
Aggregation, Transmission, and Toxicity of the Microtubule-Associated Protein Tau: A Complex Comprehension

Abstract
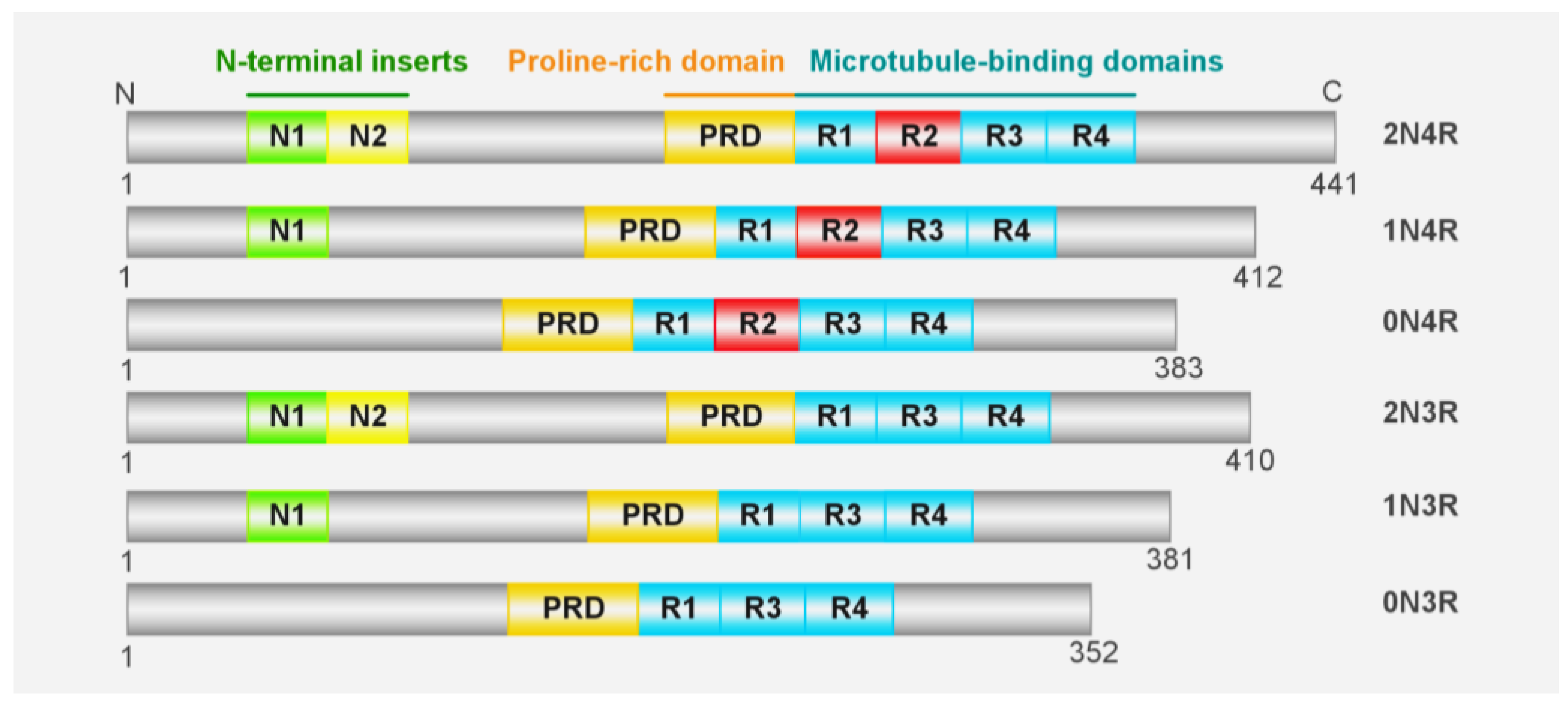
:1. Introduction
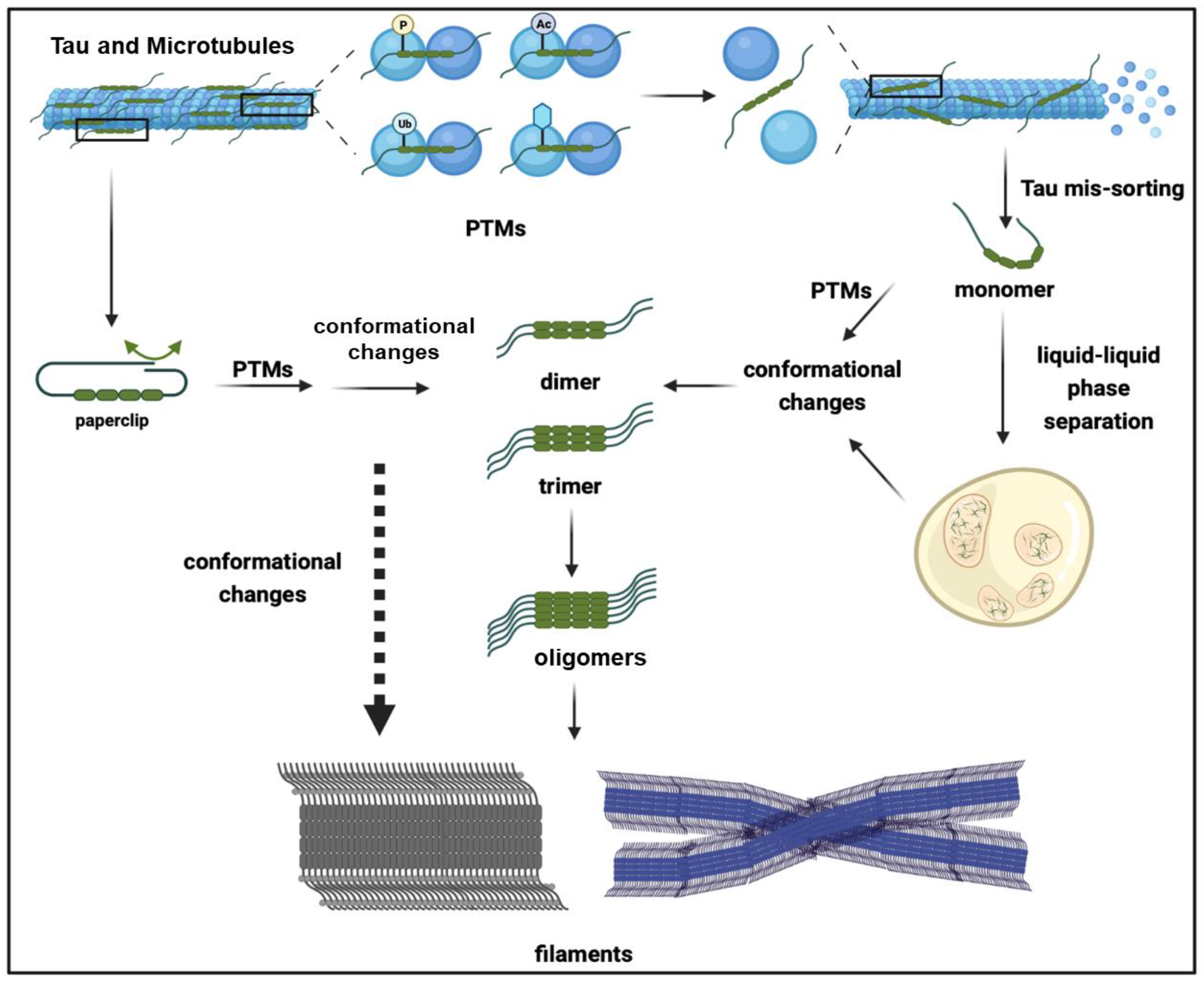
2. Tau Aggregates
3. Factors Facilitating Tau Aggregation
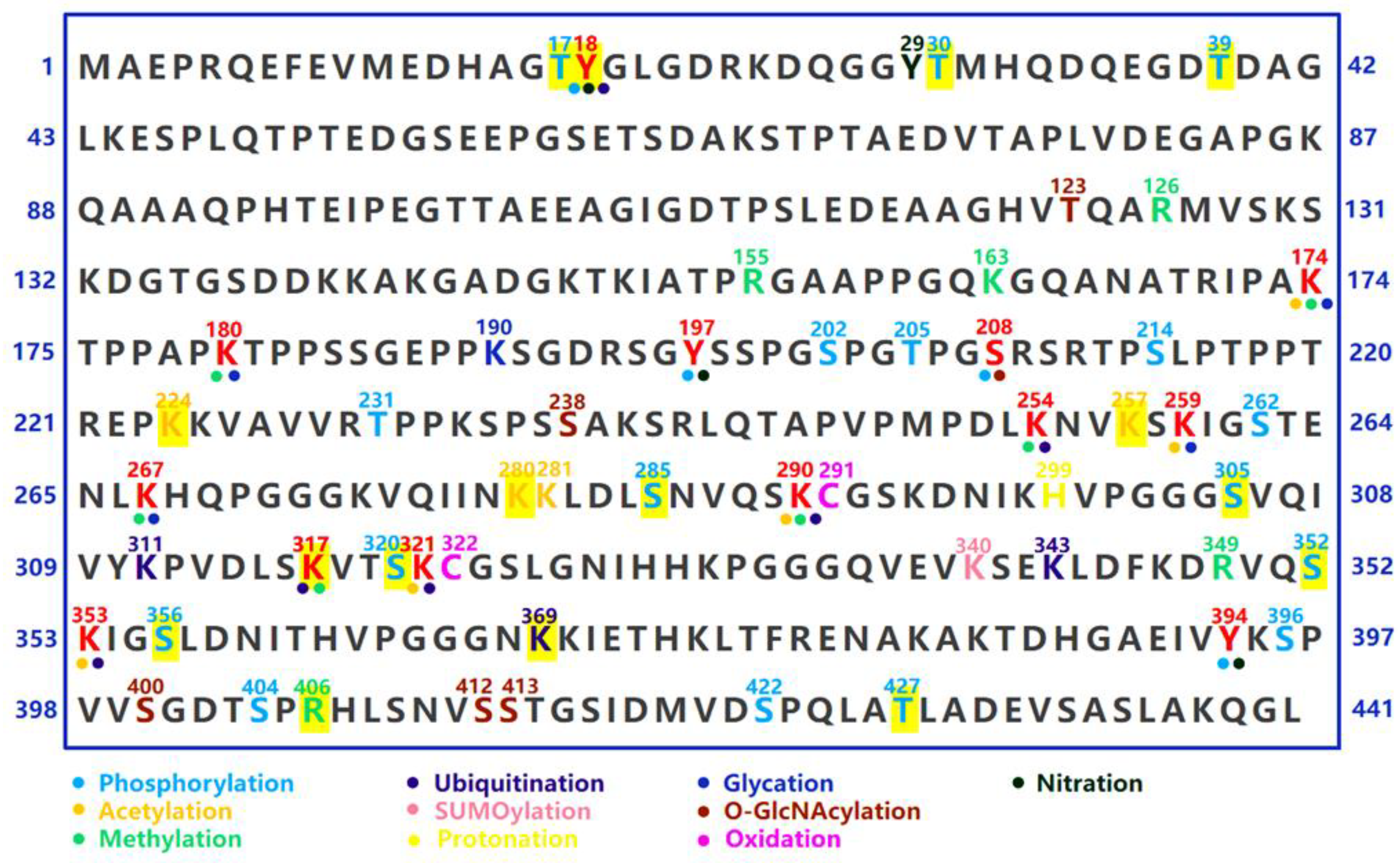
4. Post-Translational Modifications
5. Heavy Metal Elements
6. Phase Separation
7. RNAs and RNA-Binding Proteins
8. Interplay between Tau Protein and Membrane Architecture (Membrane Binding)
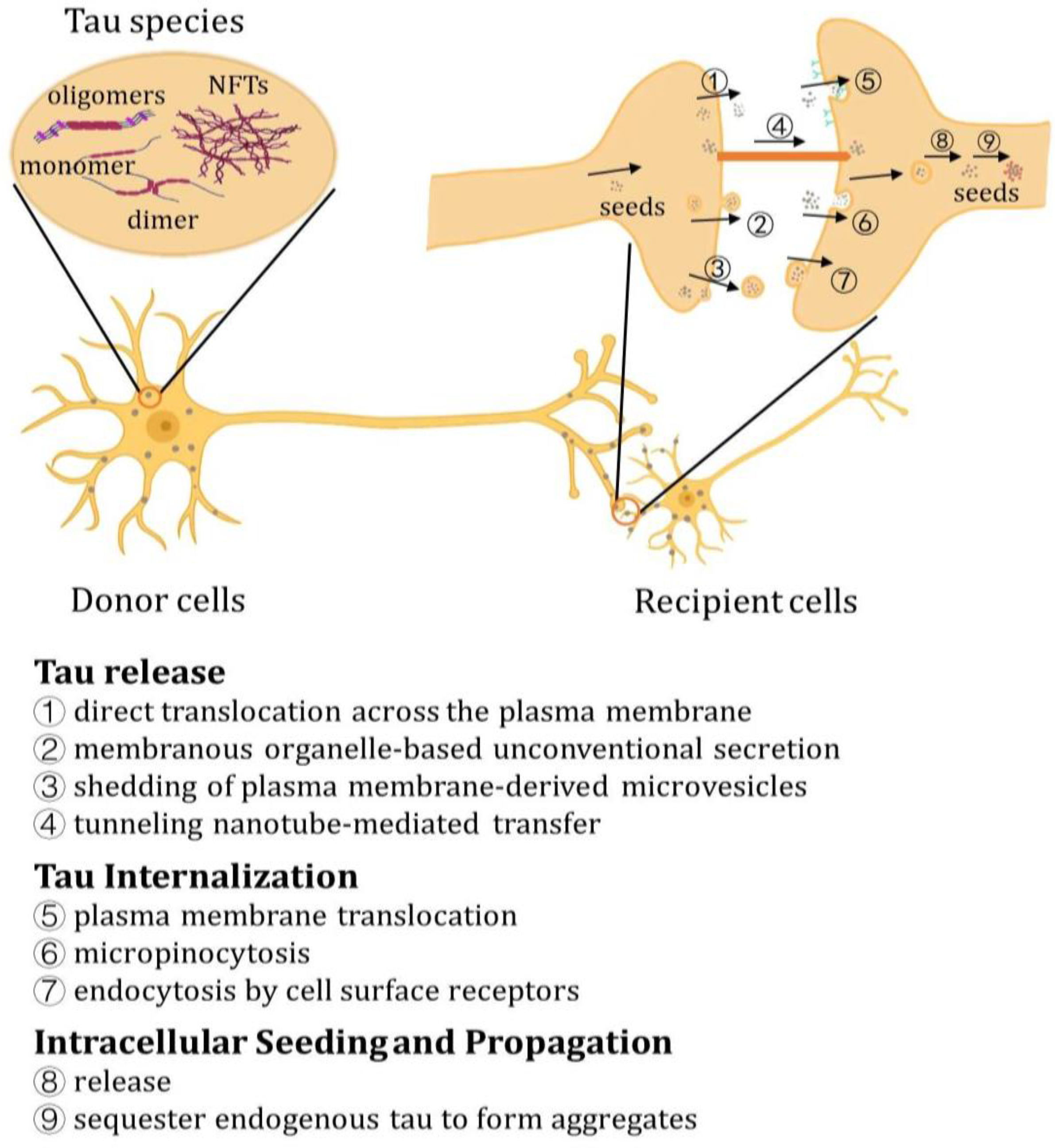
9. Propagation and Transmission of Tau
10. Release of Tau
11. Tau Internalization
12. Intracellular Seeding and Propagation of Tau
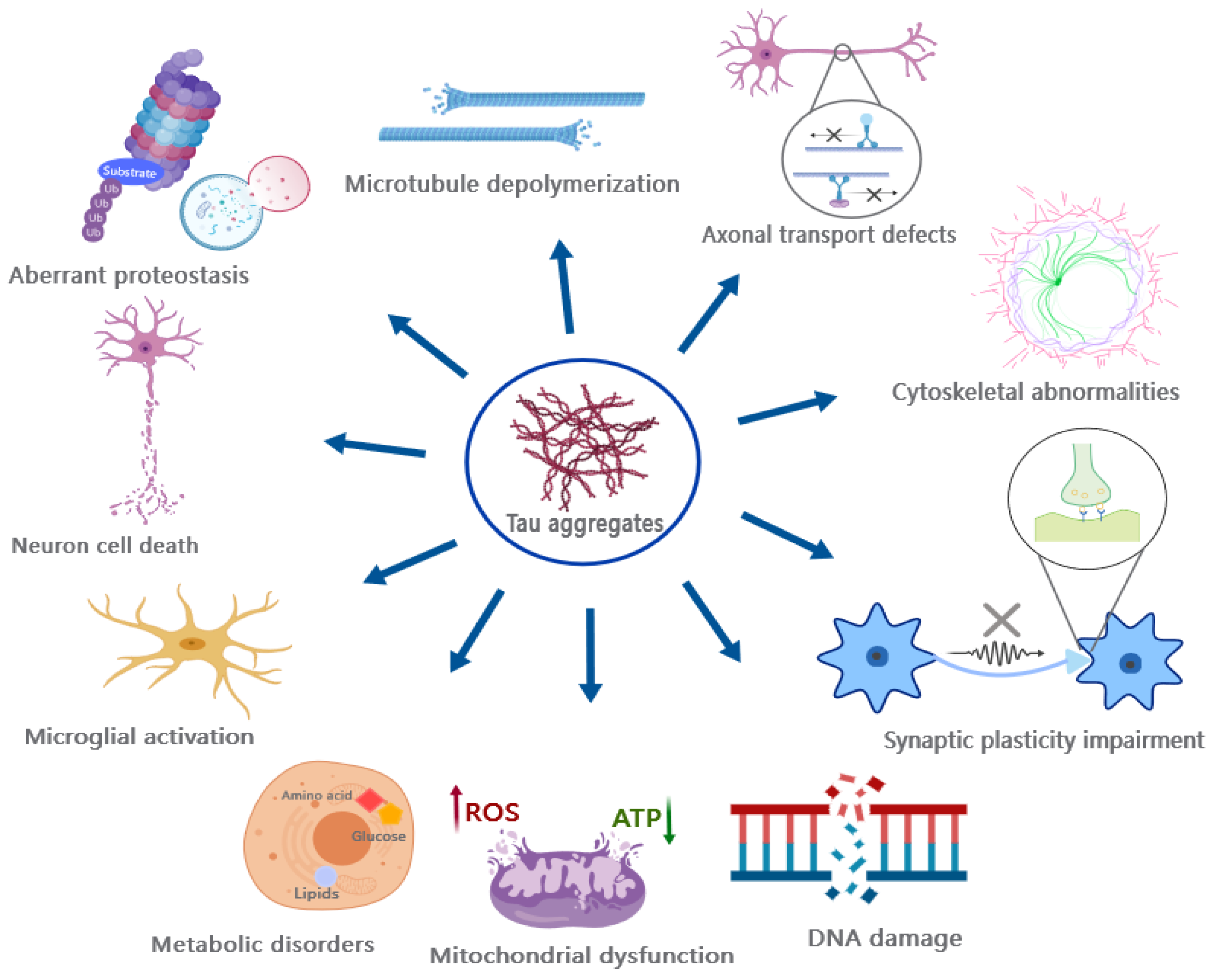
13. Revisiting the Pathogenic Mechanisms of Tau Toxicity
14. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| AFM | Atomic force microscopy |
| Aβ | Amyloid-beta |
| CBD | Corticobasal degeneration |
| ER | Endoplasmic reticulum |
| FTD | Frontotemporal dementia |
| FTDP-17 | Frontotemporal dementia with parkinsonism linked to chromosome 17 |
| HSPGs | Heparan sulphate proteoglycans |
| iNs | Induced neurons |
| LLPS | Liquid-liquid phase separation |
| LRP1 | Low-density lipoprotein receptor-related protein 1 |
| MAPS | Misfolding-associated protein secretion |
| MAPT | Microtubule-associated protein tau |
| MOBS | Membranous organelle-based unconventional secretion |
| MTBR | Microtubule-binding region |
| MTs | Microtubules |
| NFTs | Neurofibrillary tangles |
| PC | Phosphatidylcholine |
| PHFs | Paired helical filaments |
| SFs | Straight filaments |
| PiD | Pick’s disease |
| PQC | Protein quality control |
| PSP | Progressive supranuclear palsy |
| PTMs | Protein translational modifications |
| SFs | Straight filaments |
| UPS | Unconventional protein secretory |
References
- Esmaeli-Azad, B.; McCarty, J.H.; Feinstein, S.C. Sense and antisense transfection analysis of tau function: Tau influences net microtubule assembly, neurite outgrowth and neuritic stability. J. Cell Sci. 1994, 107 Pt 4, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Siano, G.; Varisco, M.; Caiazza, M.C.; Quercioli, V.; Mainardi, M.; Ippolito, C.; Cattaneo, A.; Di Primio, C. Tau Modulates VGluT1 Expression. J. Mol. Biol. 2019, 431, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Biundo, F.; Del Prete, D.; Zhang, H.; Arancio, O.; D’Adamio, L. A role for tau in learning, memory and synaptic plasticity. Sci. Rep. 2018, 8, 3184. [Google Scholar] [PubMed]
- Kent, S.A.; Spires-Jones, T.L.; Durrant, C.S. Durrant, The physiological roles of tau and Aβ: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020, 140, 417–447. [Google Scholar]
- Sjoberg, M.K.; Shestakova, E.; Mansuroglu, Z.; Maccioni, R.B.; Bonnefoy, E. Tau protein binds to pericentromeric DNA: A putative role for nuclear tau in nucleolar organization. J. Cell Sci. 2006, 119, 2025–2034. [Google Scholar] [CrossRef]
- Qi, H.; Cantrelle, F.X.; Benhelli-Mokrani, H.; Smet-Nocca, C.; Buee, L.; Lippens, G.; Bonnefoy, E.; Galas, M.C.; Landrieu, I. Nuclear magnetic resonance spectroscopy characterization of interaction of Tau with DNA and its regulation by phosphorylation. Biochemistry 2015, 54, 1525–1533. [Google Scholar] [CrossRef]
- Mansuroglu, Z.; Benhelli-Mokrani, H.; Marcato, V.; Sultan, A.; Violet, M.; Chauderlier, A.; Delattre, L.; Loyens, A.; Talahari, S.; Begard, S.; et al. Loss of Tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci. Rep. 2016, 6, 33047. [Google Scholar] [CrossRef]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef]
- Gómez-Ramos, P.; Morán, M.A. Ultrastructural aspects of neurofibrillary tangle formation in aging and Alzheimer’s disease. Microsc. Res. Tech. 1998, 43, 49–58. [Google Scholar] [CrossRef]
- Murayama, S.; Mori, H.; Ihara, Y.; Tomonaga, M. Immunocytochemical and ultrastructural studies of Pick’s disease. Ann. Neurol. 1990, 27, 394–405. [Google Scholar] [CrossRef]
- Ferrer, I.; López-González, I.; Carmona, M.; Arregui, L.; Dalfó, E.; Torrejón-Escribano, B.; Diehl, R.; Kovacs, G.G. Glial and neuronal tau pathology in tauopathies: Characterization of disease-specific phenotypes and tau pathology progression. J. Neuropathol. Exp. Neurol. 2014, 73, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Crary, J.F.; Trojanowski, J.Q.; Schneider, J.A.; Abisambra, J.F.; Abner, E.L.; Alafuzoff, I.; Arnold, S.E.; Attems, J.; Beach, T.G.; Bigio, E.H.; et al. Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol. 2014, 128, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Garringer, H.J.; Murrell, J.; D’Adamio, L.; Ghetti, B.; Vidal, R. Modeling familial British and Danish dementia. Brain Struct. Funct. 2010, 214, 235–244. [Google Scholar] [CrossRef] [PubMed]
- VanItallie, T.B. Traumatic brain injury (TBI) in collision sports: Possible mechanisms of transformation into chronic traumatic encephalopathy (CTE). Metabolism 2019, 100s, 153943. [Google Scholar] [CrossRef]
- Nishimura, T.; Ikeda, K.; Akiyama, H.; Kondo, H.; Kato, M.; Li, F.; Iseki, E.; Kosaka, K. Immunohistochemical investigation of tau-positive structures in the cerebral cortex of patients with progressive supranuclear palsy. Neurosci. Lett. 1995, 201, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Bigio, E.H.; Lipton, A.M.; Yen, S.H.; Hutton, M.L.; Baker, M.; Nacharaju, P.; White, C.L., 3rd; Davies, P.; Lin, W.; Dickson, D.W. Frontal lobe dementia with novel tauopathy: Sporadic multiple system tauopathy with dementia. J. Neuropathol. Exp. Neurol. 2001, 60, 328–341. [Google Scholar] [CrossRef]
- Komori, T.; Arai, N.; Oda, M.; Nakayama, H.; Mori, H.; Yagishita, S.; Takahashi, T.; Amano, N.; Murayama, S.; Murakami, S.; et al. Astrocytic plaques and tufts of abnormal fibers do not coexist in corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. 1998, 96, 401–408. [Google Scholar] [CrossRef]
