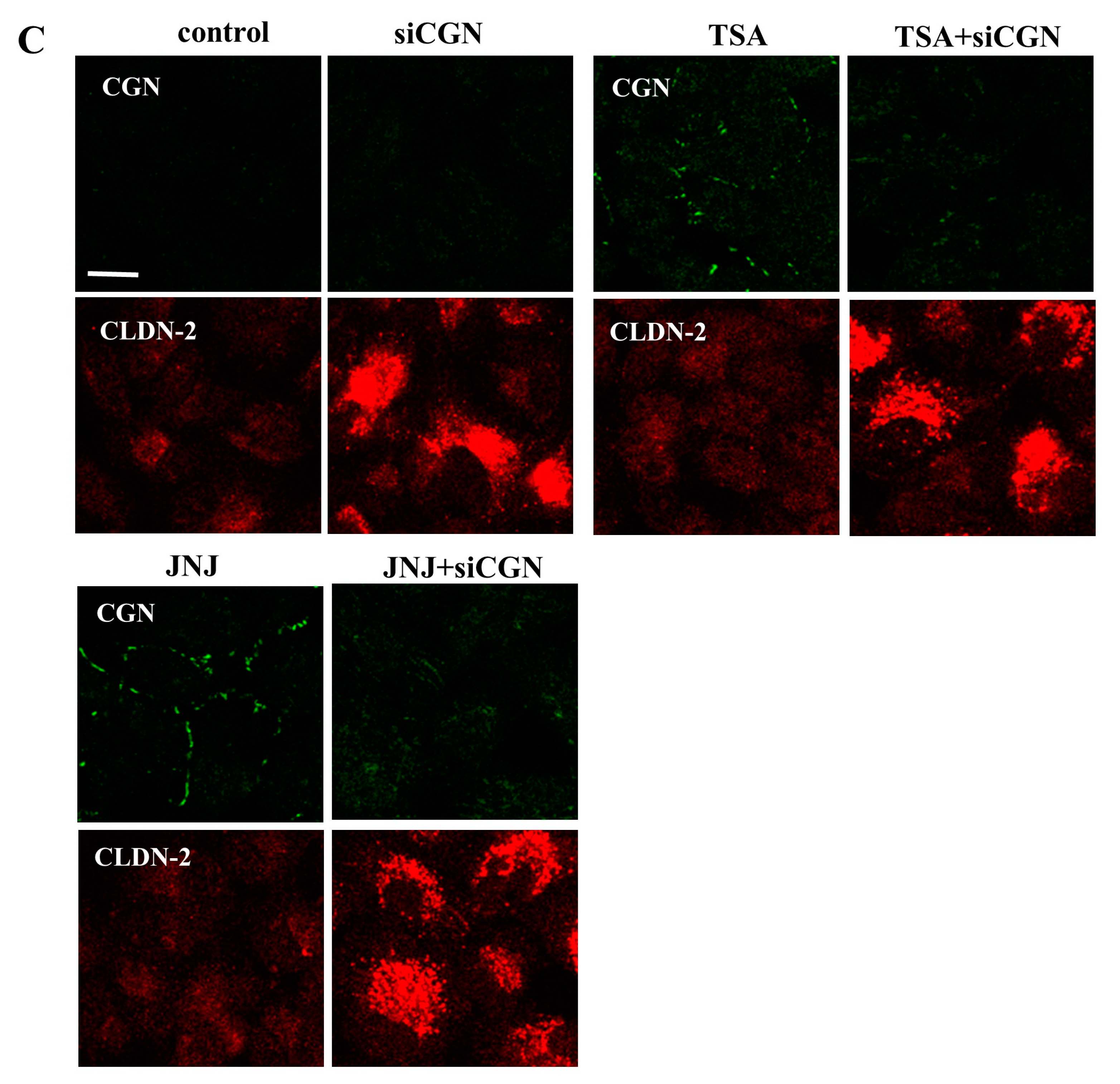
2.3. Knockdown of CGN Increased Expression of CLDN-2 in A549 Cells
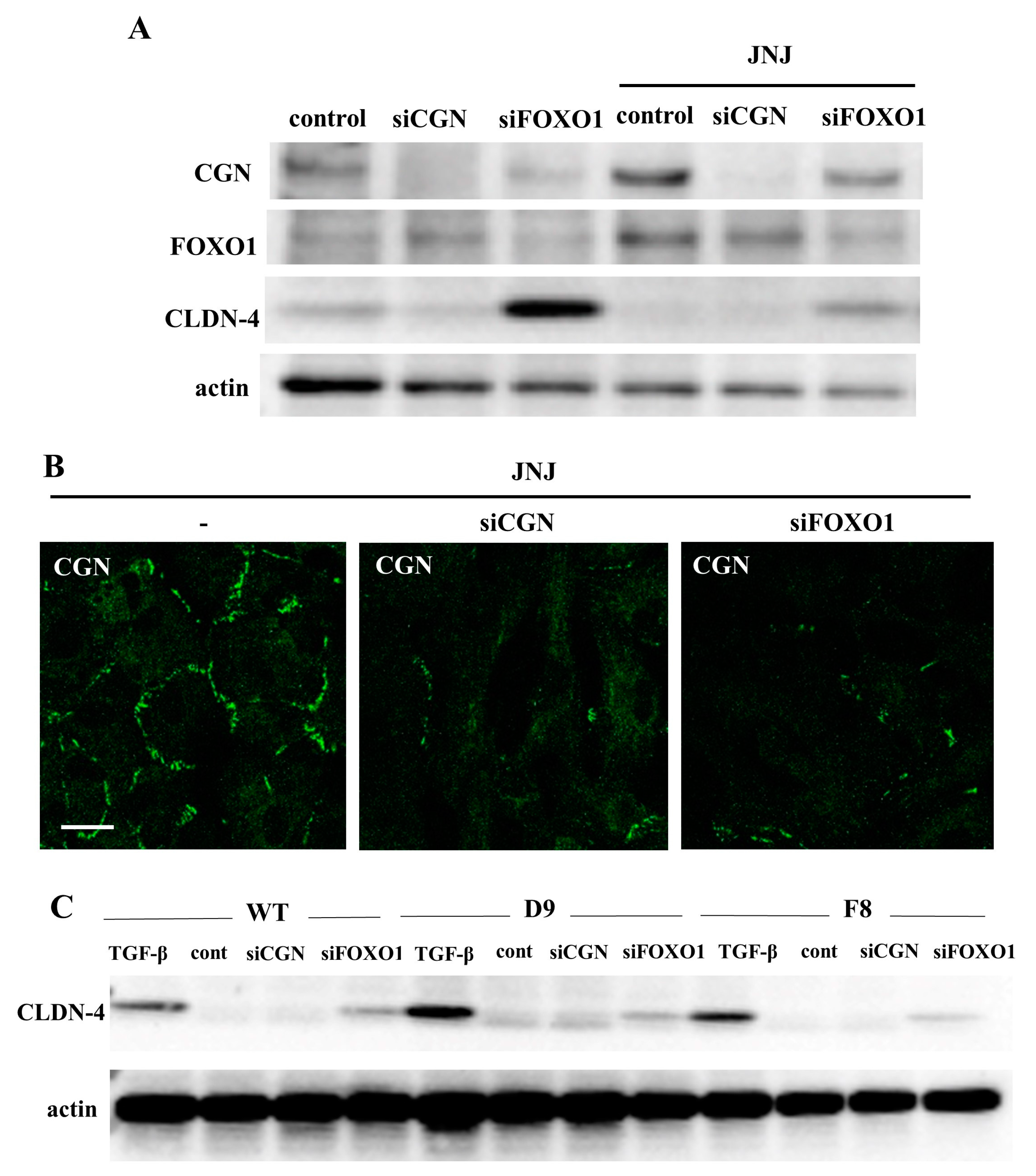
2.4. Knockdown of FOXO1 Increased Expression of CLDN-4 and Decreased Expression of CGN in A549 Cells
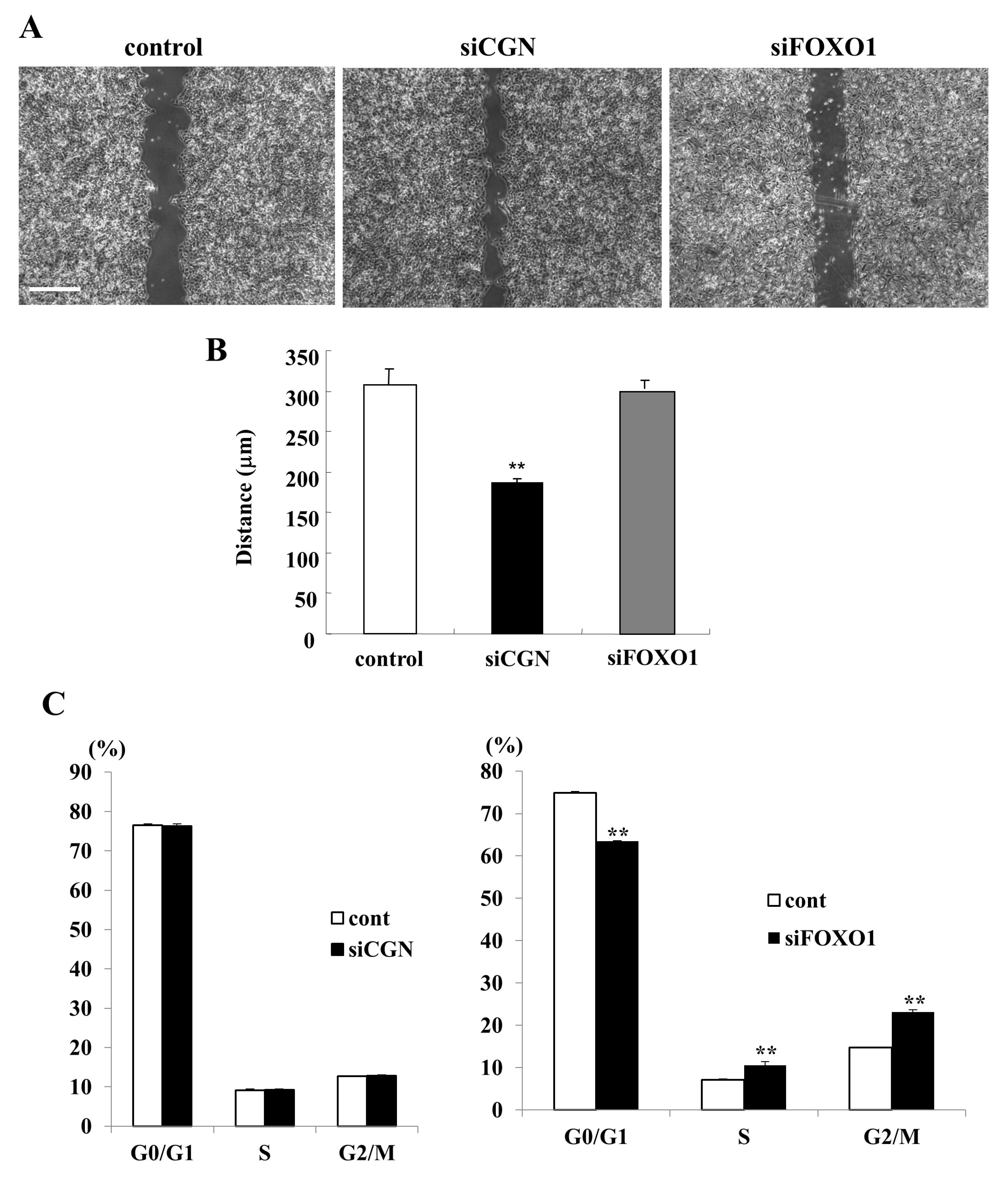
2.5. Knockdown of CGN Induced Cell Migration but Not Cell Proliferation and Knockdown of FOXO1 Induced Cell Proliferation but Not Cell Migration in A549 Cells
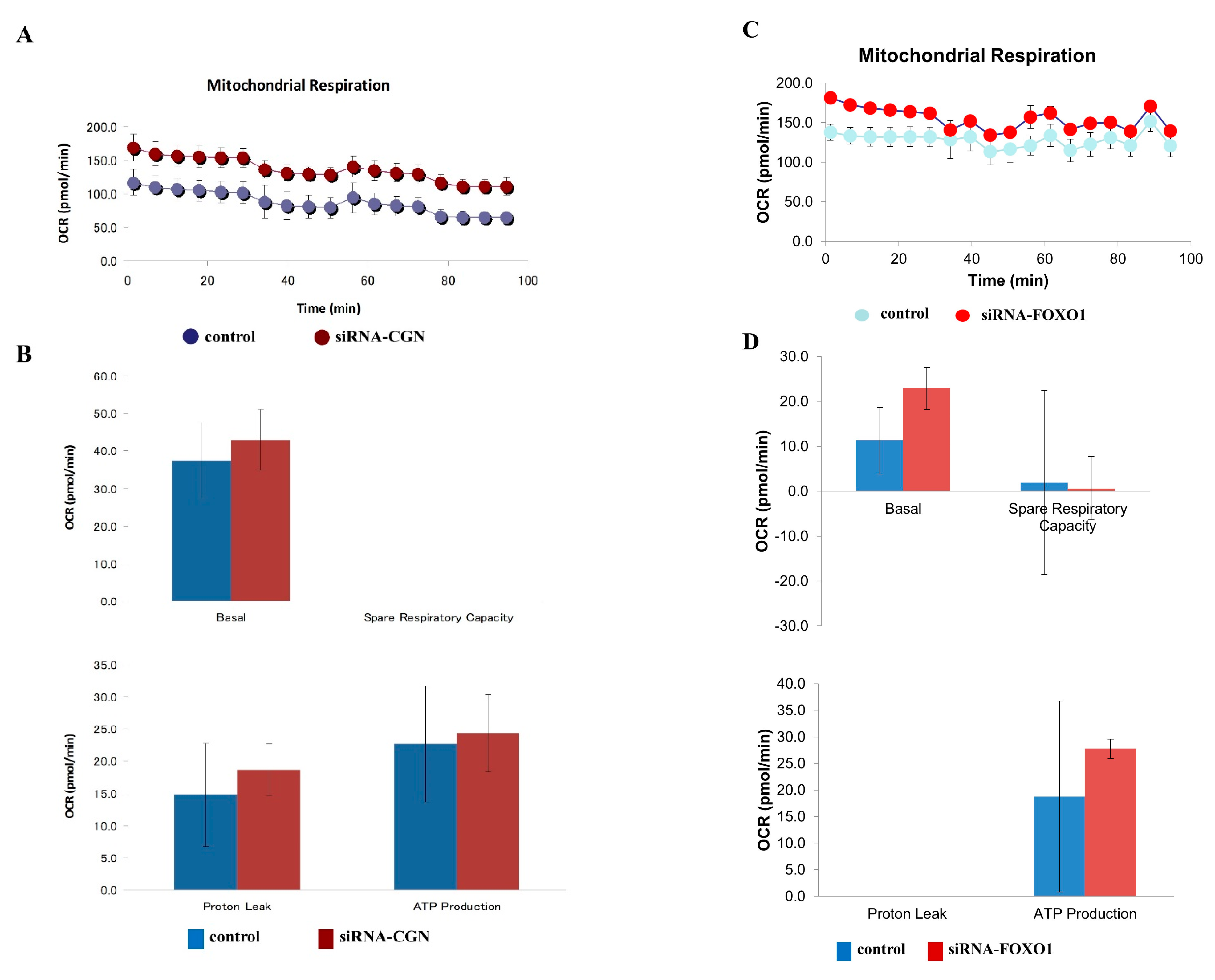
2.6. Knockdown of CGN and FOXO1 Induced Cell Metabolism in A549 Cells
2.7. HDAC Inhibitors Upregulated Expression of CGN and CLDN-4 and Knockdown of CGN and FOXO1 Upregulated Expression of CLDN-4 in Normal Human Lung Epithelial Cells (HLE Cells)
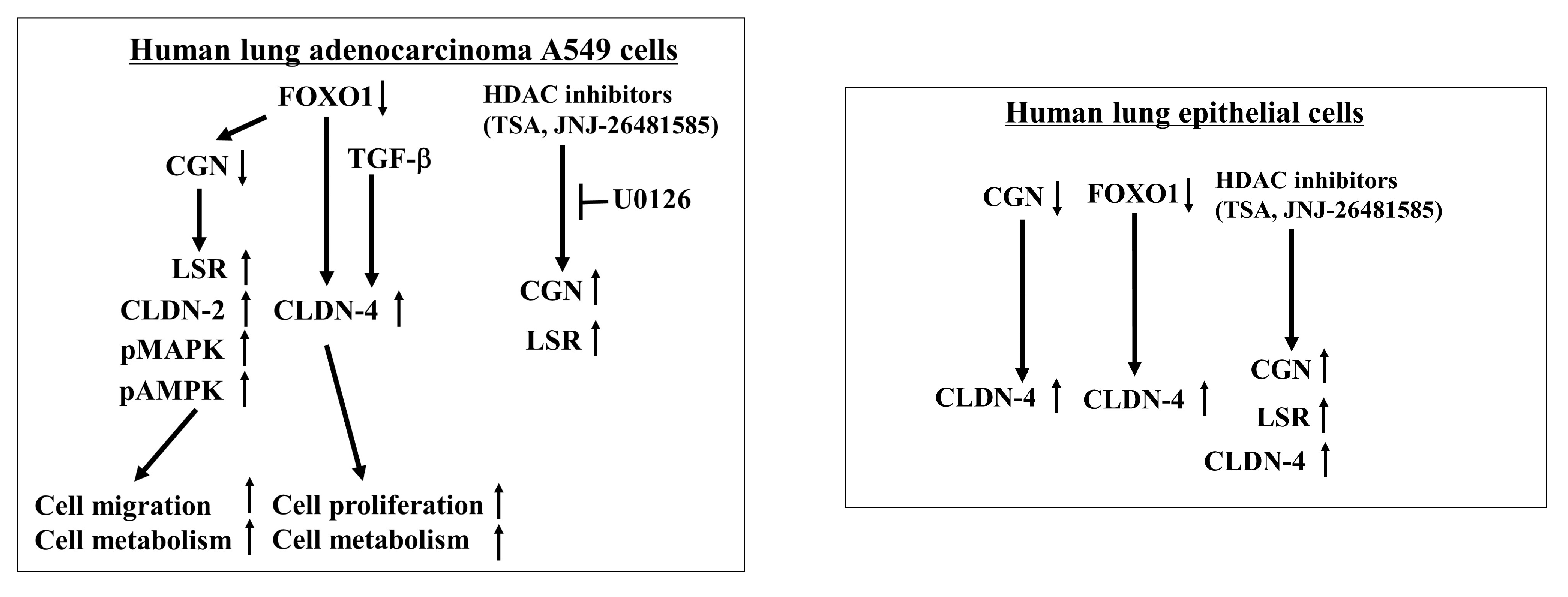
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Antibodies and Reagents
4.3. Immunohistochemical Analysis
4.4. Cell Line Culture and Treatment
4.5. Isolation and Culture of Human Lung Epithelial (HLE) Cells
4.6. RNA Interference and Transfection RNA Interference and Transfection
4.7. Immunocytochemical Staining
4.8. Western Blot Analysis
4.9. Migration Assay
4.10. Cell Cycle Assay
4.11. XF96 Extracellular Flux Measurements
4.12. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CGN | cingulin |
| FOXO1 | forkhead box protein O1 |
| HDAC | histone deacetylase |
| NSCLC | non-small cell lung cancer |
| HLE | human lung epithelial |
| TSA | trichostatin A |
| CLDN | claudin |
| TJ | tight junction |
| TKIs | tyrosine kinase inhibitors |
| LSR | lipolysis-stimulated lipoprotein receptor |
| TRIC | tricellulin |
| MAPK | mitogen-activated protein kinase |
| AMPK | adenosine monophosphate-activated protein kinase |
