
Mitochondrially Targeted Gene Therapy Rescues Visual Loss in a Mouse Model of Leber’s Hereditary Optic Neuropathy
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
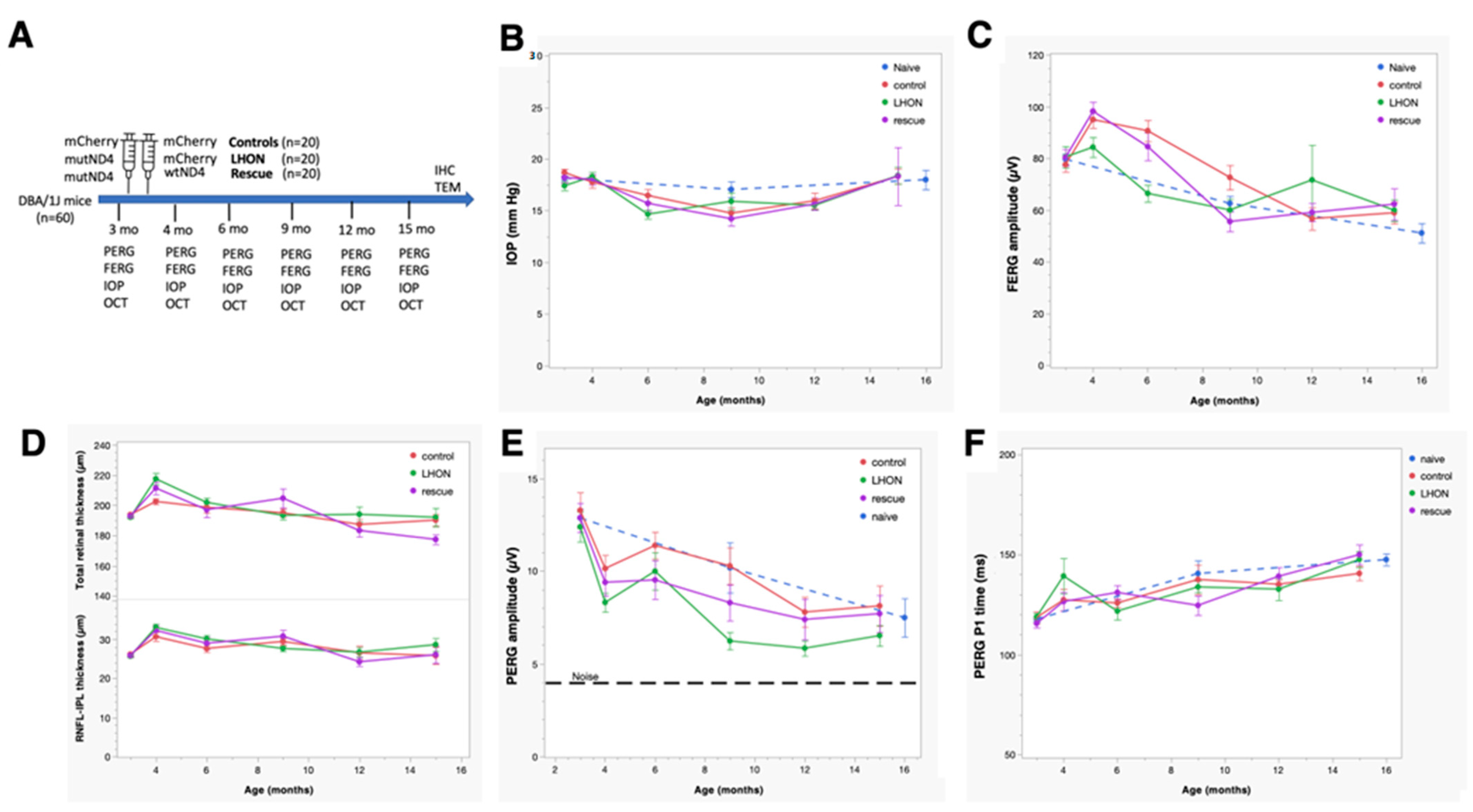
2.1. Wildtype hND4 Rescues RGC Dysfunction Induced by Mutant hND4
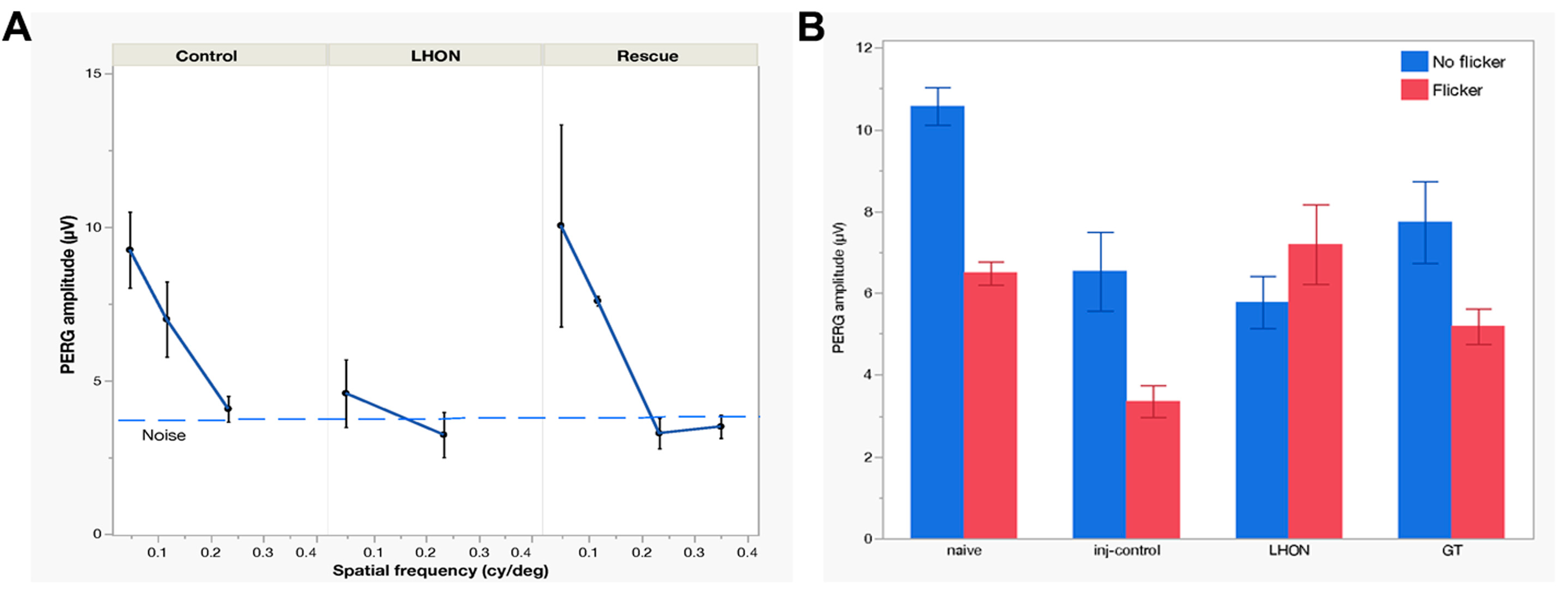
2.2. Wildtype hND4 Rescues Loss of Visual Acuity Induced by Mutant hND4
2.3. Wildtype hND4 Rescues Loss of RGC Metabolic Autoregulation Induced by Mutant hND4
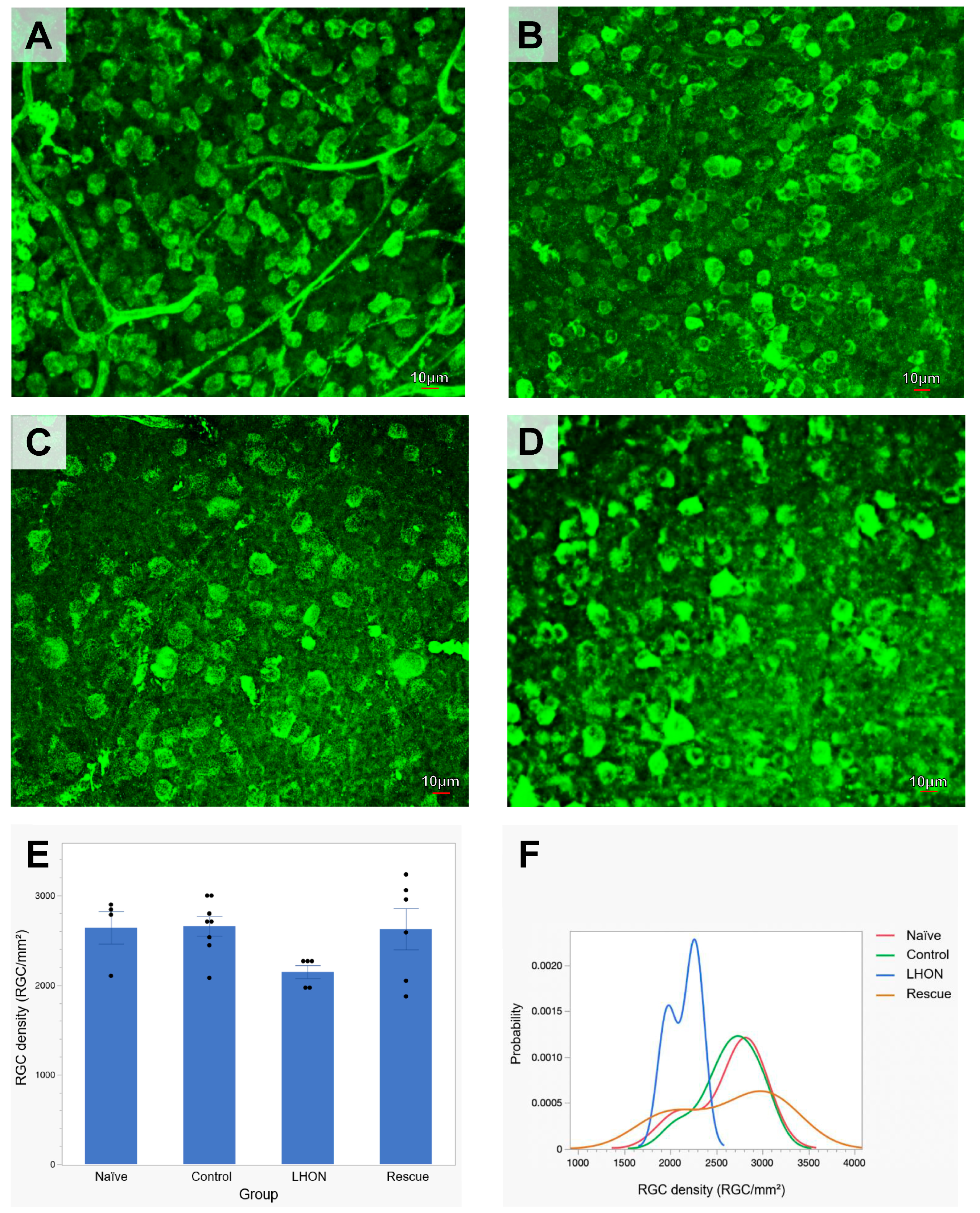
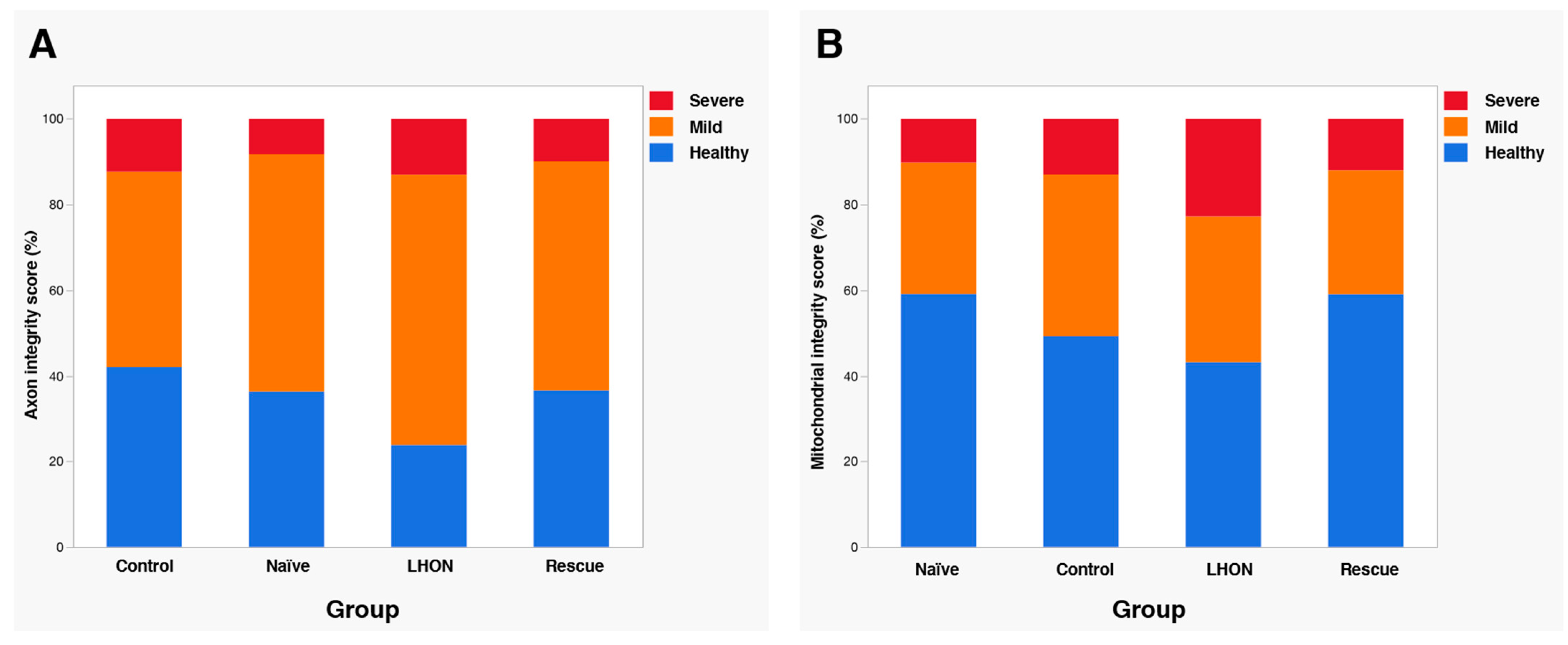
2.4. Wildtype hND4 Rescues Loss of RGCs Induced by Mutant hND4
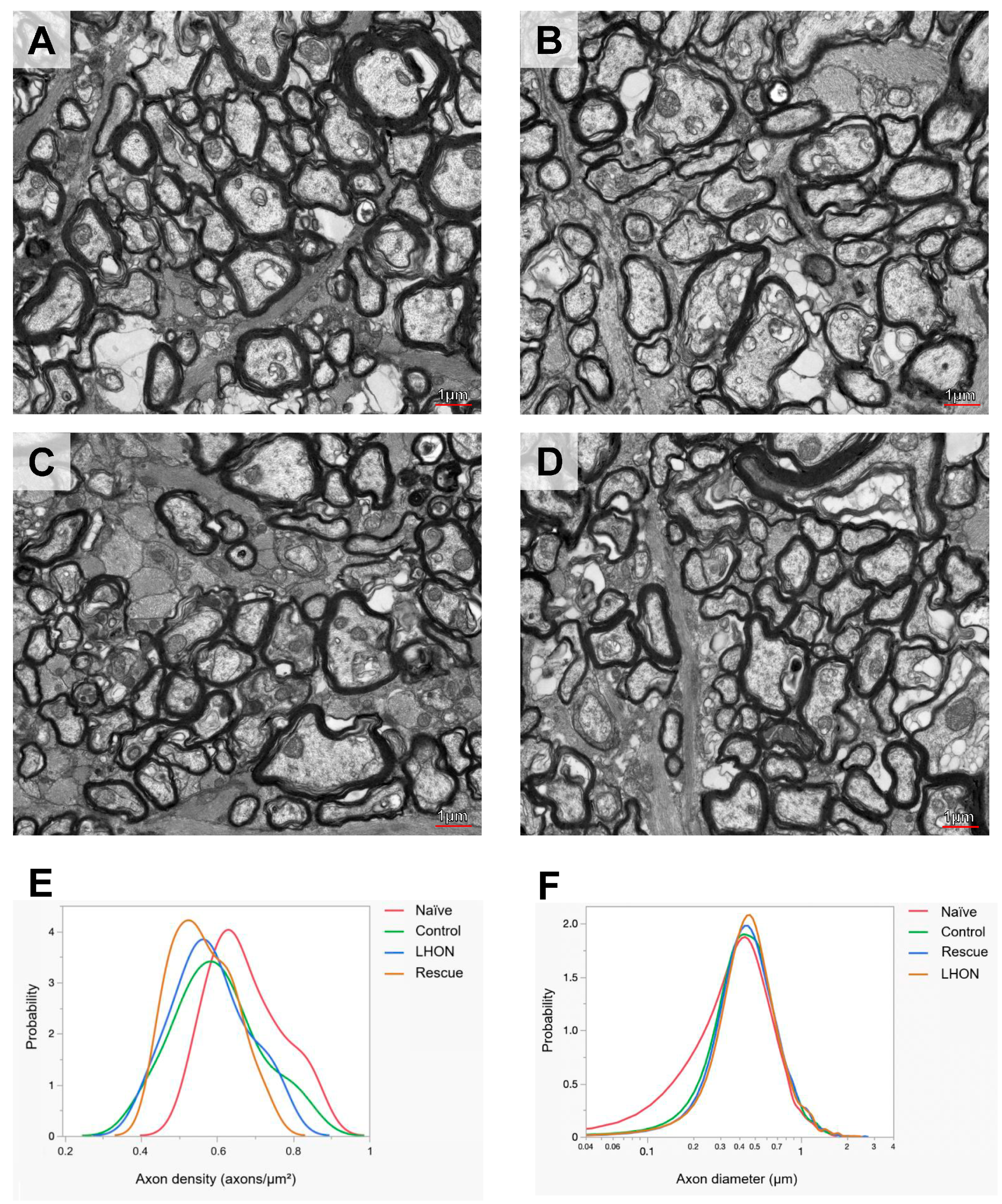
2.5. Wildtype hND4 Rescues Optic Atrophy Induced by Mutant hND4
3. Discussion
4. Materials and Methods
4.1. Plasmids and AAVs
4.2. Animals and Intravitreal Injections (IVI)
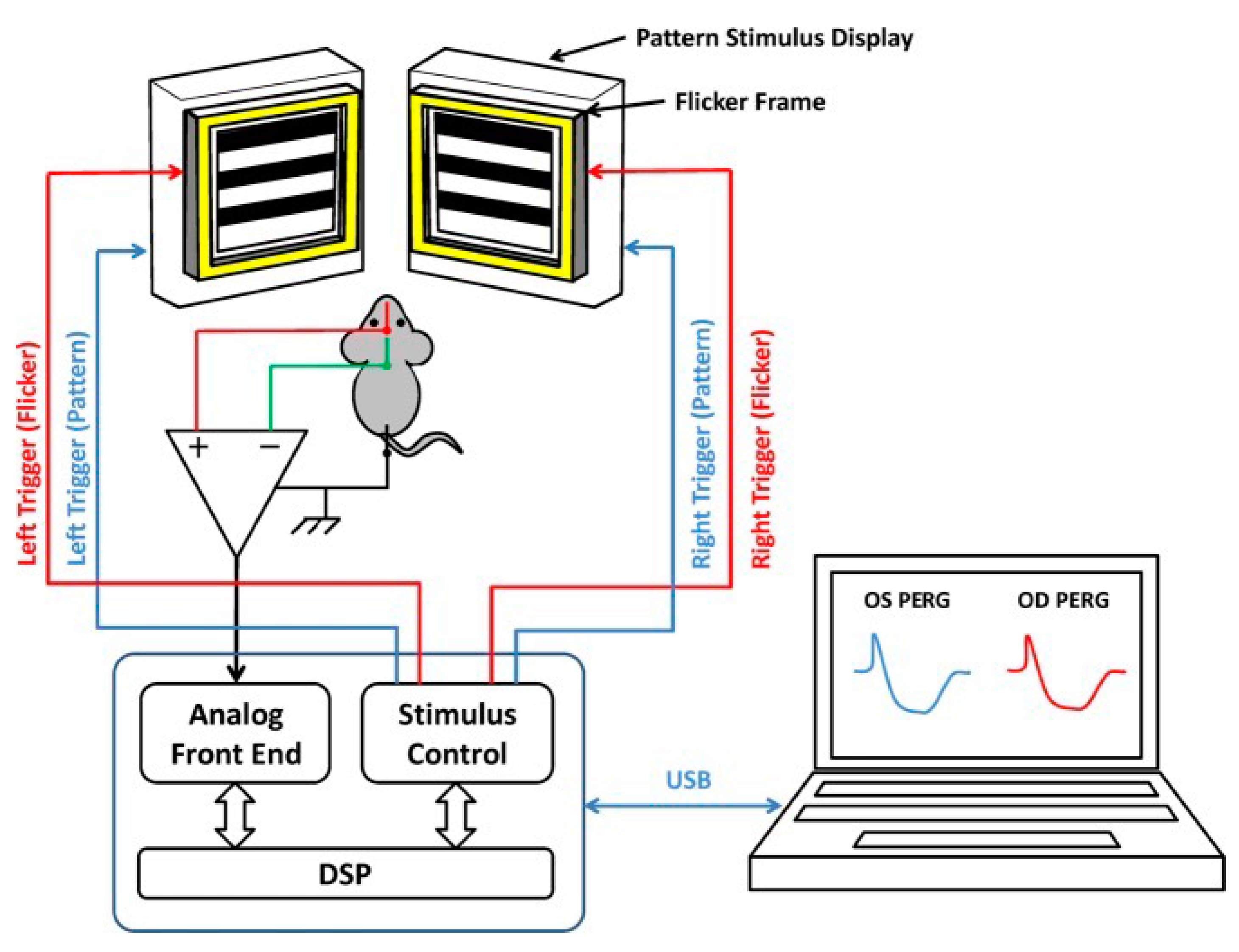
4.3. IOP, PERG, FERG, and SD-OCT
4.4. PERG-Based Visual Acuity
4.5. Flicker-Induced PERG Adaptation
4.6. Immunostaining
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kauppila, J.H.K.; Baines, H.L.; Bratic, A.; Simard, M.L.; Freyer, C.; Mourier, A.; Stamp, C.; Filograna, R.; Larsson, N.G.; Greaves, L.C.; et al. A Phenotype-Driven Approach to Generate Mouse Models with Pathogenic mtDNA Mutations Causing Mitochondrial Disease. Cell Rep. 2016, 16, 2980–2990. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial diseases in man and mouse. Science 1999, 283, 1482–1488. [Google Scholar] [CrossRef]
- Moraes, C.T.; Ricci, E.; Petruzzella, V.; Shanske, S.; DiMauro, S.; Schon, E.A.; Bonilla, E. Molecular analysis of the muscle pathology associated with mitochondrial DNA deletions. Nat. Genet. 1992, 1, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Johns, D.R. Seminars in medicine of the Beth Israel Hospital, Boston. Mitochondrial DNA and disease. N. Engl. J. Med. 1995, 333, 638–644. [Google Scholar] [CrossRef]
- Shoffner, J.M.; Wallace, D.C. Heart disease and mitochondrial DNA mutations. Heart Dis. Stroke 1992, 1, 235–241. [Google Scholar] [PubMed]
