
Phylogenetic and Structure-Function Analyses of ENA ATPases: A Case Study of the ENA1 Protein from the Fungus Neurospora crassa


Abstract
:1. Introduction
2. Results
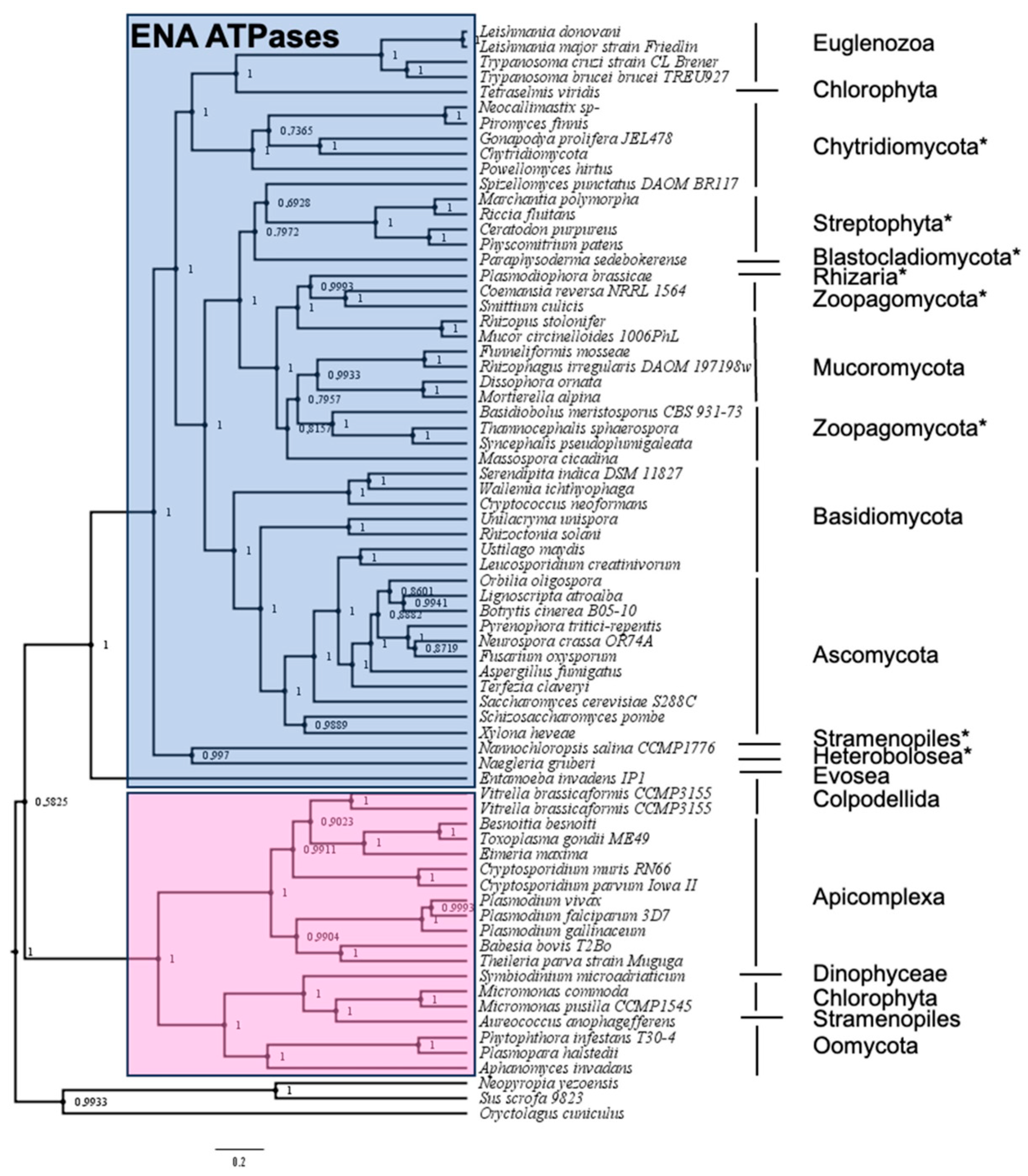
2.1. Identification of ENA Proteins in New Groups of Organisms Using the Hidden Markov Model

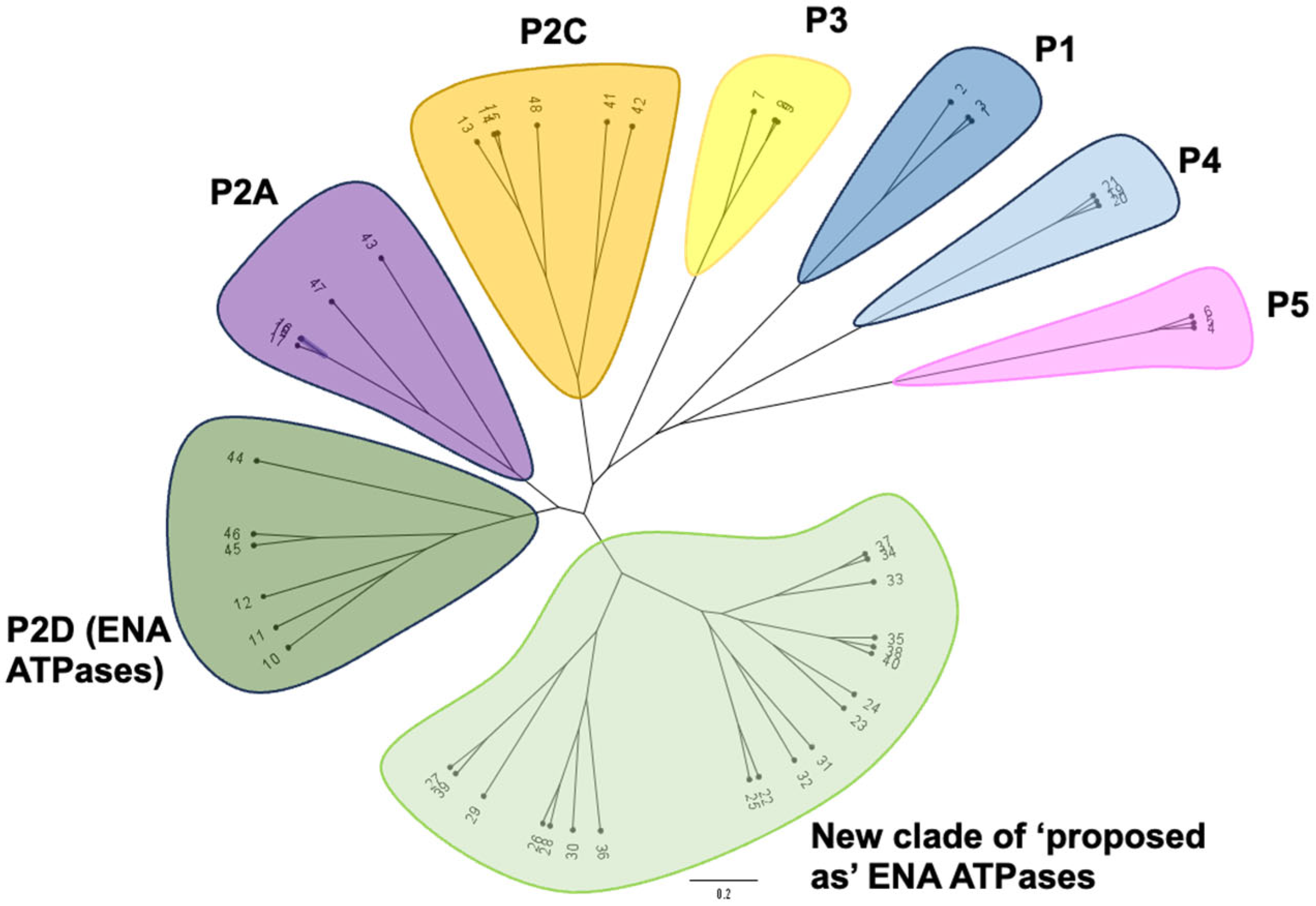
2.2. Phylogenetic Analysis of ‘Newly Identified’ and ‘Proposed as’ ENA ATPases
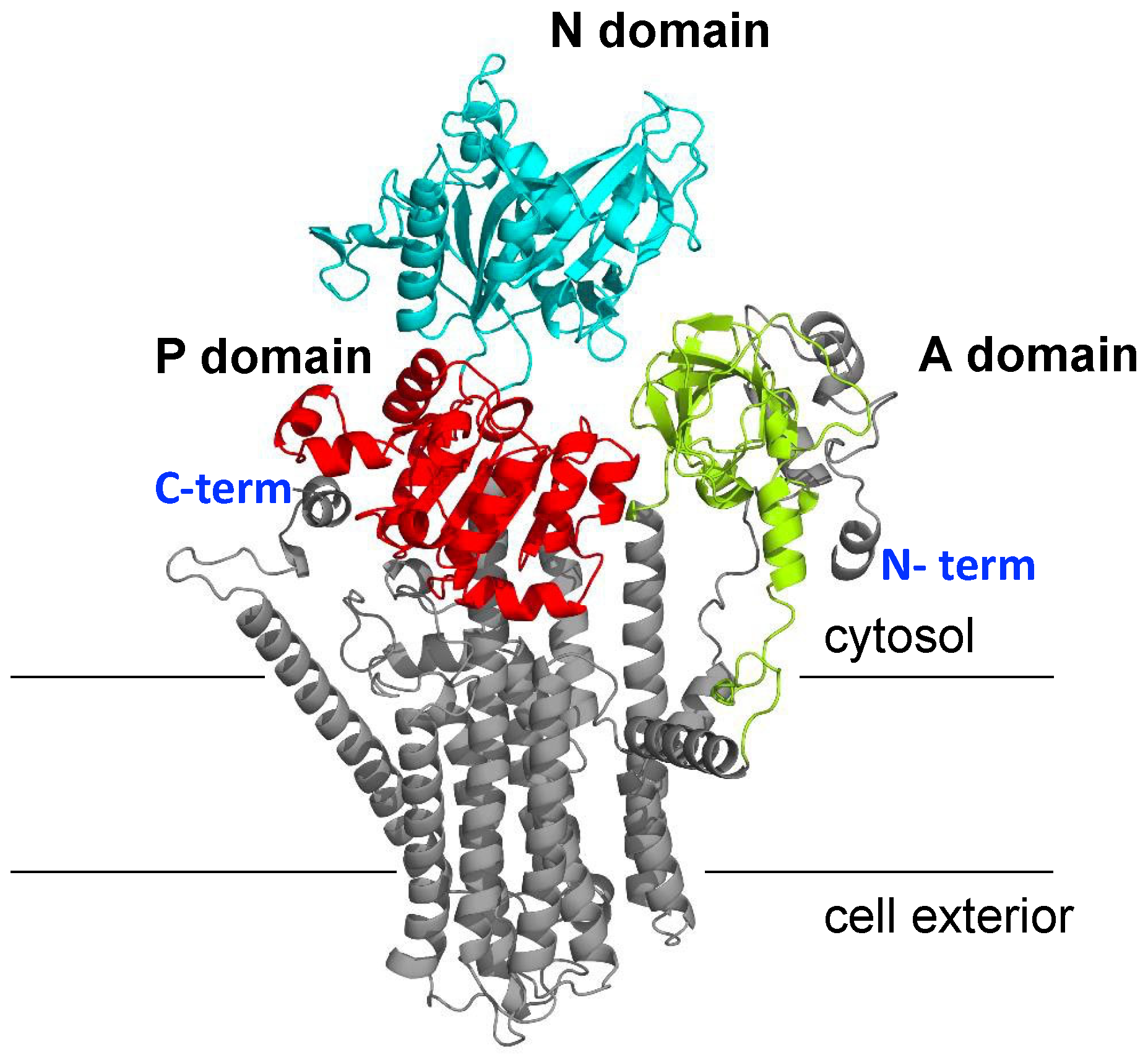
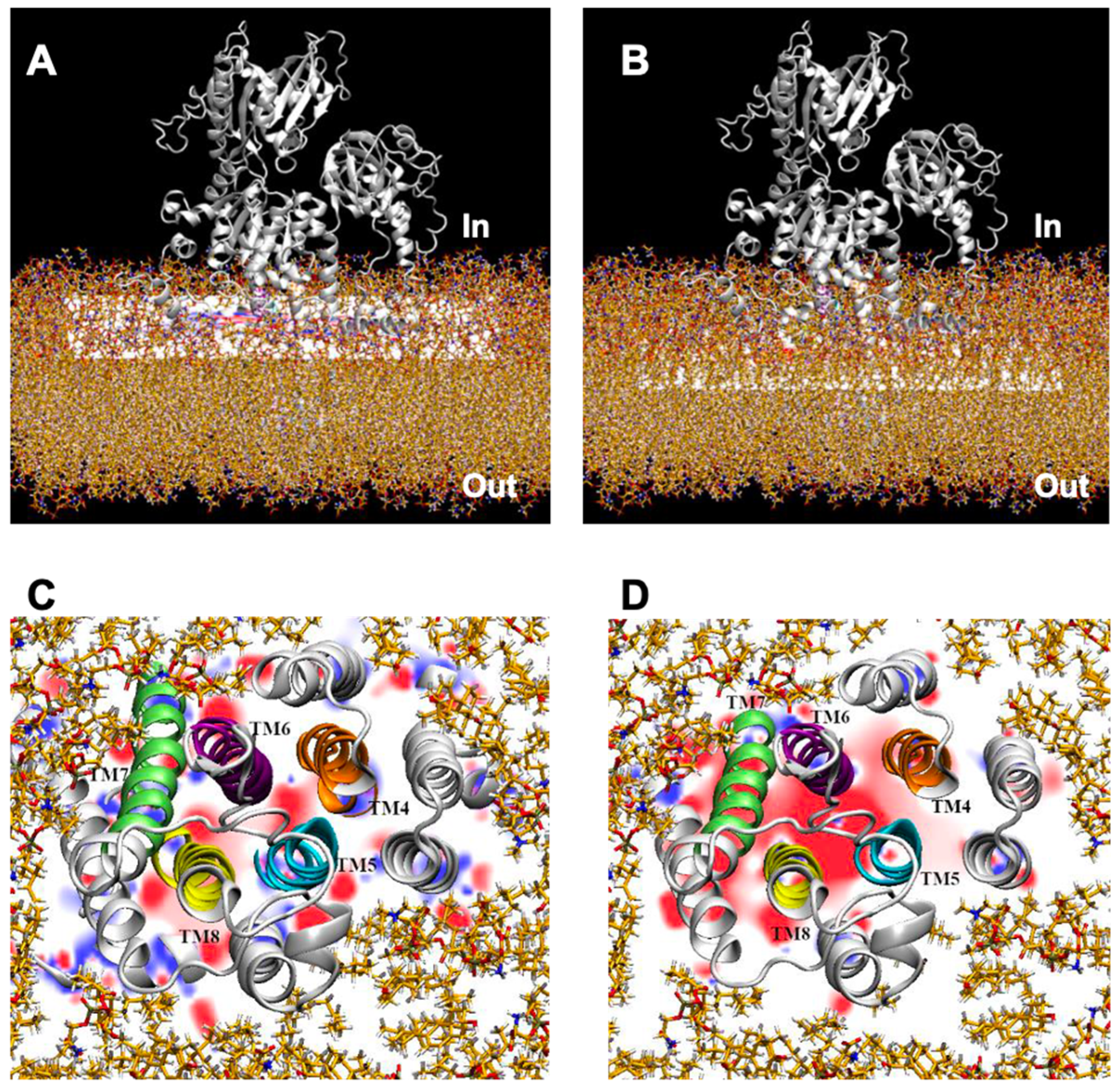
2.3. Prediction of NcENA1 ATPase Structure
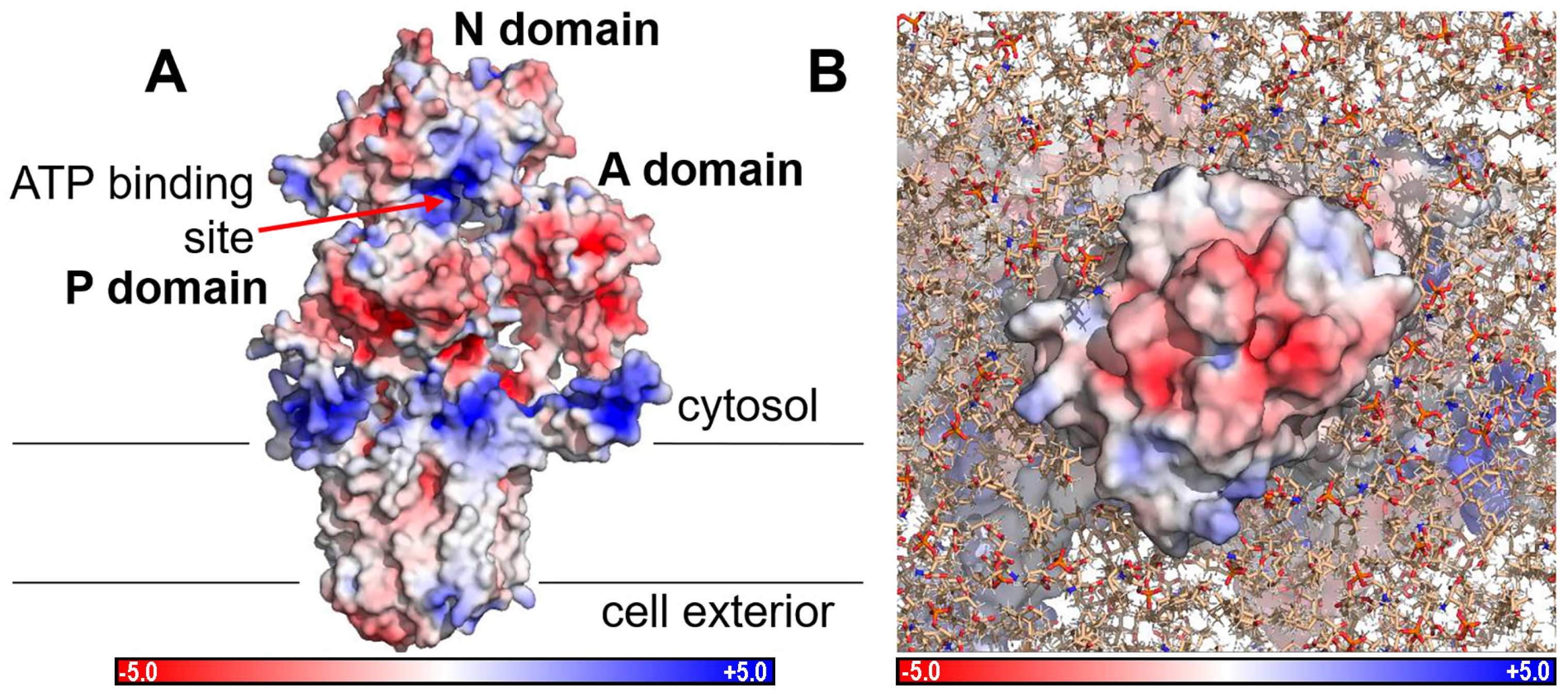
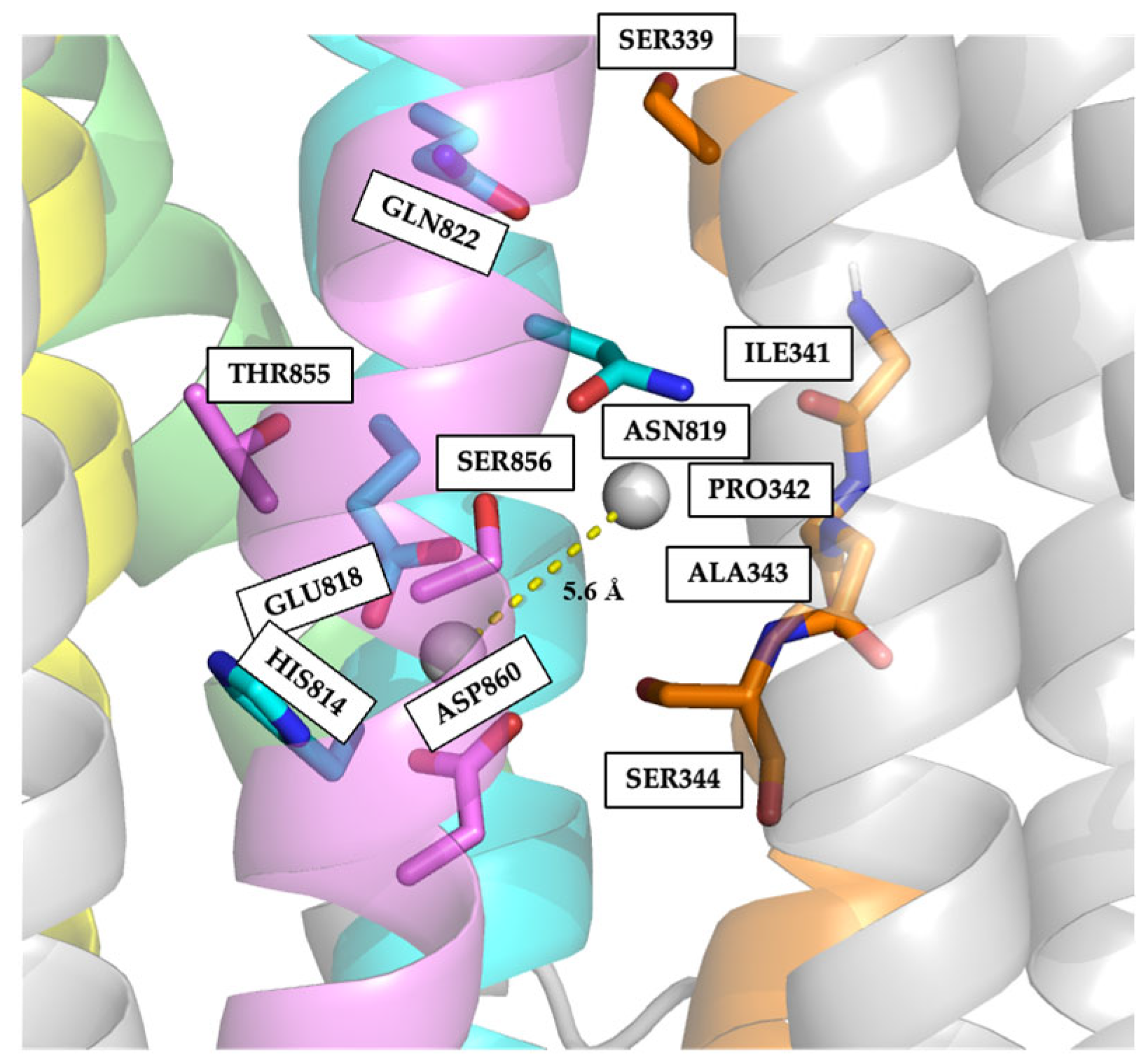
2.4. Electrostatic Potentials and Possible Na+ and/or K+ Binding Sites of NcENA1
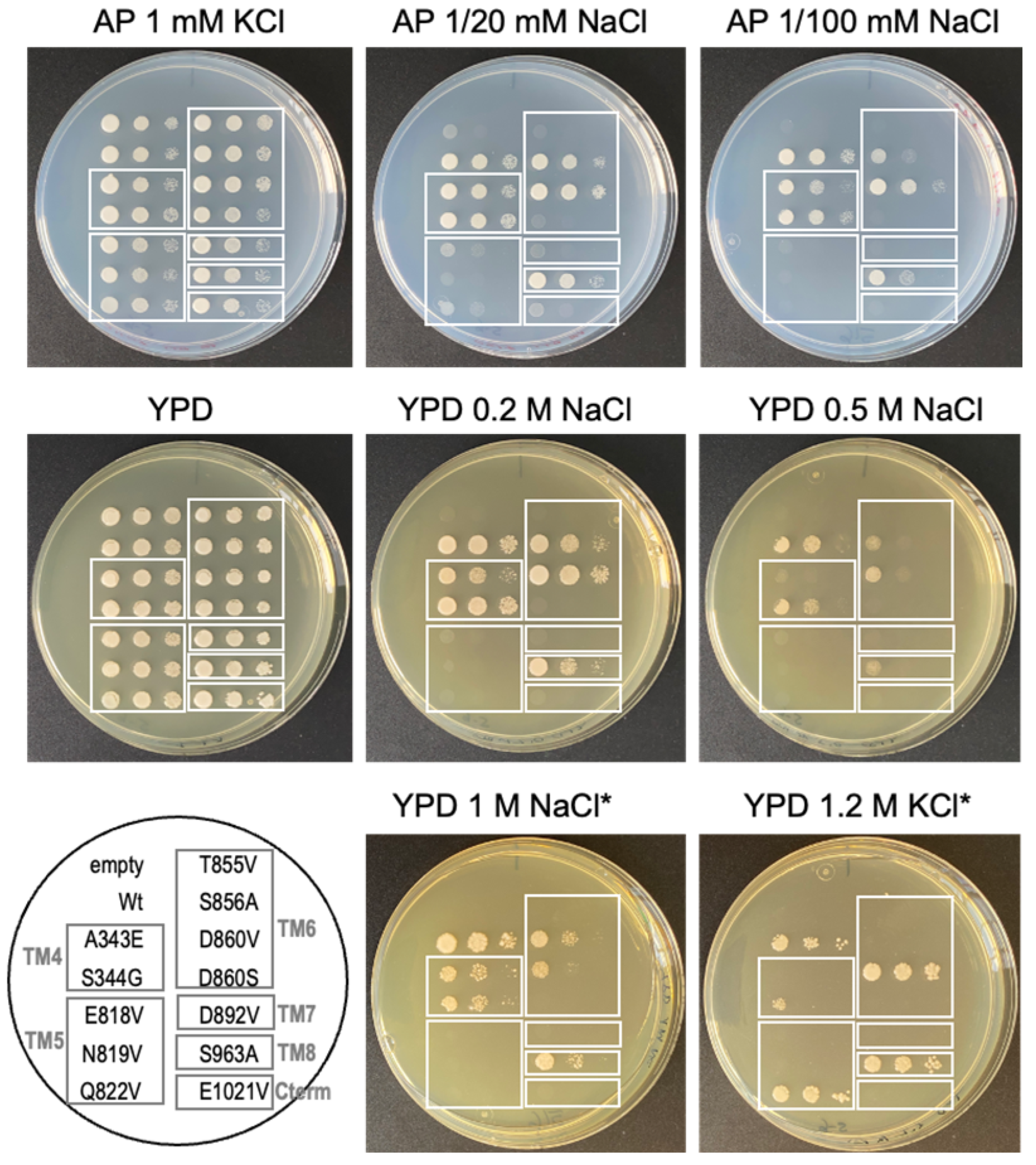
2.5. Identification of Relevant Functional Residues in NcENA1 ATPase by Site-Directed Mutagenesis
3. Discussion
3.1. Emergence of a New Clade of P-Type ATPases, Probably Involved in Na+ Transport
3.2. Structure of ENA ATPases and Proposed Na+ Binding Sites
3.3. Targeted Mutagenesis and Yeast Complementation Studies as a Useful Strategy for Advancing in the Structure-Function of ENA ATPases
4. Materials and Methods
4.1. Construction of the Profile of the Hidden Markov Model
4.2. Construction of Phylogenetic Trees
4.3. Protein Structure Prediction and Three-Dimensional Modeling of the NcENA1 Protein
4.4. Calculations of Poisson-Boltzmann (PB) Electrostatic Potentials (EPs)
4.5. Site-Directed Mutagenesis of NcENA1 ATPase
4.6. Yeast Transformation for Phenotypic Characterization of the NcENA1 Mutants
4.7. Salt Tolerance Assay of Yeast Transformants with Mutated NcENA1 Constructs