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Blandin Knight, S.; Crosbie, P.A.; Balata, H.; Chudziak, J.; Hussell, T.; Dive, C. Progress and prospects of early detection in lung cancer. Open Biol. 2017, 7, 170070. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Chris Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Matter, K.; Balda, M.S. Signalling to and from tight junctions. Nat. Rev. Mol. Cell Biol. 2003, 4, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Balda, M.S.; Matter, K. Tight junctions and the regulation of gene expression. Biochim. Biophys. Acta 2009, 1788, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Li, J. Targeting claudins in cancer: Diagnosis, prognosis and therapy. Am. J. Cancer Res. 2021, 11, 3406–3424. [Google Scholar] [PubMed]
- Ikari, A.; Sato, T.; Watanabe, R.; Yamazaki, Y.; Sugatani, J. Increase in claudin-2 expression by an EGFR/MEK/ERK/c-Fos pathway in lung adenocarcinoma A549 cells. Biochim. Biophys. Acta 2012, 1823, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Xin, S.; Huixin, C.; Benchang, S.; Aiping, B.; Jinhui, W.; Xiaoyan, L.; Yu, W.B.; Minhu, C. Expression of Cdx2 and claudin-2 in the multistage tissue of gastric carcinogenesis. Oncology 2007, 73, 357–365. [Google Scholar] [CrossRef]
- Okada, T.; Konno, T.; Kohno, T.; Shimada, H.; Saito, K.; Satohisa, S.; Saito, T.; Kojima, T. Possibility of targeting claudin-2 in therapy for human endometrioid endometrial carcinoma. Reprod. Sci. 2020, 27, 2092–2103. [Google Scholar] [CrossRef]
- Moldvay, J.; Jäckel, M.; Páska, C.; Soltész, I.; Schaff, Z.; Kiss, A. Distinct claudin expression profile in histologic subtypes of lung cancer. Lung Cancer 2007, 57, 159–167. [Google Scholar] [CrossRef]
- Luo, J.; Wang, H.; Chen, H.; Gan, G.; Zheng, Y. CLDN4 silencing promotes proliferation and reduces chemotherapy sensitivity of gastric cancer cells through activation of the PI3K/Akt signalling pathway. Exp. Physiol. 2020, 105, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Konno, T.; Kojima, T. Role of tricellular tight junction protein lipolysis-stimulated lipoprotein receptor (LSR) in cancer cells. Int. J. Mol. Sci. 2019, 20, 3555. [Google Scholar] [CrossRef] [PubMed]