- Nissanka, N.; Minczuk, M.; Moraes, C.T. Mechanisms of Mitochondrial DNA Deletion Formation. Trends Genet. 2019, 35, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Fontana, G.A.; Gahlon, H.L. Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res. 2020, 48, 11244–11258. [Google Scholar] [CrossRef]
- Herbst, A.; Wanagat, J.; Cheema, N.; Widjaja, K.; McKenzie, D.; Aiken, J.M. Latent mitochondrial DNA deletion mutations drive muscle fiber loss at old age. Aging Cell 2016, 15, 1132–1139. [Google Scholar] [CrossRef]
- Rocha, M.C.; Rosa, H.S.; Grady, J.P.; Blakely, E.L.; He, L.; Romain, N.; Haller, R.G.; Newman, J.; McFarland, R.; Ng, Y.S.; et al. Pathological mechanisms underlying single large-scale mitochondrial DNA deletions. Ann. Neurol. 2018, 83, 115–130. [Google Scholar] [CrossRef]
- Jurkute, N.; Yu-Wai-Man, P. Leber hereditary optic neuropathy: Bridging the translational gap. Curr. Opin. Ophthalmol. 2017, 28, 403–409. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Griffiths, P.G.; Hudson, G.; Chinnery, P.F. Inherited mitochondrial optic neuropathies. J. Med. Genet. 2009, 46, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Karaarslan, C. Leber’s Hereditary Optic Neuropathy as a Promising Disease for Gene Therapy Development. Adv. Ther. 2019, 36, 3299–3307. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Chinnery, P.F. Leber Hereditary Optic Neuropathy. In GeneReviews(R); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2021. [Google Scholar]
- Zuccarelli, M.; Vella-Szijj, J.; Serracino-Inglott, A.; Borg, J.J. Treatment of Leber’s hereditary optic neuropathy: An overview of recent developments. Eur. J. Ophthalmol. 2020, 30, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Bacman, S.R.; Williams, S.L.; Pinto, M.; Peralta, S.; Moraes, C.T. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat. Med. 2013, 19, 1111–1113. [Google Scholar] [CrossRef]