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palmgren, M.G.; Nissen, P. P-Type ATPases. Annu. Rev. Biophys. 2011, 40, 243–266. [Google Scholar] [CrossRef]
- Pedersen, P.L.; Carafoli, C. Ion motive ATPases. I. Ubiquity, properties, and significance to cell function. Trends Biochem. Sci. 1987, 12, 146–150. [Google Scholar] [CrossRef]
- Albers, R.W. Biochemical aspects of active transport. Annu. Rev. Biochem. 1967, 36, 727–756. [Google Scholar] [CrossRef]
- Post, R.L.; Hegyvary, C.; Kume, S. Activation by adenosine triphosphate in the phosphorylation kinetics of sodium and potassium ion transport adenosine triphosphatase. J. Biol. Chem. 1972, 247, 6530–6540. [Google Scholar] [CrossRef]
- Toyoshima, C.; Nakasako, M.; Nomura, H.; Ogawa, H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature 2000, 405, 647–655. [Google Scholar] [CrossRef]
- Toyoshima, C. Structural aspects of ion pumping by Ca2+-ATPase of sarcoplasmic reticulum. Arch. Biochem. Biophys. 2008, 476, 3–11. [Google Scholar] [CrossRef]
- Nyblom, M.; Poulsen, H.; Gourdon, P.; Reinhard, L.; Andersson, M.; Lindahl, E.; Fedosova, N.; Nissen, P. Crystal structure of Na+, K+-ATPase in the Na+-bound state. Science 2013, 342, 123–127. [Google Scholar] [CrossRef]
- Pedersen, B.P.; Buch-Pedersen, M.J.; Preben Morth, J.; Palmgren, M.G.; Nissen, P. Crystal structure of the plasma membrane proton pump. Nature 2007, 450, 1111–1114. [Google Scholar] [CrossRef]
- Dyla, M.; Terry, D.S.; Kjaergaard, M.; Sørensen, T.L.M.; Andersen, J.L.; Andersen, J.P.; Knudsen, C.R.; Altman, R.B.; Nissen, P.; Blanchard, S.C. Dynamics of P-type ATPase transport revealed by single-molecule FRET. Nature 2017, 551, 346–351. [Google Scholar] [CrossRef]