- Dickson, D.W. Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J. Neurol. 1999, 246 (Suppl. 2), II6–II15. [Google Scholar] [CrossRef]
- Ikeda, K.; Akiyama, H.; Kondo, H.; Haga, C. A study of dementia with argyrophilic grains. Possible cytoskeletal abnormality in dendrospinal portion of neurons and oligodendroglia. Acta Neuropathol. 1995, 89, 409–414. [Google Scholar] [CrossRef]
- Tolnay, M.; Spillantini, M.G.; Goedert, M.; Ulrich, J.; Langui, D.; Probst, A. Argyrophilic grain disease: Widespread hyperphosphorylation of tau protein in limbic neurons. Acta Neuropathol. 1997, 93, 477–484. [Google Scholar] [CrossRef]
- McCann, H.; Durand, B.; Shepherd, C.E. Aging-Related Tau Astrogliopathy in Aging and Neurodegeneration. Brain Sci. 2021, 11, 927. [Google Scholar] [CrossRef]
- Didonna, A. Tau at the interface between neurodegeneration and neuroinflammation. Genes Immun. 2020, 21, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Tau gene mutations and their effects. Mov. Disord. 2005, 20 (Suppl. 12), S45–S52. [Google Scholar] [CrossRef]
- Tatebayashi, Y.; Miyasaka, T.; Chui, D.H.; Akagi, T.; Mishima, K.; Iwasaki, K.; Fujiwara, M.; Tanemura, K.; Murayama, M.; Ishiguro, K.; et al. Tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc. Natl. Acad. Sci. USA 2002, 99, 13896–13901. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, M.; Masuda-Suzukake, M.; Shitara, H.; Shimozawa, A.; Suzuki, G.; Kondo, H.; Nonaka, T.; Campbell, W.; Arai, T.; Hasegawa, M. Development of a novel tau propagation mouse model endogenously expressing 3 and 4 repeat tau isoforms. Brain 2022, 145, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Bennett, R.E.; Amaral, A.S.; Hyman, B.T. Studying tau protein propagation and pathology in the mouse brain using adeno-associated viruses. Methods Cell Biol. 2017, 141, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Colom-Cadena, M.; Davies, C.; Sirisi, S.; Lee, J.E.; Simzer, E.M.; Tzioras, M.; Querol-Vilaseca, M.; Sánchez-Aced, É.; Chang, Y.Y.; Holt, K.; et al. Synaptic oligomeric tau in Alzheimer’s disease—A potential culprit in the spread of tau pathology through the brain. Neuron 2023, 111, 2170–2183. [Google Scholar] [CrossRef]
- Zabik, N.L.; Imhof, M.M.; Martic-Milne, S. Structural evaluations of tau protein conformation: Methodologies and approaches. Biochem. Cell Biol. 2017, 95, 338–349. [Google Scholar] [CrossRef]
- Eschmann, N.A.; Georgieva, E.R.; Ganguly, P.; Borbat, P.P.; Rappaport, M.D.; Akdogan, Y.; Freed, J.H.; Shea, J.E.; Han, S. Signature of an aggregation-prone conformation of tau. Sci. Rep. 2017, 7, 44739. [Google Scholar] [CrossRef]
- Chen, D.; Drombosky, K.W.; Hou, Z.; Sari, L.; Kashmer, O.M.; Ryder, B.D.; Perez, V.A.; Woodard, D.R.; Lin, M.M.; Diamond, M.I.; et al. Tau local structure shields an amyloid-forming motif and controls aggregation propensity. Nat. Commun. 2019, 10, 2493. [Google Scholar] [CrossRef]
- Avila, J.; Jiménez, J.S.; Sayas, C.L.; Bolós, M.; Zabala, J.C.; Rivas, G.; Hernández, F. Tau Structures. Front. Aging Neurosci. 2016, 8, 262. [Google Scholar] [CrossRef]
- Zeng, Y.; Yang, J.; Zhang, B.; Gao, M.; Su, Z.; Huang, Y. The structure and phase of tau: From monomer to amyloid filament. Cell Mol. Life Sci. 2021, 78, 1873–1886. [Google Scholar] [CrossRef] [PubMed]
- Jeganathan, S.; von Bergen, M.; Brutlach, H.; Steinhoff, H.J.; Mandelkow, E. Global hairpin folding of tau in solution. Biochemistry 2006, 45, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009, 7, e34. [Google Scholar] [CrossRef] [PubMed]
- Mirbaha, H.; Chen, D.; Morazova, O.A.; Ruff, K.M.; Sharma, A.M.; Liu, X.; Goodarzi, M.; Pappu, R.V.; Colby, D.W.; Mirzaei, H.; et al. Inert and seed-competent tau monomers suggest structural origins of aggregation. Elife 2018, 7, e36584. [Google Scholar] [CrossRef]
- Hou, Z.; Chen, D.; Ryder, B.D.; Joachimiak, L.A. Biophysical properties of a tau seed. Sci. Rep. 2021, 11, 13602. [Google Scholar] [CrossRef]
- Mirbaha, H.; Holmes, B.B.; Sanders, D.W.; Bieschke, J.; Diamond, M.I. Tau Trimers Are the Minimal Propagation Unit Spontaneously Internalized to Seed Intracellular Aggregation. J. Biol. Chem. 2015, 290, 14893–14903. [Google Scholar] [CrossRef]
- Takashima, A. Tauopathies and tau oligomers. J. Alzheimers Dis. 2013, 37, 565–568. [Google Scholar] [CrossRef]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, S.; Ikai, A.; Takashima, A. Increased levels of granular tau oligomers: An early sign of brain aging and Alzheimer’s disease. Neurosci. Res. 2006, 54, 197–201. [Google Scholar] [CrossRef]
- Zhang, X.; Vigers, M.; McCarty, J.; Rauch, J.N.; Fredrickson, G.H.; Wilson, M.Z.; Shea, J.E.; Han, S.; Kosik, K.S. The proline-rich domain promotes Tau liquid-liquid phase separation in cells. J. Cell Biol. 2020, 219, e202006054. [Google Scholar] [CrossRef]
- Kadavath, H.; Hofele, R.V.; Biernat, J.; Kumar, S.; Tepper, K.; Urlaub, H.; Mandelkow, E.; Zweckstetter, M. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. USA 2015, 112, 7501–7506. [Google Scholar] [CrossRef] [PubMed]
- Gyparaki, M.T.; Arab, A.; Sorokina, E.M.; Santiago-Ruiz, A.N.; Bohrer, C.H.; Xiao, J.; Lakadamyali, M. Tau forms oligomeric complexes on microtubules that are distinct from tau aggregates. Proc. Natl. Acad. Sci. USA 2021, 118, e2021461118. [Google Scholar] [CrossRef] [PubMed]
- Sahara, N.; Maeda, S.; Murayama, M.; Suzuki, T.; Dohmae, N.; Yen, S.H.; Takashima, A. Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur. J. Neurosci. 2007, 25, 3020–3029. [Google Scholar] [CrossRef]
- Congdon, E.E.; Kim, S.; Bonchak, J.; Songrug, T.; Matzavinos, A.; Kuret, J. Nucleation-dependent tau filament formation: The importance of dimerization and an estimation of elementary rate constants. J. Biol. Chem. 2008, 283, 13806–13816. [Google Scholar] [CrossRef] [PubMed]