- Chadderton, N.; Palfi, A.; Millington-Ward, S.; Gobbo, O.; Overlack, N.; Carrigan, M.; O’Reilly, M.; Campbell, M.; Ehrhardt, C.; Wolfrum, U.; et al. Intravitreal delivery of AAV-NDI1 provides functional benefit in a murine model of Leber hereditary optic neuropathy. Eur. J. Hum. Genet. 2013, 21, 62–68. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P. Therapeutic Approaches to Inherited Optic Neuropathies. Semin. Neurol. 2015, 35, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Bahr, T.; Welburn, K.; Donnelly, J.; Bai, Y. Emerging model systems and treatment approaches for Leber’s hereditary optic neuropathy: Challenges and opportunities. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165743. [Google Scholar] [CrossRef]
- Liu, H.L.; Yuan, J.J.; Zhang, Y.; Tian, Z.; Li, X.; Wang, D.; Du, Y.Y.; Song, L.; Li, B. Factors associated with rapid improvement in visual acuity in patients with Leber’s hereditary optic neuropathy after gene therapy. Acta Ophthalmol. 2020, 98, e730–e733. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Yuan, J.; Tian, Z.; Liu, H.; Wang, D.; Li, B. Prognostic factors for visual acuity in patients with Leber’s hereditary optic neuropathy after rAAV2-ND4 gene therapy. Clin. Exp. Ophthalmol. 2019, 47, 774–778. [Google Scholar] [CrossRef]
- Wan, X.; Pei, H.; Zhao, M.J.; Yang, S.; Hu, W.K.; He, H.; Ma, S.Q.; Zhang, G.; Dong, X.Y.; Chen, C.; et al. Efficacy and Safety of rAAV2-ND4 Treatment for Leber’s Hereditary Optic Neuropathy. Sci. Rep. 2016, 6, 21587. [Google Scholar] [CrossRef]
- Yang, S.; Ma, S.Q.; Wan, X.; He, H.; Pei, H.; Zhao, M.J.; Chen, C.; Wang, D.W.; Dong, X.Y.; Yuan, J.J.; et al. Long-term outcomes of gene therapy for the treatment of Leber’s hereditary optic neuropathy. EBioMedicine 2016, 10, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Newman, N.J.; Carelli, V.; Moster, M.L.; Biousse, V.; Sadun, A.A.; Klopstock, T.; Vignal-Clermont, C.; Sergott, R.C.; Rudolph, G.; et al. Bilateral visual improvement with unilateral gene therapy injection for Leber hereditary optic neuropathy. Sci. Transl. Med. 2020, 12, eaaz7423. [Google Scholar] [CrossRef]