- Axelsen, K.B.; Palmgren, M.G. Evolution of substrate specificities in the P-type ATPase superfamily. J. Mol. Evol. 1998, 46, 84–101. [Google Scholar] [CrossRef]
- Sørensen, D.M.; Holen, H.W.; Holemans, T.; Vangheluwe, P.; Palmgren, M.G. Towards defining the substrate of orphan P5A-ATPases. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 524–535. [Google Scholar] [CrossRef]
- Møller, J.V.; Juul, B.; le Maire, M. Structural organization, ion transport, and energy transduction of P-type ATPases. Biochim. Biophys. Acta 1996, 1286, 1–51. [Google Scholar] [CrossRef]
- Haro, R.; Garciadeblas, B.; Rodriguez-Navarro, A. A novel P-type ATPase from yeast involved in sodium transport. FEBS Lett. 1991, 291, 189–191. [Google Scholar] [CrossRef]
- Benito, B.; Quintero, F.J.; Rodríguez-Navarro, A. Overexpression of the sodium ATPase of Saccharomyces cerevisiae: Conditions for phosphorylation from ATP and p(i). Biochim. Biophys. Acta Biomembr. 1997, 1328, 214–226. [Google Scholar] [CrossRef]
- Benito, B.; Garciadeblás, B.; Rodríguez-Navarro, A. Molecular cloning of the calcium and sodium ATPases in Neurospora crassa. Mol. Microbiol. 2000, 35, 1079–1088. [Google Scholar] [CrossRef]
- Benito, B.; Garciadeblás, B.; Rodríguez-Navarro, A. Potassium- or sodium-efflux ATPase, a key enzyme in the evolution of fungi. Microbiology 2002, 148, 933–941. [Google Scholar] [CrossRef]