- Patterson, K.R.; Remmers, C.; Fu, Y.; Brooker, S.; Kanaan, N.M.; Vana, L.; Ward, S.; Reyes, J.F.; Philibert, K.; Glucksman, M.J.; et al. Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J. Biol. Chem. 2011, 286, 23063–23076. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Takashima, A. Tau Oligomers. Adv. Exp. Med. Biol. 2019, 1184, 373–380. [Google Scholar] [CrossRef]
- Sengupta, U.; Portelius, E.; Hansson, O.; Farmer, K.; Castillo-Carranza, D.; Woltjer, R.; Zetterberg, H.; Galasko, D.; Blennow, K.; Kayed, R. Tau oligomers in cerebrospinal fluid in Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2017, 4, 226–235. [Google Scholar] [CrossRef]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, M.; Yoshiike, Y.; Kim, H.; Miyasaka, T.; Murayama, S.; Ikai, A.; Takashima, A. Granular tau oligomers as intermediates of tau filaments. Biochemistry 2007, 46, 3856–3861. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef]
- Zhang, W.; Tarutani, A.; Newell, K.L.; Murzin, A.G.; Matsubara, T.; Falcon, B.; Vidal, R.; Garringer, H.J.; Shi, Y.; Ikeuchi, T.; et al. Novel tau filament fold in corticobasal degeneration. Nature 2020, 580, 283–287. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, W.; Yang, Y.; Murzin, A.G.; Falcon, B.; Kotecha, A.; van Beers, M.; Tarutani, A.; Kametani, F.; Garringer, H.J.; et al. Structure-based classification of tauopathies. Nature 2021, 598, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.; Zhang, W.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Vidal, R.; Crowther, R.A.; Ghetti, B.; Scheres, S.H.W.; Goedert, M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018, 561, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Zempel, H.; Mandelkow, E. Lost after translation: Missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014, 37, 721–732. [Google Scholar] [CrossRef]
- García-Escudero, V.; Ruiz-Gabarre, D.; Gargini, R.; Pérez, M.; García, E.; Cuadros, R.; Hernández, I.H.; Cabrera, J.R.; García-Escudero, R.; Lucas, J.J.; et al. A new non-aggregative splicing isoform of human Tau is decreased in Alzheimer’s disease. Acta Neuropathol. 2021, 142, 159–177. [Google Scholar] [CrossRef]
- Hernández, F.; Ferrer, I.; Pérez, M.; Zabala, J.C.; Del Rio, J.A.; Avila, J. Tau Aggregation. Neuroscience 2023, 518, 64–69. [Google Scholar] [CrossRef]
- Cowan, C.M.; Mudher, A. Are tau aggregates toxic or protective in tauopathies? Front. Neurol. 2013, 4, 114. [Google Scholar] [CrossRef] [PubMed]
- Makrides, V.; Shen, T.E.; Bhatia, R.; Smith, B.L.; Thimm, J.; Lal, R.; Feinstein, S.C. Microtubule-dependent oligomerization of tau. Implications for physiological tau function and tauopathies. J. Biol. Chem. 2003, 278, 33298–33304. [Google Scholar] [CrossRef]
- Wegmann, S.; Biernat, J.; Mandelkow, E. A current view on Tau protein phosphorylation in Alzheimer’s disease. Curr. Opin. Neurobiol. 2021, 69, 131–138. [Google Scholar] [CrossRef]
- Pevalova, M.; Filipcik, P.; Novak, M.; Avila, J.; Iqbal, K. Post-translational modifications of tau protein. Bratisl. Lekárske Listy 2006, 107, 346–353. [Google Scholar]
- Alquezar, C.; Arya, S.; Kao, A.W. Tau Post-translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation. Front. Neurol. 2020, 11, 595532. [Google Scholar] [CrossRef]
- Sontag, E.; Nunbhakdi-Craig, V.; Lee, G.; Bloom, G.S.; Mumby, M.C. Regulation of the phosphorylation state and microtubule-binding activity of Tau by protein phosphatase 2A. Neuron 1996, 17, 1201–1207. [Google Scholar] [CrossRef]
- Pei, J.-J.; Björkdahl, C.; Zhang, H.; Zhou, X.; Winblad, B. p70 S6 kinase and tau in Alzheimer’s disease. J. Alzheimers Dis. 2008, 14, 385–392. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, X.; Yao, Z.; Shi, Y.; Xiong, J.; Zhou, J.; Su, Z.; Huang, Y. 14-3-3/Tau Interaction and Tau Amyloidogenesis. J. Mol. Neurosci. 2019, 68, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Haase, C.; Stieler, J.T.; Arendt, T.; Holzer, M. Pseudophosphorylation of tau protein alters its ability for self-aggregation. J. Neurochem. 2004, 88, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, B.; Tung, E.J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur. J. Neurosci. 2007, 26, 3429–3436. [Google Scholar] [CrossRef] [PubMed]
- Bramblett, G.T.; Goedert, M.; Jakes, R.; Merrick, S.E.; Trojanowski, J.Q.; Lee, V.M. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron 1993, 10, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Tatebayashi, Y.; Akagi, T.; Chui, D.H.; Murayama, M.; Miyasaka, T.; Planel, E.; Tanemura, K.; Sun, X.; Hashikawa, T.; et al. Aberrant tau phosphorylation by glycogen synthase kinase-3β and JNK3 induces oligomeric tau fibrils in COS-7 cells. J. Biol. Chem. 2002, 277, 42060–42065. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.X.; Zhang, Y.; Saman, D.; Haider, A.M.; De, S.; Sang, J.C.; Brown, K.; Jiang, K.; Humphrey, J.; Julian, L.; et al. Hyperphosphorylated tau self-assembles into amorphous aggregates eliciting TLR4-dependent responses. Nat. Commun. 2022, 13, 2692. [Google Scholar] [CrossRef]
- Xia, Y.; Prokop, S.; Gorion, K.M.; Kim, J.D.; Sorrentino, Z.A.; Bell, B.M.; Manaois, A.N.; Chakrabarty, P.; Davies, P.; Giasson, B.I. Tau Ser208 phosphorylation promotes aggregation and reveals neuropathologic diversity in Alzheimer’s disease and other tauopathies. Acta Neuropathol. Commun. 2020, 8, 88. [Google Scholar] [CrossRef]
- Wickramasinghe, S.P.; Lempart, J.; Merens, H.E.; Murphy, J.; Huettemann, P.; Jakob, U.; Rhoades, E. Polyphosphate Initiates Tau Aggregation through Intra- and Intermolecular Scaffolding. Biophys. J. 2019, 117, 717–728. [Google Scholar] [CrossRef]
- Drazic, A.; Myklebust, L.M.; Ree, R.; Arnesen, T. The world of protein acetylation. Biochim. Biophys. Acta 2016, 1864, 1372–1401. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Guan, L. Unraveling the Influence of K280 Acetylation on the Conformational Features of Tau Core Fragment: A Molecular Dynamics Simulation Study. Front. Mol. Biosci. 2021, 8, 801577. [Google Scholar] [CrossRef] [PubMed]