- Biousse, V.; Newman, N.J.; Yu-Wai-Man, P.; Carelli, V.; Moster, M.L.; Vignal-Clermont, C.; Klopstock, T.; Sadun, A.A.; Sergott, R.C.; Hage, R.; et al. Long-Term Follow-Up After Unilateral Intravitreal Gene Therapy for Leber Hereditary Optic Neuropathy: The RESTORE Study. J. Neuroophthalmol. 2021, 41, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J.; Yu-Wai-Man, P.; Carelli, V.; Moster, M.L.; Biousse, V.; Vignal-Clermont, C.; Sergott, R.C.; Klopstock, T.; Sadun, A.A.; Barboni, P.; et al. Efficacy and Safety of Intravitreal Gene Therapy for Leber Hereditary Optic Neuropathy Treated within 6 Months of Disease Onset. Ophthalmology 2021, 128, 649–660. [Google Scholar] [CrossRef]
- Vignal-Clermont, C.; Girmens, J.F.; Audo, I.; Said, S.M.; Errera, M.H.; Plaine, L.; O’Shaughnessy, D.; Taiel, M.; Sahel, J.A. Safety of Intravitreal Gene Therapy for Treatment of Subjects with Leber Hereditary Optic Neuropathy due to Mutations in the Mitochondrial ND4 Gene: The REVEAL Study. BioDrugs 2021, 35, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J.; Yu-Wai-Man, P.; Carelli, V.; Biousse, V.; Moster, M.L.; Vignal-Clermont, C.; Sergott, R.C.; Klopstock, T.; Sadun, A.A.; Girmens, J.F.; et al. Intravitreal Gene Therapy vs. Natural History in Patients with Leber Hereditary Optic Neuropathy Carrying the m.11778G>A ND4 Mutation: Systematic Review and Indirect Comparison. Front. Neurol. 2021, 12, 662838. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.L.; Feuer, W.J.; Davis, J.L.; Porciatti, V.; Yu, H.; Levy, R.B.; Vanner, E.; Guy, J. Leber Hereditary Optic Neuropathy Gene Therapy: Adverse Events and Visual Acuity Results of all Patient Groups. Am. J. Ophthalmol. 2022, 241, 262–271. [Google Scholar] [CrossRef]
- Kaeppel, C.; Beattie, S.; Fronza, R.; van Logtenstein, R.; Salmon, F.; Schmidt, S.; Wolf, S.; Nowrouzi, A.; Glimm, H.; von Kalle, C.; et al. AAV Integrates Randomly into the Nuclear and Mitochondrial Genome after LPLD Gene Therapy. Mol. Ther. 2013, 21, S104–S105. [Google Scholar]