- Arai, W.; Konno, T.; Kohno, T.; Kodera, Y.; Tsujiwaki, M.; Shindo, Y.; Chiba, H.; Miyajima, M.; Sakuma, Y.; Watanabe, A.; et al. Downregulation of angulin-1/LSR induces malignancy via upregulation of EGF-dependent claudin-2 and TGF-β-dependent cell metabolism in human lung adenocarcinoma A549 cells. Oncotarget 2023, 14, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zeng, H.; Lei, L.; Tong, X.; Yang, L.; Yang, Y.; Li, S.; Zhou, Y.; Luo, L.; Huang, J.; et al. Tight junctions and their regulation by non-coding RNAs. Int. J. Biol. Sci. 2021, 17, 712–727. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Ohwada, K.; Shindo, Y.; Konno, T.; Kohno, T.; Kikuchi, S.; Tsujiwaki, M.; Ishii, D.; Nishida, S.; Kakuki, T.; et al. Inhibition of HDAC and signal transduction pathways induces tight junctions and promotes differentiation in p63-positive salivary duct adenocarcinoma. Cancers 2022, 14, 2584. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Guo, W.; Fang, S.; He, B.; Li, X.; Fan, L. miR-1270 enhances the proliferation, migration, and invasion of osteosarcoma via targeting cingulin. Eur. J. Histochem. 2021, 65, 3237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, T.; Han, Y.N.; Ge, M.; Wang, P.; Sun, L.; Liu, H.; Cao, T.; Nie, Y.; Fan, D.; et al. miR-125b promotes colorectal cancer migration and invasion by dual-targeting CFTR and CGN. Cancers 2021, 13, 5710. [Google Scholar] [CrossRef]
- Accili, D.; Arden, K.C. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 2004, 117, 421–426. [Google Scholar] [CrossRef]
- Jiramongkol, Y.; Lam, E.W. FOXO transcription factor family in cancer and metastasis. Cancer Metastasis Rev. 2020, 39, 681–709. [Google Scholar] [CrossRef]