- Rodriguez-Navarro, A.; Benito, B. Sodium or potassium efflux ATPase A fungal, bryophyte, and protozoal ATPase. Biochim. Biophys. Acta Biomembr. 2010, 1798, 1841–1853. [Google Scholar] [CrossRef]
- Benito, B.; Garciadeblas, B.; Pérez-Martín, J.; Rodríguez-Navarro, A. Growth at High pH and Sodium and Potassium Tolerance in Media above the Cytoplasmic pH Depend on ENA ATPases in Ustilago maydis. Eukaryot. Cell 2009, 8, 821–829. [Google Scholar] [CrossRef]
- Benito, B.; Rodriguez-Navarro, A. Molecular cloning and characterization of a sodium-pump ATPase of the moss Physcomitrella patens. Plant J. 2003, 36, 382–389. [Google Scholar] [CrossRef]
- Idnurm, A.; Walton, F.J.; Floyd, A.; Reedy, J.L.; Heitman, J. Identification of ENA1 as a virulence gene of the human pathogenic fungus Cryptococcus neoformans through signature-tagged insertional mutagenesis. Eukaryot. Cell 2009, 8, 315–326. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, 29–37. [Google Scholar] [CrossRef]
- Nordberg, H.; Cantor, M.; Dusheyko, S.; Hua, S.; Poliakov, A.; Shabalov, I.; Smirnova, T.; Grigoriev, I.V.; Dubchak, I. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res. 2014, 42, 26–31. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar]
- Spillman, N.J.; Allen, R.J.W.; McNamara, C.W.; Yeung, B.K.S.; Winzeler, E.A.; Diagana, T.T.; Kirk, K. Na+ regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the spiroindolone antimalarials. Cell Host Microbe 2013, 13, 227–237. [Google Scholar] [CrossRef]
- Ramanathan, A.A.; Morrisey, J.M.; Daly, T.M.; Bergman, L.W.; Mather, M.W.; Vaidya, A.B. Oligomerization of the antimalarial drug target PfATP4 is essential for parasite survival. bioRxiv 2019, 1–23. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. IDDT: A local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef]