- Yu, H.; Koilkonda, R.D.; Chou, T.H.; Porciatti, V.; Ozdemir, S.S.; Chiodo, V.; Boye, S.L.; Boye, S.E.; Hauswirth, W.W.; Lewin, A.S.; et al. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber’s hereditary optic neuropathy in a mouse model. Proc. Natl. Acad. Sci. USA 2012, 109, E1238–E1247. [Google Scholar] [CrossRef]
- Yu, H.; Koilkonda, R.D.; Chou, T.H.; Porciatti, V.; Mehta, A.; Hentall, I.D.; Chiodo, V.A.; Boye, S.L.; Hauswirth, W.W.; Lewin, A.S.; et al. Consequences of zygote injection and germline transfer of mutant human mitochondrial DNA in mice. Proc. Natl. Acad. Sci. USA 2015, 112, E5689–E5698. [Google Scholar] [CrossRef]
- Liu, Y.; Eastwood, J.D.; Alba, D.E.; Velmurugan, S.; Sun, N.; Porciatti, V.; Lee, R.K.; Hauswirth, W.W.; Guy, J.; Yu, H. Gene therapy restores mitochondrial function and protects retinal ganglion cells in optic neuropathy induced by a mito-targeted mutant ND1 gene. Gene Ther. 2022, 29, 368–378. [Google Scholar] [CrossRef]
- Yu, H.; Sant, D.W.; Wang, G.; Guy, J. Mitochondrial Transfer of the Mutant Human ND6T14484C Gene Causes Visual Loss and Optic Neuropathy. Transl. Vis. Sci. Technol. 2020, 9, 1. [Google Scholar] [CrossRef]
- Velmurugan, S.; Chou, T.H.; Eastwood, J.D.; Porciatti, V.; Liu, Y.; Hauswirth, W.W.; Guy, J.; Yu, H. Comparison of different gene-therapy methods to treat Leber hereditary optic neuropathy in a mouse model. Front. Neurosci. 2023, 17, 1119724. [Google Scholar] [CrossRef] [PubMed]
- Lietze, A. The role of particulate insoluble substances in food allergy. 3. Heat labile antibody to wheat starch in sera of wheat sensitive patients. Ann. Allergy 1969, 27, 9–12. [Google Scholar] [PubMed]
- Sadowsky, C.; Muhl, Z.F.; Sakols, E.I.; Sommerville, J.M. Temporomandibular joint sounds related to orthodontic therapy. J. Dent. Res. 1985, 64, 1392–1395. [Google Scholar] [CrossRef]
- Shi, C.; Yuan, X.; Chang, K.; Cho, K.S.; Xie, X.S.; Chen, D.F.; Luo, G. Optimization of Optomotor Response-based Visual Function Assessment in Mice. Sci. Rep. 2018, 8, 9708. [Google Scholar] [CrossRef]