- Benayoun, B.A.; Caburet, S.; Veitia, R.A. Forkhead transcription factors: Key players in health and disease. Trends Genet. 2011, 27, 224–232. [Google Scholar] [CrossRef]
- Habashy, H.O.; Rakha, E.A.; Aleskandarany, M.; Ahmed, M.A.; Green, A.R.; Ellis, I.O.; Powe, D.G. FOXO3a nuclear localisation is associated with good prognosis in luminal-like breast cancer. Breast Cancer Res. Treat. 2011, 129, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Bullock, M.D.; Bruce, A.; Sreekumar, R.; Curtis, N.; Cheung, T.; Reading, I.; Primrose, J.N.; Ottensmeier, C.; Packham, G.K.; Thomas, G.; et al. FOXO3 expression during colorectal cancer progression: Biomarker potential reflects a tumour suppressor role. Br. J. Cancer 2013, 109, 387–394. [Google Scholar] [CrossRef]
- Su, B.; Gao, L.; Baranowski, C.; Gillard, B.; Wang, J.; Ransom, R.; Ko, H.K.; Gelman, I.H. A genome-wide RNAi screen identifies FOXO4 as a metastasis-suppressor through counteracting PI3K/AKT signal pathway in prostate cancer. PLoS ONE 2014, 9, e101411. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Liu, R.; Ye, N.; Liu, C.; Li, X.; Guo, X.; Zhang, Z.; Li, X.; Yao, Y.; Jiang, X. FOXO1 inhibits tumor cell migration via regulating cell surface morphology in non-small cell lung cancer cells. Cell Physiol. Biochem. 2018, 48, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Luo, J.; Li, J.; Kim, G.; Chen, E.S.; Xiao, S.; Snapper, S.B.; Bao, B.; An, D.; Blumberg, R.S.; et al. Foxo1 controls gut homeostasis and commensalism by regulating mucus secretion. J. Exp. Med. 2021, 218, e20210324. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J.; Wu, Y.L.; Paz-Ares, L. Lung cancer: Current therapies and new targeted treatments. Lancet 2017, 389, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Tsao, A.S.; Scagliotti, G.V.; Bunn, P.A., Jr.; Carbone, D.P.; Warren, G.W.; Bai, C.; de Koning, H.J.; Yousaf-Khan, A.U.; McWilliams, A.; Tsao, M.S.; et al. Scientific advances in lung cancer 2015. J. Thorac. Oncol. 