- Carvacho, I.; Gonzalez, W.; Torres, Y.P.; Brauchi, S.; Alvarez, O.; Gonzalez-Nilo, F.D.; Latorre, R. Intrinsic electrostatic potential in the BK channel pore: Role in determining single channel conductance and block. J. Gen. Physiol. 2008, 131, 147–161. [Google Scholar] [CrossRef]
- Lee, A.G. Ca2+-ATPase structure in the E1 and E2 conformations: Mechanism, helix-helix and helix-lipid interactions. Biochim. Biophys. Acta Biomembr. 2002, 1565, 246–266. [Google Scholar] [CrossRef]
- Palmgren, M. P-type ATPases: Many more enigmas left to solve. J. Biol. Chem. 2023, 299, 105352. [Google Scholar] [CrossRef]
- Rosling, J.E.O.; Ridgway, M.C.; Summers, R.L.; Kirk, K.; Lehane, A.M. Biochemical characterization and chemical inhibition of PfATP4-associated Na-ATPase activity in Plasmodium falciparum membranes. J. Biol. Chem. 2018, 293, 13327–13337. [Google Scholar] [CrossRef] [PubMed]
- Drew, D.P.; Hrmova, M.; Lunde, C.; Jacobs, A.K.; Tester, M.; Fincher, G.B. Structural and functional analyses of PpENA1 provide insights into cation binding by type IID P-type ATPases in lower plants and fungi. Biochim. Biophys. Acta Biomembr. 2011, 1808, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Shimono, Y.; Tsuji, H.; Tamai, Y. Role of the glutamic and aspartic residues in Na+-ATPase function in the ZrENA1 gene of Zygosaccharomyces rouxii. FEMS Microbiol. Lett. 2002, 209, 37–41. [Google Scholar] [CrossRef]
