
Forsythiaside A Activates AMP-Activated Protein Kinase and Regulates Oxidative Stress via Nrf2 Signaling
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
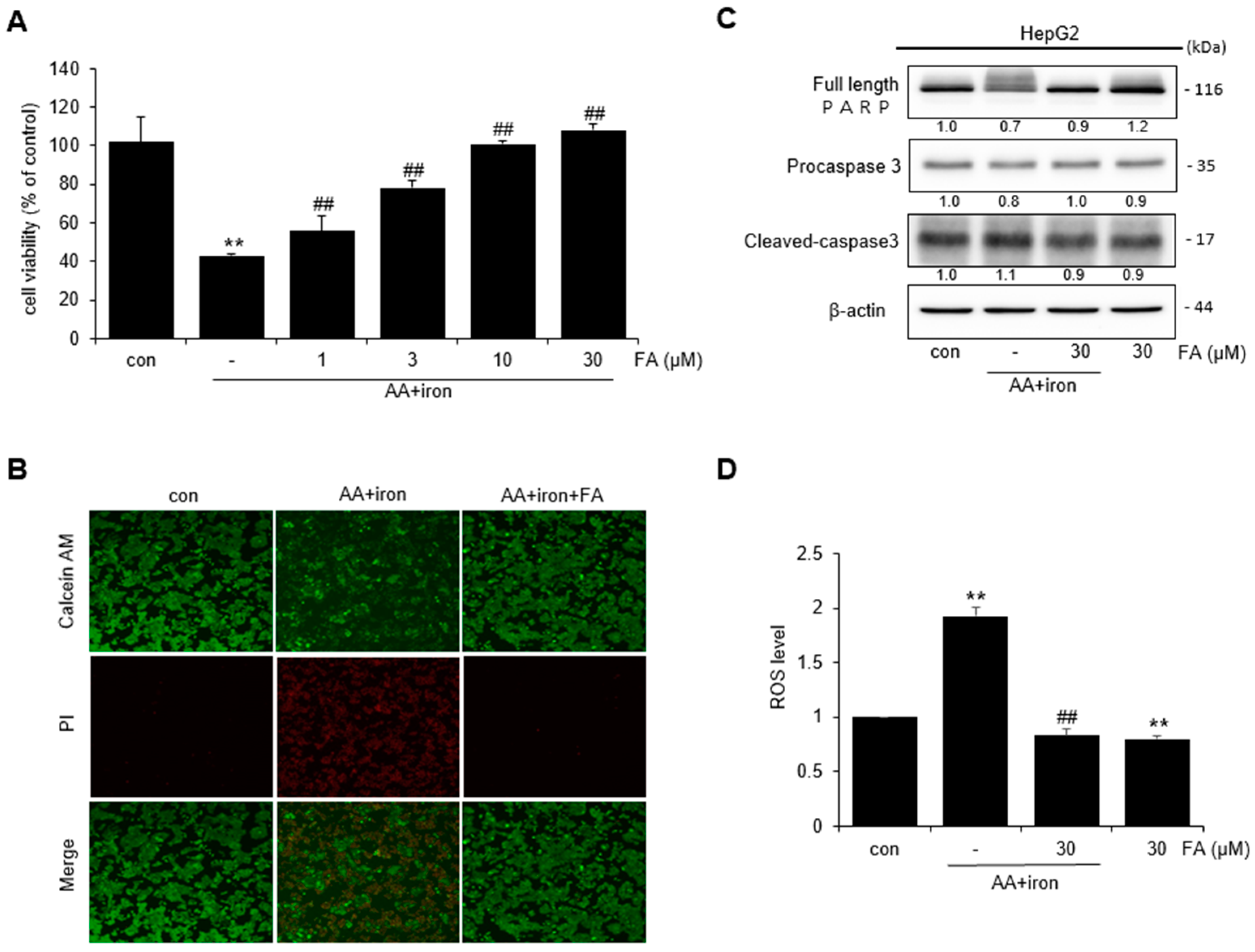
2.1. Effect of FA on AA + Iron-Induced Cytotoxicity
2.2. Effect of FA on AA + Iron ROS Generation and Mitochondrial Dysfunction
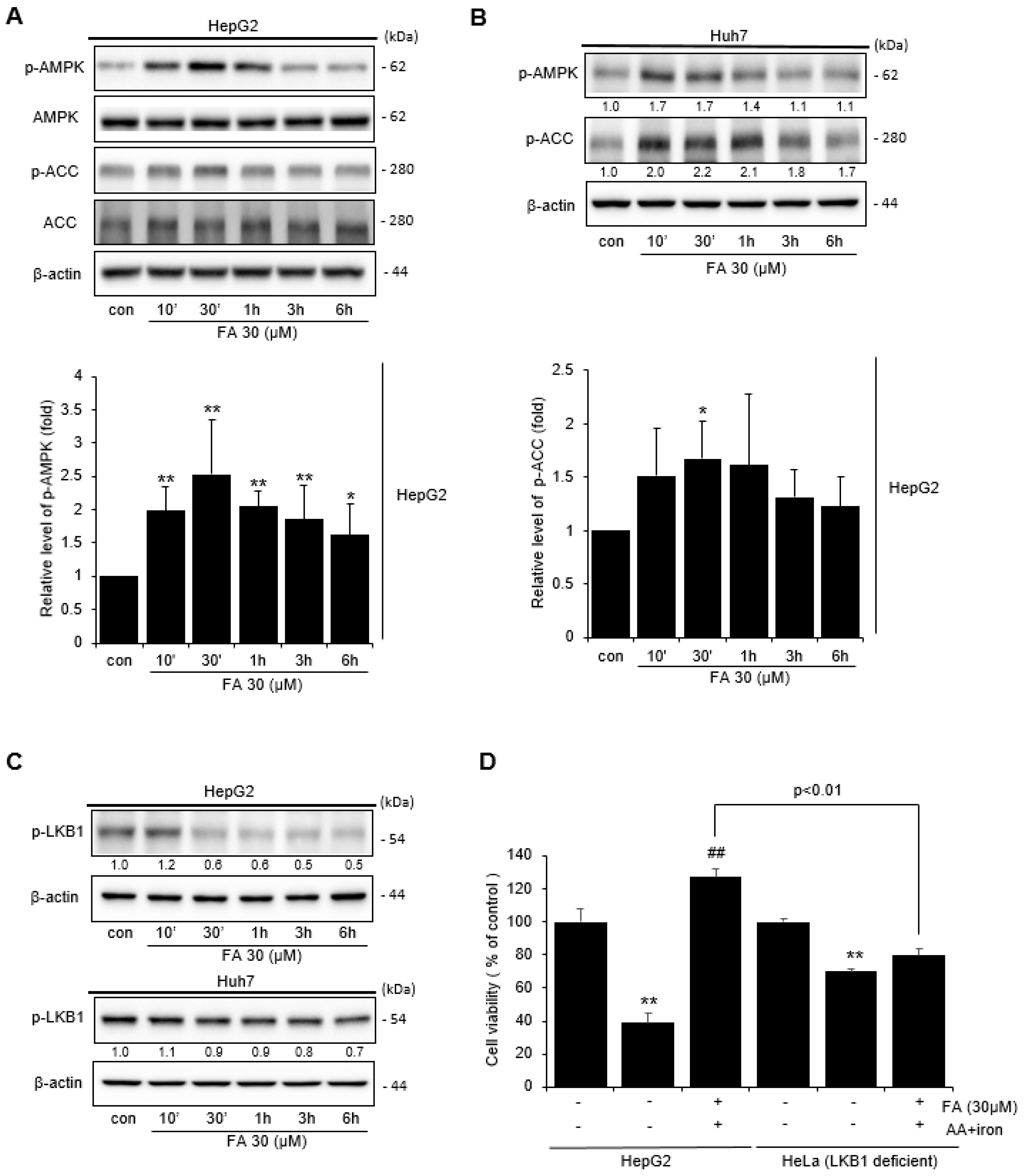
2.3. Effect of FA on AMPK Activation
2.4. Effect of FA on Nuclear Nrf2 Activation
2.5. Effect of FA on ERK Activation
2.6. Effect of FA on Carbon Tetrachloride (CCl₄)-Induced Liver Injury
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. MTT Assay
4.4. Measurement of ROS Production
4.5. Flow Cytometric Analysis
4.6. FACS Analysis of Annexin V and PI Staining
4.7. Mitochondrial Membrane Potential (ΔΨm) Analysis
4.8. Measurement of Superoxide in Mitochondria
4.9. Preparation of Whole-Cell Lysate and Nuclear Extracts
4.10. Western Blot Analysis
4.11. Real-Time RT-PCR Analysis
4.12. Animals and Treatment
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sánchez-Valle, V.; Chávez-Tapia, N.C.; Uribe, M.; Méndez-Sánchez, N. Role of oxidative stress and molecular changes in liver fibrosis: A review. Curr. Med. Chem. 2012, 19, 4850–4860. [Google Scholar] [CrossRef] [PubMed]
- Cichoż-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.L.; Baek, S.Y.; Lee, E.H.; Lee, J.H.; Lee, S.G.; Kim, K.Y.; Jang, M.H.; Park, M.H.; Kim, J.H.; Kim, K.J.; et al. Liqustri lucidi Fructus inhibits hepatic injury and functions as an antioxidant by activation of AMP-activated protein kinase in vivo and in vitro. Chem.-Biol. Interact. 2017, 262, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Chen, Q.L.; Song, Y.N.; Sun, Y.; Wei, B.; Li, X.Y.; Hu, Y.Y.; Liu, P.; Su, S.B. Mechanisms of CCl4-induced liver fibrosis with combined transcriptomic and proteomic analysis. J. Toxicol. Sci. 2016, 41, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Li, A.N.; Li, S.; Zhang, Y.J.; Xu, X.R.; Chen, Y.M.; Li, H.B. Resources and biological activities of natural polyphenols. Nutrients 2014, 6, 6020–6047. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; Lee, S.M.; Shin, S.M.; Hwang, S.J.; Brooks, J.S.; Kang, H.E.; Lee, M.G.; Kim, S.C.; Kim, S.G. Efficacy of sauchinone as a novel AMPK-activating lignan for preventing iron-induced oxidative stress and liver injury. Free Radic. Biol. Med. 2009, 47, 1082–1092. [Google Scholar] [CrossRef]