2016, 11, 613–638. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, H.; Lin, L.; Takahashi, T.; Nomura, M.; Suzuki, M.; Wistuba, I.I.; Fong, K.M.; Lee, H.; Toyooka, S.; Shimizu, N.; et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J. Natl. Cancer Inst. 2005, 97, 339–346. [Google Scholar] [CrossRef]
- Tsao, M.S.; Sakurada, A.; Cutz, J.C.; Zhu, C.Q.; Kamel-Reid, S.; Squire, J.; Lorimer, I.; Zhang, T.; Liu, N.; Daneshmand, M.; et al. Erlotinib in lung cancer-molecular and clinical predictors of outcome. N. Engl. J. Med. 2005, 353, 133–144. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crinó, L.; Ahn, M.J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef]
- Liu, X.; Wang, P.; Zhang, C.; Ma, Z. Epidermal growth factor receptor (EGFR): A rising star in the era of precision medicine of lung cancer. Oncotarget 2017, 8, 50209–50220. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gallinari, P.; Di Marco, S.; Jones, P.; Pallaoro, M.; Steinkühler, C. HDACs, histone deacetylation and gene transcription: From molecular biology to cancer therapeutics. Cell Res. 2007, 17, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Ma, K.; Zhu, H.; Wang, S.; Liu, M.; Zhang, W.; Liang, S.; Xu, N. Molecular, biological characterization and drug sensitivity of chidamide-resistant non-small cell lung cancer cells. Oncol. Lett. 2017, 14, 6869–6875. [Google Scholar] [CrossRef] [PubMed]
- Bartling, B.; Hofmann, H.S.; Boettger, T.; Hansen, G.; Burdach, S.; Silber, R.E.; Simm, A. Comparative application of antibody and gene array for expression profiling in human squamous cell lung carcinoma. Lung Cancer 2005, 49, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Chikamatsu, K.; Ishii, H.; Murata, T.; Sakakura, K.; Shino, M.; Toyoda, M.; Takahashi, K.; Masuyama, K. Alteration