- Benito, B.; Haro, R.; Amtmann, A.; Cuin, T.A.; Dreyer, I. The twins K+ and Na+ in plants. J. Plant Physiol. 2014, 171, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Serrano, R. Characterization of the plasma membrane ATPase of Saccharomyces cerevisiae. Mol. Cell. Biochem. 1978, 22, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Sweadner, K.J. Chapter 10 Colorimetric Assays of Na, K-ATPase. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; Volume 1377, pp. 89–104. ISBN 9781493931798. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Steinegger, M.; Söding, J. MMseqs2 desktop and local web server app for fast, interactive sequence searches. Bioinformatics 2019, 35, 2856–2858. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Von Den Driesch, L.; Galiez, C.; Martin, M.J.; Soding, J.; Steinegger, M. Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 2017, 45, D170–D176. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-, E.M.; Qi, Y.; Lee, J.; Monje-galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder Toward Realistic Biological Membrane Simulations. J. Comput. Chem. 2015, 35, 1997–2004. [Google Scholar] [CrossRef]
- Florek, O.B.; Clifton, L.A.; Wilde, M.; Arnold, T.; Green, R.J.; Frazier, R.A. Lipid composition in fungal membrane models: Effect of lipid fluidity. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 1233–1244. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, 665–667. [Google Scholar] [CrossRef]