- Kretschmer, F.; Sajgo, S.; Kretschmer, V.; Badea, T.C. A system to measure the Optokinetic and Optomotor response in mice. J. Neurosci. Methods 2015, 256, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, G.; Brem, G.; Montoliu, L. Correction of retinal abnormalities found in albinism by introduction of a functional tyrosinase gene in transgenic mice and rabbits. Brain Res. Dev. Brain Res. 1997, 99, 95–102. [Google Scholar] [CrossRef]
- Porciatti, V.; Pizzorusso, T.; Cenni, M.C.; Maffei, L. The visual response of retinal ganglion cells is not altered by optic nerve transection in transgenic mice overexpressing Bcl-2. Proc. Natl. Acad. Sci. USA 1996, 93, 14955–14959. [Google Scholar] [CrossRef]
- Chou, T.H.; Toft-Nielsen, J.; Porciatti, V. Adaptation of retinal ganglion cell function during flickering light in the mouse. Sci. Rep. 2019, 9, 18396. [Google Scholar] [CrossRef]
- Riva, C.E.; Logean, E.; Falsini, B. Visually evoked hemodynamical response and assessment of neurovascular coupling in the optic nerve and retina. Prog. Retin. Eye Res. 2005, 24, 183–215. [Google Scholar] [CrossRef] [PubMed]
- Albanna, W.; Kotliar, K.; Luke, J.N.; Alpdogan, S.; Conzen, C.; Lindauer, U.; Clusmann, H.; Hescheler, J.; Vilser, W.; Schneider, T.; et al. Non-invasive evaluation of neurovascular coupling in the murine retina by dynamic retinal vessel analysis. PLoS ONE 2018, 13, e0204689. [Google Scholar] [CrossRef] [PubMed]
- Cepurna, W.O.; Kayton, R.J.; Johnson, E.C.; Morrison, J.C. Age related optic nerve axonal loss in adult Brown Norway rats. Exp. Eye Res. 2005, 80, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Hedberg-Buenz, A.; Soukup, D.A.; Taghizadeh, S.; Wang, K.; Anderson, M.G.; Garvin, M.K. AxonDeep: Automated Optic Nerve Axon Segmentation in Mice with Deep Learning. Transl. Vis. Sci. Technol. 2021, 10, 22. [Google Scholar] [CrossRef]