of cancer stem cell-like phenotype by histone deacetylase inhibitors in squamous cell carcinoma of the head and neck. Cancer Sci. 2013, 104, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Liu, H.T.; Wang, X.Y.; Wu, S.C.; Chen, L.C.; Liu, Y.W. Trichostatin A induces bladder cancer cell death via intrinsic apoptosis at the early phase and Sp1-survivin downregulation at the late phase of treatment. Oncol. Rep. 2017, 38, 1587–1596. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wu, J.Q.; Yu, X.F.; Yang, X.S.; Yang, Y. Trichostatin A inhibits proliferation of triple negative breast cancer cells by inducing cell cycle arrest and apoptosis. Neoplasma 2018, 65, 898–906. [Google Scholar] [CrossRef]
- Arts, J.; King, P.; Mariën, A.; Floren, W.; Beliën, A.; Janssen, L.; Pilatte, I.; Roux, B.; Decrane, L.; Gilissen, R.; et al. JNJ-26481585, a novel “second-generation” oral histone deacetylase inhibitor, shows broad-spectrum preclinical antitumoral activity. Clin. Cancer Res. 2009, 15, 6841–6851. [Google Scholar] [CrossRef]
- Shindo, Y.; Arai, W.; Konno, T.; Kohno, T.; Kodera, Y.; Chiba, H.; Miyajima, M.; Sakuma, Y.; Watanabe, A.; Kojima, T. Effects of histone deacetylase inhibitors Tricostatin a and Quisinostat on tight junction proteins of human lung adenocarcinoma A549 cells and normal lung epithelial cells. Histochem. Cell Biol. 2021, 155, 637–653. [Google Scholar] [CrossRef]
- Ikari, A.; Watanabe, R.; Sato, T.; Taga, S.; Shimobaba, S.; Yamaguchi, M.; Yamazaki, Y.; Endo, S.; Matsunaga, T.; Sugatani, J. Nuclear distribution of claudin-2 increases cell proliferation in human lung adenocarcinoma cells. Biochim. Biophys. Acta 2014, 1843, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Nasako, H.; Akizuki, R.; Takashina, Y.; Eguchi, H.; Matsunaga, T.; Yoshino, Y.; Endo, S.; Ikari, A. Elevation of chemosensitivity of lung adenocarcinoma A549 spheroid cells by claudin-2 knockdown through activation of glucose transport and inhibition of Nrf2 signal. Int. J. Mol. Sci. 2021, 22, 6582. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Lin, F.; Wang, X.; Zhang, J.; Xia, J.; Sun, Y.; Wen, M.; Li, X.; Zhang, Z.; Zhao, J. STYK1/NOK promotes metastasis and epithelial-mesenchymal transition in non-small cell lung cancer by suppressing FoxO1 signaling. Front. Cell Dev. Biol. 2021, 9, 621147. [Google Scholar] [CrossRef]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight junctions: From simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef] [PubMed]
- Runkle, E.A.; Mu, D. Tight junction proteins: From barrier to tumorigenesis. Cancer Lett. 2013, 337, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Hichino, A.; Okamoto, M.; Taga, S.; Akizuki, R.; Endo, S.; Matsunaga, T.; Ikari, A. Down-regulation of Claudin-2 Expression and Proliferation by Epigenetic Inhibitors in Human Lung Adenocarcinoma A549 Cells. J. Biol. Chem. 2017, 292, 2411–2421. [Google Scholar] [CrossRef] [PubMed]
- Kodera, Y.; Chiba, H.; Konno, T.; Kohno, T.; Takahashi, H.; Kojima, T. HMGB1-downregulated angulin-1/LSR induces epithelial barrier disruption via claudin-2 and cellular metabolism via AMPK in airway epithelial Calu-3 cells. Biochem. Biophys. Res. Commun. 2020, 52, 553–560. [Google Scholar] [CrossRef]
- Luo, Y.; Kishi, S.; Sasaki, T.; Ohmori, H.; Fujiwara-Tani, R.; Mori, S.; Goto, K.; Nishiguchi, Y.; Mori, T.; Kawahara, I.; et al. Targeting claudin-4 enhances chemosensitivity in breast cancer. Cancer Sci. 2020, 111, 1840–1850. [Google Scholar] [CrossRef]
- Zhang, L.; Liang, B.; Xu, H.; Gong, Y.; Hu, W.; Jin, Z.; Wu, X.; Chen, X.; Li, M.; Shi, L.; et al. Cinobufagin induces FOXO1-regulated apoptosis, proliferation, migration, and invasion by inhibiting G9a in non-small-cell lung cancer A549 cells. J. Ethnopharmacol. 2022, 291, 115095. [Google Scholar] [CrossRef]