- Zhou, J.; Zheng, Q.; Chen, Z. The Nrf2 Pathway in Liver Diseases. Front. Cell Dev. Biol. 2022, 10, 826204. [Google Scholar] [CrossRef]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef]
- Mansouri, A.; Reiner, Ž.; Ruscica, M.; Tedeschi-Reiner, E.; Radbakhsh, S.; Bagheri Ekta, M.; Sahebkar, A. Antioxidant Effects of Statins by Modulating Nrf2 and Nrf2/HO-1 Signaling in Different Diseases. J. Clin. Med. 2022, 11, 1313. [Google Scholar] [CrossRef]
- Sherif, I.O. The effect of natural antioxidants in cyclophosphamide-induced hepatotoxicity: Role of Nrf2/HO-1 pathway. Int. Immunopharmacol. 2018, 61, 29–36. [Google Scholar] [CrossRef]
- Silva-Palacios, A.; Ostolga-Chavarría, M.; Zazueta, C.; Königsberg, M. Nrf2: Molecular and epigenetic regulation during aging. Ageing Res. Rev. 2018, 47, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Shao, M.; Zhu, Y. Gastrodin inhibits high glucose-induced inflammation, oxidative stress and apoptosis in podocytes by activating the AMPK/Nrf2 signaling pathway. Exp. Ther. Med. 2022, 23, 168. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, C.; Zhang, L.; Zhao, Y.; Duan, C.; Zhang, X.; Gao, L.; Li, S. Lactobacillus plantarum NA136 improves the non-alcoholic fatty liver disease by modulating the AMPK/Nrf2 pathway. Appl. Microbiol. Biotechnol. 2019, 103, 5843–5850. [Google Scholar] [CrossRef]
- Gong, L.; Zhou, H.; Wang, C.; He, L.; Guo, C.; Peng, C.; Li, Y. Hepatoprotective effect of forsythiaside a against acetaminophen-induced liver injury in zebrafish: Coupling network pharmacology with biochemical pharmacology. J. Ethnopharmacol. 2021, 271, 113890. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Wang, C.; Zhou, H.; Ma, C.; Zhang, Y.; Peng, C.; Li, Y. A review of pharmacological and pharmacokinetic properties of Forsythiaside A. Pharmacol. Res. 2019, 169, 105690. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Shi, Y.L.; Wang, Y.L. Forsythiaside A protects against focal cerebral ischemic injury by mediating the activation of the Nrf2 and endoplasmic reticulum stress pathways. Mol. Med. Rep. 2019, 20, 1313–1320. [Google Scholar] [CrossRef]
- Cheng, L.; Li, F.; Ma, R.; Hu, X. Forsythiaside inhibits cigarette smoke-induced lung inflammation by activation of Nrf2 and inhibition of NF-κB. Int. Immunopharmacol. 2015, 28, 494–499. [Google Scholar] [CrossRef]