- Ho, S.N.; Hunt, H.D.; Horton, R.M.; Pullen, J.K.; Pease, L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 1989, 77, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Rentsch, D.; Laloi, M.; Rouhara, I.; Schmelzer, E.; Delrot, S.; Frommer, W.B. NTR1 encodes a high affinity oligopeptide transporter in Arabidopsis. FEBS Lett. 1995, 370, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Banuelos, M.A.; Sychrova, H.; Bleykasten-Grosshans, C.; Souciet, J.; Potier, S. The Nha1 antiporter of Saccharomyces cerevisiae mediates sodium and potassium efflux. Microbiology 1998, 144, 2749–2758. [Google Scholar]
- Elble, R.C. A simple and efficient procedure for transformation of yeasts. Biotechniques 1992, 13, 18–20. [Google Scholar]
- Sherman, F. Getting started with yeast. Methods Enzymol. 2002, 350, 3–41. [Google Scholar]
- Rodriguez-Navarro, A.; Ramos, J. Dual system for potassium transport in Saccharomyces cerevisiae. J. Bacteriol. 1984, 159, 940–945. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Name | Protein Sequence | TM Location | Growth in Na+ | Growth in K+ | Comments on Phenotype |
|---|---|---|---|---|---|
| NcENA1(wt) | I P A S L | TM4 | ++ | + | Na+-efflux/slight K+ efflux activity |
| A343E | I P E S L | “ | + | −− | ↓ Na+-efflux/Loss of K+ efflux |
| S344G | I P A G L | “ | ++ | − | ↓ K+ efflux |
| NcENA1(wt) | E N I A Q | TM5 | ++ | + | Na+-efflux/slight K+ efflux activity |
| E818V | V N I A Q | “ | −− | −− | Loss of Na+ and K+ efflux function |
| N819V | E V I A Q | “ | −− | −− | Loss of Na+ and K+ efflux function |
| Q822V | E N I A V | “ | −− | ++ | Loss of Na+ efflux/ ↑ K+ efflux |
| NcENA1(wt) | T S G L P D | TM6 | ++ | + | Na+-efflux/slight K+ efflux activity |
| T855V | V S G L P D | “ | −− | −− | Loss of Na+ and K+ efflux function |
| S856A | T A G L P D | “ | + | −− | ↓ Na+-efflux/Loss of K+ efflux |