- Zeng, X.; Liu, D.; Peng, G.; Liu, J.; Yang, H. MiroRNA-31-3p promotes the invasion and metastasis of non-small-cell lung cancer cells by targeting forkhead box 1 (FOXO1). Comput. Math. Methods Med. 2022, 2022, 4597087. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Benny, S.; Mishra, R.; Manojkumar, M.K.; Aneesh, T.P. From warburg effect to reverse warburg effect; the new horizons of anti-cancer therapy. Med. Hypotheses 2020, 144, 110216. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishii, D.; Shindo, Y.; Arai, W.; Konno, T.; Kohno, T.; Honda, K.; Miyajima, M.; Watanabe, A.; Kojima, T. The Roles and Regulatory Mechanisms of Tight Junction Protein Cingulin and Transcription Factor Forkhead Box Protein O1 in Human Lung Adenocarcinoma A549 Cells and Normal Lung Epithelial Cells. Int. J. Mol. Sci. 2024, 25, 1411. https://doi.org/10.3390/ijms25031411
Ishii D, Shindo Y, Arai W, Konno T, Kohno T, Honda K, Miyajima M, Watanabe A, Kojima T. The Roles and Regulatory Mechanisms of Tight Junction Protein Cingulin and Transcription Factor Forkhead Box Protein O1 in Human Lung Adenocarcinoma A549 Cells and Normal Lung Epithelial Cells. International Journal of Molecular Sciences. 2024; 25(3):1411. https://doi.org/10.3390/ijms25031411
Chicago/Turabian StyleIshii, Daichi, Yuma Shindo, Wataru Arai, Takumi Konno, Takayuki Kohno, Kazuya Honda, Masahiro Miyajima, Atsushi Watanabe, and Takashi Kojima. 2024. "The Roles and Regulatory Mechanisms of Tight Junction Protein Cingulin and Transcription Factor Forkhead Box Protein O1 in Human Lung Adenocarcinoma A549 Cells and Normal Lung Epithelial Cells" International Journal of Molecular Sciences 25, no. 3: 1411. https://doi.org/10.3390/ijms25031411