- Shin, H.S.; Park, S.Y.; Song, H.G.; Hwang, E.; Lee, D.G.; Yi, T.H. The Androgenic Alopecia Protective Effects of Forsythiaside-A and the Molecular Regulation in a Mouse Model. Phytother. Res. PTR 2015, 29, 870–876. [Google Scholar] [CrossRef]
- Chen, Y.; Wei, W.; Fu, J.; Zhang, T.; Zhao, J.; Ma, T. Forsythiaside A ameliorates sepsis-induced acute kidney injury via anti-inflammation and antiapoptotic effects by regulating endoplasmic reticulum stress. BMC Complement. Med. Ther. 2023, 23, 35. [Google Scholar] [CrossRef]
- Pan, C.W.; Zhou, G.Y.; Chen, W.L.; Zhuge, L.; Jin, L.X.; Zheng, Y.; Lin, W.; Pan, Z.Z. Protective effect of forsythiaside A on lipopolysaccharide/d-galactosamine-induced liver injur. Int. Immunopharmacol. 2015, 26, 80–85. [Google Scholar] [CrossRef]
- Gong, L.; Zhou, H.; Zhang, S.; Wang, C.; Fu, K.; Ma, C.; Zhang, Y.; Peng, C.; Li, Y. CD44-Targeting Drug Delivery System of Exosomes Loading Forsythiaside a Combats Liver Fibrosis via Regulating NLRP3-Mediated Pyroptosis. Adv. Healthc. Mater. 2023, 12, e2202228. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.; Ma, C.; Wang, C.; Zhou, H.; Gong, L.; Zhang, Y.; Li, Y. Forsythiaside A alleviated carbon tetrachloride-induced liver fibrosis by modulating gut microbiota composition to increase short-chain fatty acids and restoring bile acids metabolism disorder. Biomed. Pharmacother. 2022, 151, 113185. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.; Dai, S.; Li, Y.; Ma, C.; Xue, X.; Zhang, S.; Wang, C.; Zhou, H.; Zhang, Y.; Li, Y. The protective effect of forsythiaside A on 3,5-diethoxycarbonyl-1,4-dihydrocollidine-induced cholestatic liver injury in mice: Based on targeted metabolomics and molecular biology technology. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2023, 1869, 166822. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Miao, H.; Yan, H.; Sheng, Y.; Ji, L. Hepatoprotective effect of Forsythiae Fructus water extract against carbon tetrachloride-induced liver fibrosis in mice. J. Ethnopharmacol. 2018, 218, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Jiang, Y.F.; Ponnusamy, M.; Diallo, M. Role of Nrf2 in chronic liver disease. World J. Gastoenterol. 2014, 20, 13079–13087. [Google Scholar] [CrossRef]