| D860V | T S G L P V | “ | + | ++ | ↓ Na+-efflux/ ↑ K+ efflux |
| D860S | T S G L P S | “ | −− | −− | Loss of Na+ and K+ efflux function |
| NcENA1(wt) | F I I D M I F Y | TM7 | ++ | + | Na+-efflux/slight K+ efflux activity |
| D892V | F I I V M I F Y | “ | −− | −− | Loss of Na+ and K+ efflux function |
| NcENA1(wt) | F L A W E L V D M R R S | TM8 | ++ | + | Na+-efflux/slight K+ efflux activity |
| S963A | F L A W E L V D M R R A | “ | + | ++ | ↓ Na+-efflux/ ↑ K+ efflux |
| NcENA1(wt) | W E W G I V | C-term | ++ | + | Na+-efflux/slight K+ efflux activity |
| E1021V | W V W G I V | “ | −− | −− | Loss of Na+ and K+ efflux function |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilella, M.; Garciadeblás, B.; Fernández Pacios, L.; Benito, B. Phylogenetic and Structure-Function Analyses of ENA ATPases: A Case Study of the ENA1 Protein from the Fungus Neurospora crassa. Int. J. Mol. Sci. 2024, 25, 514. https://doi.org/10.3390/ijms25010514
Aguilella M, Garciadeblás B, Fernández Pacios L, Benito B. Phylogenetic and Structure-Function Analyses of ENA ATPases: A Case Study of the ENA1 Protein from the Fungus Neurospora crassa. International Journal of Molecular Sciences. 2024; 25(1):514. https://doi.org/10.3390/ijms25010514
Chicago/Turabian StyleAguilella, Marcos, Blanca Garciadeblás, Luis Fernández Pacios, and Begoña Benito. 2024. "Phylogenetic and Structure-Function Analyses of ENA ATPases: A Case Study of the ENA1 Protein from the Fungus Neurospora crassa" International Journal of Molecular Sciences 25, no. 1: 514. https://doi.org/10.3390/ijms25010514