- Zhu, Y.; Pappas, A.C.; Wang, R.; Seifert, P.; Sun, D.; Jakobs, T.C. Ultrastructural Morphology of the Optic Nerve Head in Aged and Glaucomatous Mice. Invest. Ophthalmol. Vis. Sci. 2018, 59, 3984–3996. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, Y.; Liu, H.; Wang, D.; Du, Y.; Tian, Z.; Li, X.; Yang, S.; Pei, H.; Wan, X.; et al. Seven-Year Follow-up of Gene Therapy for Leber’s Hereditary Optic Neuropathy. Ophthalmology 2020, 127, 1125–1127. [Google Scholar] [CrossRef]
- Johnston, I.G.; Williams, B.P. Evolutionary Inference across Eukaryotes Identifies Specific Pressures Favoring Mitochondrial Gene Retention. Cell Syst. 2016, 2, 101–111. [Google Scholar] [CrossRef]
- Allen, J.F. Why chloroplasts and mitochondria retain their own genomes and genetic systems: Colocation for redox regulation of gene expression. Proc. Natl. Acad. Sci. USA 2015, 112, 10231–10238. [Google Scholar] [CrossRef]
- Adams, K.L.; Palmer, J.D. Evolution of mitochondrial gene content: Gene loss and transfer to the nucleus. Mol. Phylogenetics Evol. 2003, 29, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Braha, M.; Porciatti, V.; Chou, T.H. Retinal and cortical visual acuity in a common inbred albino mouse. PLoS ONE 2021, 16, e0242394. [Google Scholar] [CrossRef]
- Pello, R.; Martin, M.A.; Carelli, V.; Nijtmans, L.G.; Achilli, A.; Pala, M.; Torroni, A.; Gomez-Duran, A.; Ruiz-Pesini, E.; Martinuzzi, A.; et al. Mitochondrial DNA background modulates the assembly kinetics of OXPHOS complexes in a cellular model of mitochondrial disease. Hum. Mol. Genet. 2008, 17, 4001–4011. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Valletti, A.; Longo, G.; Bisceglia, L.; Montoya, J.; Emperador, S.; Guerriero, S.; Petruzzella, V. Mitochondrial DNA copy number in affected and unaffected LHON mutation carriers. BMC Res. Notes 2018, 11, 911. [Google Scholar] [CrossRef] [PubMed]
- Howell, N.; Xu, M.; Halvorson, S.; Bodis-Wollner, I.; Sherman, J. A heteroplasmic LHON family: Tissue distribution and transmission of the 11778 mutation. Am. J. Hum. Genet. 1994, 55, 203–206. [Google Scholar] [PubMed]
- Vilkki, J.; Savontaus, M.L.; Nikoskelainen, E.K. Segregation of mitochondrial genomes in a heteroplasmic lineage with Leber hereditary optic neuroretinopathy. Am. J. Hum. Genet. 1990, 47, 95–100. [Google Scholar]