- Puntarulo, S. Iron, oxidative stress and human health. Mol. Asp. Med. 2005, 26, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.Z.; Lee, J.H.; Ki, S.H.; Yang, J.H.; Cho, I.J.; Kang, S.H.; Zhao, R.J.; Kim, S.C.; Kim, Y.W. AMPK activation by isorhamnetin protects hepatocytes against oxidative stress and mitochondrial dysfunction. Eur. J. Pharmacol. 2014, 740, 634–640. [Google Scholar] [CrossRef]
- Tobe, E.H. Mitochondrial dysfunction, oxidative stress, and major depressive disorder. Neuropsychiatr. Dis. Treat. 2013, 9, 567–573. [Google Scholar] [CrossRef]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef]
- Neustadt, J.; Pieczenik, S.R. Medication-induced mitochondrial damage and disease. Mol. Nutr. Food Res. 2008, 52, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.H.; Lu, C.Y.; Lee, H.C.; Pang, C.Y.; Ma, Y.S. Oxidative damage and mutation to mitochondrial DNA and age-dependent decline of mitochondrial respiratory function. Ann. N. Y. Acad. Sci. 1998, 854, 155–170. [Google Scholar] [CrossRef]
- Duchen, M.R. Mitochondria in health and disease: Perspectives on a new mitochondrial biology. Mol. Asp. Med. 2004, 25, 365–451. [Google Scholar] [CrossRef] [PubMed]
- Green, K.; Brand, M.D.; Murphy, M.P. Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes 2004, 53 (Suppl. S1), S110–S118. [Google Scholar] [CrossRef]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef]
- Hardie, D.G. New roles for the LKB1→AMPK pathway. Curr. Opin. Cell Biol. 2005, 17, 167–173. [Google Scholar] [CrossRef]
- Wu, S.B.; Wu, Y.T.; Wu, T.P.; Wei, Y.H. Role of AMPK-mediated adaptive responses in human cells with mitochondrial dysfunction to oxidative stress. Biochim. Biophys. Acta BBA Gen. Subj. 2014, 1840, 1331–1344. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef]
- Li, B.; Nasser, M.I.; Masood, M.; Adlat, S.; Huang, Y.; Yang, B.; Luo, C.; Jiang, N. Efficiency of Traditional Chinese medicine targeting the Nrf2/HO-1 signaling pathway. Biomed. Pharmacother. 2020, 126, 110074. [Google Scholar] [CrossRef]
- Kaspar, J.W.; Niture, S.K.; Jaiswal, A.K. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 2009, 47, 1304–1309. [Google Scholar] [CrossRef]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Asp. Med. 2011, 32, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Chen, X.; Chen, D.; Yu, B.; Zheng, P.; He, J.; Chen, H.; Yan, H.; Luo, Y.; Huang, Z. Dihydromyricetin Enhances Intestinal Antioxidant Capacity of Growing-Finishing Pigs by Activating ERK/Nrf2/HO-1 Signaling Pathway. Antioxidants 2022, 11, 704. [Google Scholar] [CrossRef] [PubMed]