- Trzeciakiewicz, H.; Tseng, J.H.; Wander, C.M.; Madden, V.; Tripathy, A.; Yuan, C.X.; Cohen, T.J. A Dual Pathogenic Mechanism Links Tau Acetylation to Sporadic Tauopathy. Sci. Rep. 2017, 7, 44102. [Google Scholar] [CrossRef]
- Xia, Y.; Bell, B.M.; Giasson, B.I. Tau K321/K353 pseudoacetylation within KXGS motifs regulates tau-microtubule interactions and inhibits aggregation. Sci. Rep. 2021, 11, 17069. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Seto, E. Lysine acetylation: Codified crosstalk with other posttranslational modifications. Mol. Cell 2008, 31, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jiang, Y.; Wang, J.Z.; Liu, R.; Wang, X. Tau Ubiquitination in Alzheimer’s Disease. Front. Neurol. 2021, 12, 786353. [Google Scholar] [CrossRef]
- Munari, F.; Barracchia, C.G.; Franchin, C.; Parolini, F.; Capaldi, S.; Romeo, A.; Bubacco, L.; Assfalg, M.; Arrigoni, G.; D’Onofrio, M. Semisynthetic and Enzyme-Mediated Conjugate Preparations Illuminate the Ubiquitination-Dependent Aggregation of Tau Protein. Angew. Chem. Int. Ed. Engl. 2020, 59, 6607–6611. [Google Scholar] [CrossRef]
- Ye, Y.; Klenerman, D.; Finley, D. N-Terminal Ubiquitination of Amyloidogenic Proteins Triggers Removal of Their Oligomers by the Proteasome Holoenzyme. J. Mol. Biol. 2020, 432, 585–596. [Google Scholar] [CrossRef]
- Tan, J.M.; Wong, E.S.; Kirkpatrick, D.S.; Pletnikova, O.; Ko, H.S.; Tay, S.-P.; Ho, M.W.; Troncoso, J.; Gygi, S.P.; Lee, M.K. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 2008, 17, 431–439. [Google Scholar] [CrossRef]
- Arakhamia, T.; Lee, C.E.; Carlomagno, Y.; Kumar, M.; Duong, D.M.; Wesseling, H.; Kundinger, S.R.; Wang, K.; Williams, D.; DeTure, M.; et al. Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains. Cell 2021, 184, 6207–6210. [Google Scholar] [CrossRef] [PubMed]
- Dickey, C.A.; Yue, M.; Lin, W.L.; Dickson, D.W.; Dunmore, J.H.; Lee, W.C.; Zehr, C.; West, G.; Cao, S.; Clark, A.M.; et al. Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J. Neurosci. 2006, 26, 6985–6996. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, L.; Dickson, D.; Kehoe, K.; Taylor, J.; Snyder, H.; Grover, A.; De Lucia, M.; McGowan, E.; Lewis, J.; Prihar, G.; et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 2004, 13, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Puangmalai, N.; Sengupta, U.; Bhatt, N.; Gaikwad, S.; Montalbano, M.; Bhuyan, A.; Garcia, S.; McAllen, S.; Sonawane, M.; Jerez, C.; et al. Lysine 63-linked ubiquitination of tau oligomers contributes to the pathogenesis of Alzheimer’s disease. J. Biol. Chem. 2022, 298, 101766. [Google Scholar] [CrossRef] [PubMed]
- Huseby, C.J.; Hoffman, C.N.; Cooper, G.L.; Cocuron, J.C.; Alonso, A.P.; Thomas, S.N.; Yang, A.J.; Kuret, J. Quantification of Tau Protein Lysine Methylation in Aging and Alzheimer’s Disease. J. Alzheimers Dis. 2019, 71, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Funk, K.E.; Thomas, S.N.; Schafer, K.N.; Cooper, G.L.; Liao, Z.; Clark, D.J.; Yang, A.J.; Kuret, J. Lysine methylation is an endogenous post-translational modification of tau protein in human brain and a modulator of aggregation propensity. Biochem. J. 2014, 462, 77–88. [Google Scholar] [CrossRef]
- Shams, H.; Matsunaga, A.; Ma, Q.; Mofrad, M.R.K.; Didonna, A. Methylation at a conserved lysine residue modulates tau assembly and cellular functions. Mol. Cell Neurosci. 2022, 120, 103707. [Google Scholar] [CrossRef]
- Morris, M.; Knudsen, G.M.; Maeda, S.; Trinidad, J.C.; Ioanoviciu, A.; Burlingame, A.L.; Mucke, L. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 2015, 18, 1183–1189. [Google Scholar] [CrossRef]
- Kontaxi, C.; Piccardo, P.; Gill, A.C. Lysine-Directed Post-translational Modifications of Tau Protein in Alzheimer’s Disease and Related Tauopathies. Front. Mol. Biosci. 2017, 4, 56. [Google Scholar] [CrossRef]
- Thomas, S.N.; Funk, K.E.; Wan, Y.; Liao, Z.; Davies, P.; Kuret, J.; Yang, A.J. Dual modification of Alzheimer’s disease PHF-tau protein by lysine methylation and ubiquitylation: A mass spectrometry approach. Acta Neuropathol. 2012, 123, 105–117. [Google Scholar] [CrossRef]
- Islam, F.; Shohag, S.; Akhter, S.; Islam, M.R.; Sultana, S.; Mitra, S.; Chandran, D.; Khandaker, M.U.; Ashraf, G.M.; Idris, A.M.; et al. Exposure of metal toxicity in Alzheimer’s disease: An extensive review. Front. Pharmacol. 2022, 13, 903099. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yin, Y.L.; Liu, X.Z.; Shen, P.; Zheng, Y.G.; Lan, X.R.; Lu, C.B.; Wang, J.Z. Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl. Neurodegener. 2020, 9, 10. [Google Scholar] [CrossRef]
- Kim, A.C.; Lim, S.; Kim, Y.K. Metal Ion Effects on Aβ and Tau Aggregation. Int. J. Mol. Sci. 2018, 19, 128. [Google Scholar] [CrossRef]
- La Rocca, R.; Tsvetkov, P.O.; Golovin, A.V.; Allegro, D.; Barbier, P.; Malesinski, S.; Guerlesquin, F.; Devred, F. Identification of the three zinc-binding sites on tau protein. Int. J. Biol. Macromol. 2022, 209, 779–784. [Google Scholar] [CrossRef]
- Huang, Y.; Wu, Z.; Cao, Y.; Lang, M.; Lu, B.; Zhou, B. Zinc binding directly regulates tau toxicity independent of tau hyperphosphorylation. Cell Rep. 2014, 8, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Zubčić, K.; Hof, P.R.; Šimić, G.; Jazvinšćak Jembrek, M. The Role of Copper in Tau-Related Pathology in Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 572308. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Quattrocchi, C.C.; Salustri, C.; Rossini, P.M. Ceruloplasmin fragmentation is implicated in ‘free’ copper deregulation of Alzheimer’s disease. Prion 2008, 2, 23–27. [Google Scholar] [CrossRef]