- Zhu, D.P.; Economou, E.P.; Antonarakis, S.E.; Maumenee, I.H. Mitochondrial DNA mutation and heteroplasmy in type I Leber hereditary optic neuropathy. Am. J. Med. Genet. 1992, 42, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Abramoff, M.D.; Garvin, M.K.; Sonka, M. Retinal imaging and image analysis. IEEE Rev. Biomed. Eng. 2010, 3, 169–208. [Google Scholar] [CrossRef]
- Dysli, C.; Enzmann, V.; Sznitman, R.; Zinkernagel, M.S. Quantitative Analysis of Mouse Retinal Layers Using Automated Segmentation of Spectral Domain Optical Coherence Tomography Images. Transl. Vis. Sci. Technol. 2015, 4, 9. [Google Scholar] [CrossRef]
- Fan, Q.; Teo, Y.Y.; Saw, S.M. Application of advanced statistics in ophthalmology. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6059–6065. [Google Scholar] [CrossRef]
- Zeger, S.L.; Liang, K.Y. An overview of methods for the analysis of longitudinal data. Stat. Med. 1992, 11, 1825–1839. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chou, T.-H.; Hao, Z.; Alba, D.; Lazo, A.; Gallo Afflitto, G.; Eastwood, J.D.; Porciatti, V.; Guy, J.; Yu, H. Mitochondrially Targeted Gene Therapy Rescues Visual Loss in a Mouse Model of Leber’s Hereditary Optic Neuropathy. Int. J. Mol. Sci. 2023, 24, 17068. https://doi.org/10.3390/ijms242317068
Chou T-H, Hao Z, Alba D, Lazo A, Gallo Afflitto G, Eastwood JD, Porciatti V, Guy J, Yu H. Mitochondrially Targeted Gene Therapy Rescues Visual Loss in a Mouse Model of Leber’s Hereditary Optic Neuropathy. International Journal of Molecular Sciences. 2023; 24(23):17068. https://doi.org/10.3390/ijms242317068
Chicago/Turabian StyleChou, Tsung-Han, Zixuan Hao, Diego Alba, Angelina Lazo, Gabriele Gallo Afflitto, Jeremy D. Eastwood, Vittorio Porciatti, John Guy, and Hong Yu. 2023. "Mitochondrially Targeted Gene Therapy Rescues Visual Loss in a Mouse Model of Leber’s Hereditary Optic Neuropathy" International Journal of Molecular Sciences 24, no. 23: 17068. https://doi.org/10.3390/ijms242317068