- Limón-Pacheco, J.H.; Hernández, N.A.; Fanjul-Moles, M.L.; Gonsebatt, M.E. Glutathione depletion activates mitogen-activated protein kinase (MAPK) pathways that display organ-specific responses and brain protection in mice. Free Radic. Biol. Med. 2007, 43, 1335–1347. [Google Scholar] [CrossRef]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signaling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [PubMed]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Rui, W.; Guan, L.; Zhang, F.; Zhang, W.; Ding, W. PM2.5-induced oxidative stress increases adhesion molecules expression in human endothelial cells through the ERK/AKT/NF-κB-dependent pathway. J. Appl. Toxicol. 2016, 36, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.Y.; Lee, J.H.; Jegal, K.H.; Cho, I.J.; Kim, Y.W.; Kim, S.C. Oxyresveratrol abrogates oxidative stress by activating ERK-Nrf2 pathway in the liver. Chem.-Biol. Interact. 2016, 245, 110–121. [Google Scholar] [CrossRef]
- Yang, Y.C.; Lii, C.K.; Lin, A.H.; Yeh, Y.W.; Yao, H.T.; Li, C.C.; Liu, K.L.; Chen, H.W. Induction of glutathione synthesis and heme oxygenase 1 by the flavonoids butein and phloretin is mediated through the ERK/Nrf2 pathway and protects against oxidative stress. Free Radic. Biol. Med. 2011, 51, 2073–2081. [Google Scholar] [CrossRef]
- Liu, J.Y.; Chen, C.C.; Wang, W.H.; Hsu, J.D.; Yang, M.Y.; Wang, C.J. The protective effects of Hibiscus sabdariffa extract on CCl4-induced liver fibrosis in rats. Food Chem. Toxicol. 2006, 44, 336–343. [Google Scholar] [CrossRef]
- Fujii, T.; Fuchs, B.C.; Yamada, S.; Lauwers, G.Y.; Kulu, Y.; Goodwin, J.M.; Lanuti, M.; Tanabe, K.K. Mouse model of carbon tetrachloride induced liver fibrosis: Histopathological changes and expression of CD133 and epidermal growth factor. BMC Gastroenterol. 2010, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Kim, Y.W.; Shin, S.M.; Kim, C.W.; Yu, I.J.; Kim, S.G. Synergistic hepatotoxicity of N,N-dimethylformamide with carbon tetrachloride in association with endoplasmic reticulum stress. Chem.-Biol. Interact. 2010, 184, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.F.; Wang, M.Y.; Yu, D.H.; Zhao, Y.; Xu, H.M.; Zhong, S.; Sun, W.Y.; He, Y.F.; Niu, J.Q.; Gao, P.J.; et al. Therapeutic effect of chitosan on CCl4 induced hepatic fibrosis in rats. Mol. Med. Rep. 2018, 18, 3211–3218. [Google Scholar] [CrossRef]