- Ma, Q.F.; Li, Y.M.; Du, J.T.; Kanazawa, K.; Nemoto, T.; Nakanishi, H.; Zhao, Y.F. Binding of copper (II) ion to an Alzheimer’s tau peptide as revealed by MALDI-TOF MS, CD, and NMR. Biopolymers 2005, 79, 74–85. [Google Scholar] [CrossRef]
- Sadqi, M.; Hernández, F.; Pan, U.; Pérez, M.; Schaeberle, M.D.; Avila, J.; Muñoz, V. Alpha-helix structure in Alzheimer’s disease aggregates of tau-protein. Biochemistry 2002, 41, 7150–7155. [Google Scholar] [CrossRef]
- Mo, Z.Y.; Zhu, Y.Z.; Zhu, H.L.; Fan, J.B.; Chen, J.; Liang, Y. Low micromolar zinc accelerates the fibrillization of human tau via bridging of Cys-291 and Cys-322. J. Biol. Chem. 2009, 284, 34648–34657. [Google Scholar] [CrossRef]
- Roman, A.Y.; Devred, F.; Byrne, D.; La Rocca, R.; Ninkina, N.N.; Peyrot, V.; Tsvetkov, P.O. Zinc Induces Temperature-Dependent Reversible Self-Assembly of Tau. J. Mol. Biol. 2019, 431, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.; Ebralidze, I.I.; She, Z.; Kraatz, H.-B. Electrochemical studies of tau protein-iron interactions—Potential implications for Alzheimer’s Disease. Electrochim. Acta 2017, 100, 384–393. [Google Scholar] [CrossRef]
- Rametti, A.; Esclaire, F.; Yardin, C.; Cogné, N.; Terro, F. Lithium down-regulates tau in cultured cortical neurons: A possible mechanism of neuroprotection. Neurosci. Lett. 2008, 434, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Chen, D.C.; Klein, P.S.; Lee, V.M. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J. Biol. Chem. 1997, 272, 25326–25332. [Google Scholar] [CrossRef] [PubMed]
- De-Paula, V.J.; Forlenza, O.V. Lithium modulates multiple tau kinases with distinct effects in cortical and hippocampal neurons according to concentration ranges. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2022, 395, 105–113. [Google Scholar] [CrossRef]
- Brown, K.M.; Tracy, D.K. Lithium: The pharmacodynamic actions of the amazing ion. Ther. Adv. Psychopharmacol. 2013, 3, 163–176. [Google Scholar] [CrossRef]
- Lenox, R.H.; Hahn, C.G. Overview of the mechanism of action of lithium in the brain: Fifty-year update. J. Clin. Psychiatry 2000, 61 (Suppl. 9), 5–15. [Google Scholar]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid-liquid phase separation can initiate tau aggregation. Embo J. 2018, 37, e98049. [Google Scholar] [CrossRef]
- Rai, S.K.; Savastano, A.; Singh, P.; Mukhopadhyay, S.; Zweckstetter, M. Liquid-liquid phase separation of tau: From molecular biophysics to physiology and disease. Protein Sci. 2021, 30, 1294–1314. [Google Scholar] [CrossRef]
- Rane, J.S.; Kumari, A.; Panda, D. The Acetyl Mimicking Mutation, K274Q in Tau, Enhances the Metal Binding Affinity of Tau and Reduces the Ability of Tau to Protect DNA. ACS Chem. Neurosci. 2020, 11, 291–303. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Hamel, C.; Grabinski, T.; Combs, B. Liquid-liquid phase separation induces pathogenic tau conformations in vitro. Nat. Commun. 2020, 11, 2809. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Hong, L.; Krainer, G.; Yao, Q.Q.; Knowles, T.P.J.; Wu, S.; Perrett, S. Conformational Expansion of Tau in Condensates Promotes Irreversible Aggregation. J. Am. Chem. Soc. 2021, 143, 13056–13064. [Google Scholar] [CrossRef] [PubMed]
- Lester, E.; Ooi, F.K.; Bakkar, N.; Ayers, J.; Woerman, A.L.; Wheeler, J.; Bowser, R.; Carlson, G.A.; Prusiner, S.B.; Parker, R. Tau aggregates are RNA-protein assemblies that mislocalize multiple nuclear speckle components. Neuron 2021, 109, 1675–1691. [Google Scholar] [CrossRef] [PubMed]
- McMillan, P.J.; Benbow, S.J.; Uhrich, R.; Saxton, A.; Baum, M.; Strovas, T.; Wheeler, J.M.; Baker, J.; Liachko, N.F.; Keene, C.D.; et al. Tau-RNA complexes inhibit microtubule polymerization and drive disease-relevant conformation change. Brain 2023, 146, 3206–3220. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Cowan, N.; Kirschner, M. The primary structure and heterogeneity of tau protein from mouse brain. Science 1988, 239, 285–288. [Google Scholar] [CrossRef]
- Kampers, T.; Friedhoff, P.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996, 399, 344–349. [Google Scholar] [CrossRef]
- Ambadipudi, S.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 2017, 8, 275. [Google Scholar] [CrossRef]
- Abskharon, R.; Sawaya, M.R.; Boyer, D.R.; Cao, Q.; Nguyen, B.A.; Cascio, D.; Eisenberg, D.S. Cryo-EM structure of RNA-induced tau fibrils reveals a small C-terminal core that may nucleate fibril formation. Proc. Natl. Acad. Sci. USA 2022, 119, e2119952119. [Google Scholar] [CrossRef]
- Zwierzchowski-Zarate, A.N.; Mendoza-Oliva, A.; Kashmer, O.M.; Collazo-Lopez, J.E.; White, C.L., 3rd; Diamond, M.I. RNA induces unique tau strains and stabilizes Alzheimer’s disease seeds. J. Biol. Chem. 2022, 298, 102132. [Google Scholar] [CrossRef]
- Chakraborty, P.; Rivière, G.; Liu, S.; de Opakua, A.I.; Dervişoğlu, R.; Hebestreit, A.; Andreas, L.B.; Vorberg, I.M.; Zweckstetter, M. Co-factor-free aggregation of tau into seeding-competent RNA-sequestering amyloid fibrils. Nat. Commun. 2021, 12, 4231. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, Y.; Eschmann, N.A.; Zhou, H.; Rauch, J.N.; Hernandez, I.; Guzman, E.; Kosik, K.S.; Han, S. RNA stores tau reversibly in complex coacervates. PLoS Biol. 2017, 15, e2002183. [Google Scholar] [CrossRef] [PubMed]
- Ash, P.E.A.; Lei, S.; Shattuck, J.; Boudeau, S.; Carlomagno, Y.; Medalla, M.; Mashimo, B.L.; Socorro, G.; Al-Mohanna, L.F.A.; Jiang, L.; et al. TIA1 potentiates tau phase separation and promotes generation of toxic oligomeric tau. Proc. Natl. Acad. Sci. USA 2021, 118, e2014188118. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Lin, W.; Zhang, C.; Ash, P.E.A.; Verma, M.; Kwan, J.; van Vliet, E.; Yang, Z.; Cruz, A.L.; Boudeau, S.; et al. Interaction of tau with HNRNPA2B1 and N6-methyladenosine RNA mediates the progression of tauopathy. Mol Cell 2021, 81, 4209–4227. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.G.; Paula-Barbosa, M.; Roher, A. Alzheimer’s disease: Paired helical filaments and cytomembranes. Neuropathol. Appl. Neurobiol. 1987, 13, 91–110. [Google Scholar] [CrossRef]