- Ma, J.Q.; Ding, J.; Zhang, L.; Liu, C.M. Ursolic acid protects mouse liver against CCl4-induced oxidative stress and inflammation by the MAPK/NF-κB pathway. Environ. Toxicol. Pharmacol. 2014, 37, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Xie, Z.; Liu, G.; Sun, X.; Peng, G.; Lin, B.; Liao, Q. Isolation, identification and activities of natural antioxidants from Callicarpa kwangtungensis Chun. PLoS ONE 2014, 9, e93000. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Aydın, M.M.; Akçalı, K.C. Liver fibrosis. Turk. J. Gastroenterol. 2018, 29, 14–21. [Google Scholar] [CrossRef]
- Li, J.P.; Gao, Y.; Chu, S.F.; Zhang, Z.; Xia, C.Y.; Mou, Z.; Song, X.Y.; He, W.B.; Guo, N.H.; Chen, X.F. Nrf2 pathway activation contributes to anti-fibrosis effects of ginsenoside Rg1 in a rat model of alcohol- and CCl4-induced hepatic fibrosis. Acta Pharmacol. Sin. 2014, 35, 1031–1044. [Google Scholar] [CrossRef]
- Song, Y.R.; Jang, B.; Lee, S.M.; Bae, S.J.; Bak, S.B.; Kim, Y.W. Angelica gigas NAKAI and Its Active Compound, Decursin, Inhibit Cellular Injury as an Antioxidant by the Regulation of AMP-Activated Protein Kinase and YAP Signaling. Molecules 2022, 27, 1858. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.E.; Bak, S.-B.; Kim, M.-J.; Bae, S.-J.; Lee, W.-Y.; Kim, Y.W. Forsythiaside A Activates AMP-Activated Protein Kinase and Regulates Oxidative Stress via Nrf2 Signaling. Int. J. Mol. Sci. 2023, 24, 17033. https://doi.org/10.3390/ijms242317033
Kim YE, Bak S-B, Kim M-J, Bae S-J, Lee W-Y, Kim YW. Forsythiaside A Activates AMP-Activated Protein Kinase and Regulates Oxidative Stress via Nrf2 Signaling. International Journal of Molecular Sciences. 2023; 24(23):17033. https://doi.org/10.3390/ijms242317033
Chicago/Turabian StyleKim, Young Eun, Seon-Been Bak, Min-Jin Kim, Su-Jin Bae, Won-Yung Lee, and Young Woo Kim. 2023. "Forsythiaside A Activates AMP-Activated Protein Kinase and Regulates Oxidative Stress via Nrf2 Signaling" International Journal of Molecular Sciences 24, no. 23: 17033. https://doi.org/10.3390/ijms242317033