- Gellermann, G.P.; Appel, T.R.; Davies, P.; Diekmann, S. Paired helical filaments contain small amounts of cholesterol, phosphatidylcholine and sphingolipids. Biol. Chem. 2006, 387, 1267–1274. [Google Scholar] [CrossRef]
- Bok, E.; Leem, E.; Lee, B.R.; Lee, J.M.; Yoo, C.J.; Lee, E.M.; Kim, J. Role of the Lipid Membrane and Membrane Proteins in Tau Pathology. Front. Cell Dev. Biol. 2021, 9, 653815. [Google Scholar] [CrossRef]
- Ait-Bouziad, N.; Lv, G.; Mahul-Mellier, A.L.; Xiao, S.; Zorludemir, G.; Eliezer, D.; Walz, T.; Lashuel, H.A. Discovery and characterization of stable and toxic Tau/phospholipid oligomeric complexes. Nat. Commun. 2017, 8, 1678. [Google Scholar] [CrossRef]
- Sallaberry, C.A.; Voss, B.J.; Majewski, J.; Biernat, J.; Mandelkow, E.; Chi, E.Y.; Vander Zanden, C.M. Tau and Membranes: Interactions That Promote Folding and Condensation. Front. Cell Dev. Biol. 2021, 9, 725241. [Google Scholar] [CrossRef]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef]
- Wysocka, A.; Palasz, E.; Steczkowska, M.; Niewiadomska, G. Dangerous Liaisons: Tau Interaction with Muscarinic Receptors. Curr. Alzheimer. Res. 2020, 17, 224–237. [Google Scholar] [CrossRef]
- Brundin, P.; Melki, R.; Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol 2010, 11, 301–307. [Google Scholar] [CrossRef]
- Guo, J.L.; Lee, V.M. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 2011, 286, 15317–15331. [Google Scholar] [CrossRef]
- Guo, J.L.; Lee, V.M. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 2014, 20, 130–138. [Google Scholar] [CrossRef]
- Dujardin, S.; Hyman, B.T. Tau Prion-Like Propagation: State of the Art and Current Challenges. Adv. Exp. Med. Biol. 2019, 1184, 305–325. [Google Scholar] [CrossRef]
- Merezhko, M.; Uronen, R.L.; Huttunen, H.J. The Cell Biology of Tau Secretion. Front. Mol. Neurosci. 2020, 13, 569818. [Google Scholar] [CrossRef]
- Annadurai, N.; De Sanctis, J.B.; Hajdúch, M.; Das, V. Tau secretion and propagation: Perspectives for potential preventive interventions in Alzheimer’s disease and other tauopathies. Exp. Neurol. 2021, 343, 113756. [Google Scholar] [CrossRef]
- Katsinelos, T.; Zeitler, M.; Dimou, E.; Karakatsani, A.; Müller, H.M.; Nachman, E.; Steringer, J.P.; Ruiz de Almodovar, C.; Nickel, W.; Jahn, T.R. Unconventional Secretion Mediates the Trans-cellular Spreading of Tau. Cell Rep. 2018, 23, 2039–2055. [Google Scholar] [CrossRef]
- Lorente-Gea, L.; García, B.; Martín, C.; Ordiales, H.; García-Suárez, O.; Piña-Batista, K.M.; Merayo-Lloves, J.; Quirós, L.M.; Fernández-Vega, I. Heparan Sulfate Proteoglycans Undergo Differential Expression Alterations in Alzheimer Disease Brains. J. Neuropathol. Exp. Neurol. 2020, 79, 474–483. [Google Scholar] [CrossRef]
- Steringer, J.P.; Bleicken, S.; Andreas, H.; Zacherl, S.; Laussmann, M.; Temmerman, K.; Contreras, F.X.; Bharat, T.A.; Lechner, J.; Müller, H.M.; et al. Phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2)-dependent oligomerization of fibroblast growth factor 2 (FGF2) triggers the formation of a lipidic membrane pore implicated in unconventional secretion. J. Biol. Chem. 2012, 287, 27659–27669. [Google Scholar] [CrossRef]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef]
- Saman, S.; Kim, W.; Raya, M.; Visnick, Y.; Miro, S.; Saman, S.; Jackson, B.; McKee, A.C.; Alvarez, V.E.; Lee, N.C.; et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 2012, 287, 3842–3849. [Google Scholar] [CrossRef]
- Polanco, J.C.; Scicluna, B.J.; Hill, A.F.; Götz, J. Extracellular Vesicles Isolated from the Brains of rTg4510 Mice Seed Tau Protein Aggregation in a Threshold-dependent Manner. J. Biol. Chem. 2016, 291, 12445–12466. [Google Scholar] [CrossRef]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Hamano, T.; Enomoto, S.; Shirafuji, N.; Ikawa, M.; Yamamura, O.; Yen, S.H.; Nakamoto, Y. Autophagy and Tau Protein. Int. J. Mol. Sci. 2021, 22, 7475. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Wang, C.; Tang, Y.; Mok, S.-A.; Tsai, R.M.; Rojas, J.C.; Karydas, A.; Miller, B.L.; Boxer, A.L.; et al. Promoting tau secretion and propagation by hyperactive p300/CBP via autophagy-lysosomal pathway in tauopathy. Mol. Neurodegener. 2020, 15, 2. [Google Scholar] [CrossRef]
- Feng, Q.; Luo, Y.; Zhang, X.N.; Yang, X.F.; Hong, X.Y.; Sun, D.S.; Li, X.C.; Hu, Y.; Li, X.G.; Zhang, J.F.; et al. MAPT/Tau accumulation represses autophagy flux by disrupting IST1-regulated ESCRT-III complex formation: A vicious cycle in Alzheimer neurodegeneration. Autophagy 2020, 16, 641–658. [Google Scholar] [CrossRef]
- Chambraud, B.; Daguinot, C.; Guillemeau, K.; Genet, M.; Dounane, O.; Meduri, G.; Poüs, C.; Baulieu, E.E.; Giustiniani, J. Decrease of neuronal FKBP4/FKBP52 modulates perinuclear lysosomal positioning and MAPT/Tau behavior during MAPT/Tau-induced proteotoxic stress. Autophagy 2021, 17, 3491–3510. [Google Scholar] [CrossRef]
- Mohamed, N.V.; Plouffe, V.; Rémillard-Labrosse, G.; Planel, E.; Leclerc, N. Starvation and inhibition of lysosomal function increased tau secretion by primary cortical neurons. Sci. Rep. 2014, 4, 5715. [Google Scholar] [CrossRef]
- Lee, I.H.; Finkel, T. Regulation of autophagy by the p300 acetyltransferase. J. Biol. Chem. 2009, 284, 6322–6328. [Google Scholar] [CrossRef]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef]
- Spitzer, P.; Mulzer, L.M.; Oberstein, T.J.; Munoz, L.E.; Lewczuk, P.; Kornhuber, J.; Herrmann, M.; Maler, J.M. Microvesicles from cerebrospinal fluid of patients with Alzheimer’s disease display reduced concentrations of tau and APP protein. Sci. Rep. 2019, 9, 7089. [Google Scholar] [CrossRef]
- Dujardin, S.; Bégard, S.; Caillierez, R.; Lachaud, C.; Delattre, L.; Carrier, S.; Loyens, A.; Galas, M.C.; Bousset, L.; Melki, R.; et al. Ectosomes: A new mechanism for non-exosomal secretion of tau protein. PLoS ONE 2014, 9, e100760. [Google Scholar] [CrossRef]
- Yan, M.; Zheng, T. Role of the endolysosomal pathway and exosome release in tau propagation. Neurochem. Int. 2021, 145, 104988. [Google Scholar] [CrossRef]
- Rodriguez, L.; Mohamed, N.V.; Desjardins, A.; Lippé, R.; Fon, E.A.; Leclerc, N. Rab7A regulates tau secretion. J. Neurochem. 2017, 141, 592–605. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Mufson, E.J.; Counts, S.E.; Wuu, J.; Alldred, M.J.; Nixon, R.A.; Che, S. Regional selectivity of rab5 and rab7 protein upregulation in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis. 2010, 22, 631–639. [Google Scholar] [CrossRef]
- Armstrong, A.; Mattsson, N.; Appelqvist, H.; Janefjord, C.; Sandin, L.; Agholme, L.; Olsson, B.; Svensson, S.; Blennow, K.; Zetterberg, H.; et al. Lysosomal network proteins as potential novel CSF biomarkers for Alzheimer’s disease. Neuromolecular. Med. 2014, 16, 150–160. [Google Scholar] [CrossRef]
- Fontaine, S.N.; Zheng, D.; Sabbagh, J.J.; Martin, M.D.; Chaput, D.; Darling, A.; Trotter, J.H.; Stothert, A.R.; Nordhues, B.A.; Lussier, A.; et al. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 2016, 35, 1537–1549. [Google Scholar] [CrossRef]
- Cheetham, M.E.; Anderton, B.H.; Jackson, A.P. Inhibition of hsc70-catalysed clathrin uncoating by HSJ1 proteins. Biochem. J. 1996, 319 Pt 1, 103–108. [Google Scholar] [CrossRef]
- Xu, Y.; Cui, L.; Dibello, A.; Wang, L.; Lee, J.; Saidi, L.; Lee, J.G.; Ye, Y. DNAJC5 facilitates USP19-dependent unconventional secretion of misfolded cytosolic proteins. Cell Discov. 2018, 4, 11. [Google Scholar] [CrossRef]
- Lee, J.G.; Takahama, S.; Zhang, G.; Tomarev, S.I.; Ye, Y. Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat. Cell Biol. 2016, 18, 765–776. [Google Scholar] [CrossRef]
- Tai, H.C.; Serrano-Pozo, A.; Hashimoto, T.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 2012, 181, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Keck, S.; Nitsch, R.; Grune, T.; Ullrich, O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem. 2003, 85, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef]
- Gurke, S.; Barroso, J.F.; Hodneland, E.; Bukoreshtliev, N.V.; Schlicker, O.; Gerdes, H.H. Tunneling nanotube (TNT)-like structures facilitate a constitutive, actomyosin-dependent exchange of endocytic organelles between normal rat kidney cells. Exp. Cell Res. 2008, 314, 3669–3683. [Google Scholar] [CrossRef] [PubMed]
- Tardivel, M.; Bégard, S.; Bousset, L.; Dujardin, S.; Coens, A.; Melki, R.; Buée, L.; Colin, M. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 2016, 4, 117. [Google Scholar] [CrossRef]
- Abounit, S.; Wu, J.W.; Duff, K.; Victoria, G.S.; Zurzolo, C. Tunneling nanotubes: A possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion 2016, 10, 344–351. [Google Scholar] [CrossRef]
- Victoria, G.S.; Zurzolo, C. The spread of prion-like proteins by lysosomes and tunneling nanotubes: Implications for neurodegenerative diseases. J. Cell Biol. 2017, 216, 2633–2644. [Google Scholar] [CrossRef]
- Wu, J.W.; Herman, M.; Liu, L.; Simoes, S.; Acker, C.M.; Figueroa, H.; Steinberg, J.I.; Margittai, M.; Kayed, R.; Zurzolo, C.; et al. Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 2013, 288, 1856–1870. [Google Scholar] [CrossRef]
- Imoto, Y.; Raychaudhuri, S.; Ma, Y.; Fenske, P.; Sandoval, E.; Itoh, K.; Blumrich, E.M.; Matsubayashi, H.T.; Mamer, L.; Zarebidaki, F.; et al. Dynamin is primed at endocytic sites for ultrafast endocytosis. Neuron 2022, 110, 2815–2835.e2813. [Google Scholar] [CrossRef]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 is a master regulator of tau uptake and spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef]
- Heusermann, W.; Hean, J.; Trojer, D.; Steib, E.; von Bueren, S.; Graff-Meyer, A.; Genoud, C.; Martin, K.; Pizzato, N.; Voshol, J.; et al. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J. Cell Biol. 2016, 213, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Calafate, S.; Flavin, W.; Verstreken, P.; Moechars, D. Loss of Bin1 Promotes the Propagation of Tau Pathology. Cell Rep. 2016, 17, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Freeman, D.; Cedillos, R.; Choyke, S.; Lukic, Z.; McGuire, K.; Marvin, S.; Burrage, A.M.; Sudholt, S.; Rana, A.; O’Connor, C.; et al. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS ONE 2013, 8, e62143. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.H.; Ajit, A.; Tabassum, Z.; Patel, N.; Tian, X.; Chen, Y.; Prevatte, A.W.; Ling, K.; Rigo, F.; Meeker, R.B.; et al. Tau seeds are subject to aberrant modifications resulting in distinct signatures. Cell Rep. 2021, 35, 109037. [Google Scholar] [CrossRef]
- Alonso, A.D.; Grundke-Iqbal, I.; Barra, H.S.; Iqbal, K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: Sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. USA 1997, 94, 298–303. [Google Scholar] [CrossRef]
- Strang, K.H.; Croft, C.L.; Sorrentino, Z.A.; Chakrabarty, P.; Golde, T.E.; Giasson, B.I. Distinct differences in prion-like seeding and aggregation between Tau protein variants provide mechanistic insights into tauopathies. J. Biol. Chem. 2018, 293, 2408–2421. [Google Scholar] [CrossRef] [PubMed]
- Manassero, G.; Guglielmotto, M.; Monteleone, D.; Vasciaveo, V.; Butenko, O.; Tamagno, E.; Arancio, O.; Tabaton, M. Dual Mechanism of Toxicity for Extracellular Injection of Tau Oligomers versus Monomers in Human Tau Mice. J. Alzheimers Dis. 2017, 59, 743–751. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef]
- Hanger, D.P.; Lau, D.H.; Phillips, E.C.; Bondulich, M.K.; Guo, T.; Woodward, B.W.; Pooler, A.M.; Noble, W. Intracellular and extracellular roles for tau in neurodegenerative disease. J. Alzheimers Dis. 2014, 40 (Suppl. 1), S37–S45. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, K.J.; Ross, J.L.; Feinstein, H.E.; Feinstein, S.C.; Israelachvili, J. Complementary dimerization of microtubule-associated tau protein: Implications for microtubule bundling and tau-mediated pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 7445–7450. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Bera, S.; Qiao, Q.; Tang, Y.; Lao, Z.; Luo, Y.; Gazit, E.; Wei, G. Liquid–liquid phase separation of tau protein is encoded at the monomeric level. J. Phys. Chem. Lett. 2021, 12, 2576–2586. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Grundke-Iqbal, I. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.M.; Bossing, T.; Page, A.; Shepherd, D.; Mudher, A. Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol. 2010, 120, 593–604. [Google Scholar] [CrossRef]
- Man, V.H.; He, X.; Gao, J.; Wang, J. Phosphorylation of Tau R2 Repeat Destabilizes Its Binding to Microtubules: A Molecular Dynamics Simulation Study. ACS Chem. Neurosci. 2023, 14, 458–467. [Google Scholar] [CrossRef]
- Stokin, G.B.; Goldstein, L.S. Axonal transport and Alzheimer’s disease. Annu. Rev. Biochem. 2006, 75, 607–627. [Google Scholar] [CrossRef]
- Sherizadeh, H. What is axoplasmic transport? Considering the role of exercise training: A mini review. J. Exerc. Organ Cross Talk 2022, 2, 123–131. [Google Scholar]
- Venkatramani, A.; Panda, D. Regulation of neuronal microtubule dynamics by tau: Implications for tauopathies. Int. J. Biol. Macromol. 2019, 133, 473–483. [Google Scholar] [CrossRef]
- Prior, R.; Van Helleputte, L.; Benoy, V.; Van Den Bosch, L. Defective axonal transport: A common pathological mechanism in inherited and acquired peripheral neuropathies. Neurobiol. Dis. 2017, 105, 300–320. [Google Scholar] [CrossRef]
- Kesidou, E.; Theotokis, P.; Damianidou, O.; Boziki, M.; Konstantinidou, N.; Taloumtzis, C.; Sintila, S.-A.; Grigoriadis, P.; Evangelopoulos, M.E.; Bakirtzis, C. CNS Ageing in Health and Neurodegenerative Disorders. J. Clin. Med. 2023, 12, 2255. [Google Scholar] [CrossRef]
- Castellani, R.J.; Rolston, R.K.; Smith, M.A. Alzheimer disease. Disease-a-Month DM 2010, 56, 484. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomska, G.; Niewiadomski, W.; Steczkowska, M.; Gasiorowska, A. Tau Oligomers Neurotoxicity. Life 2021, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Shafiei, S.S.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Front. Aging Neurosci. 2017, 9, 83. [Google Scholar] [CrossRef]
- Ward, S.M.; Himmelstein, D.S.; Lancia, J.K.; Binder, L.I. Tau oligomers and tau toxicity in neurodegenerative disease. Biochem. Soc. Trans. 2012, 40, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Gerson, J.E.; Kayed, R. Formation and propagation of tau oligomeric seeds. Front. Neurol. 2013, 4, 93. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- de Calignon, A.; Fox, L.M.; Pitstick, R.; Carlson, G.A.; Bacskai, B.J.; Spires-Jones, T.L.; Hyman, B.T. Caspase activation precedes and leads to tangles. Nature 2010, 464, 1201–1204. [Google Scholar] [CrossRef]
- Ishihara, T.; Hong, M.; Zhang, B.; Nakagawa, Y.; Lee, M.K.; Trojanowski, J.Q.; Lee, V.M. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron 1999, 24, 751–762. [Google Scholar] [CrossRef]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef]
- Laurent, C.; Buée, L.; Blum, D. Tau and neuroinflammation: What impact for Alzheimer’s Disease and Tauopathies? Biomed. J. 2018, 41, 21–33. [Google Scholar] [CrossRef]
- Bellucci, A.; Bugiani, O.; Ghetti, B.; Spillantini, M.G. Presence of reactive microglia and neuroinflammatory mediators in a case of frontotemporal dementia with P301S mutation. Neurodegener. Dis. 2011, 8, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Schofield, E.; Kersaitis, C.; Shepherd, C.E.; Kril, J.J.; Halliday, G.M. Severity of gliosis in Pick’s disease and frontotemporal lobar degeneration: Tau-positive glia differentiate these disorders. Brain 2003, 126, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Botrán, R.; Ahmed, Z.; Crespo, F.A.; Gatenbee, C.; Gonzalez, J.; Dickson, D.W.; Litvan, I. Cytokine expression and microglial activation in progressive supranuclear palsy. Parkinsonism. Relat. Disord. 2011, 17, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shiwaku, H.; Tanaka, H.; Obita, T.; Ohuchi, S.; Yoshioka, Y.; Jin, X.; Kondo, K.; Fujita, K.; Homma, H.; et al. Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat. Commun. 2021, 12, 6565. [Google Scholar] [CrossRef]
- Chen, X.; Firulyova, M.; Manis, M.; Herz, J.; Smirnov, I.; Aladyeva, E.; Wang, C.; Bao, X.; Finn, M.B.; Hu, H.; et al. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature 2023, 615, 668–677. [Google Scholar] [CrossRef]
- Kalia, V.; Niedzwiecki, M.M.; Bradner, J.M.; Lau, F.K.; Anderson, F.L.; Bucher, M.L.; Manz, K.E.; Schlotter, A.P.; Fuentes, Z.C.; Pennell, K.D.; et al. Cross-species metabolomic analysis of tau- and DDT-related toxicity. PNAS Nexus 2022, 1, pgac050. [Google Scholar] [CrossRef]
- Pascoal, T.A.; Mathotaarachchi, S.; Mohades, S.; Benedet, A.L.; Chung, C.O.; Shin, M.; Wang, S.; Beaudry, T.; Kang, M.S.; Soucy, J.P.; et al. Amyloid-β and hyperphosphorylated tau synergy drives metabolic decline in preclinical Alzheimer’s disease. Mol. Psychiatry 2017, 22, 306–311. [Google Scholar] [CrossRef]
- Adams, J.N.; Lockhart, S.N.; Li, L.; Jagust, W.J. Relationships Between Tau and Glucose Metabolism Reflect Alzheimer’s Disease Pathology in Cognitively Normal Older Adults. Cereb. Cortex 2019, 29, 1997–2009. [Google Scholar] [CrossRef]
- Kaddurah-Daouk, R.; Zhu, H.; Sharma, S.; Bogdanov, M.; Rozen, S.G.; Matson, W.; Oki, N.O.; Motsinger-Reif, A.A.; Churchill, E.; Lei, Z.; et al. Alterations in metabolic pathways and networks in Alzheimer’s disease. Transl. Psychiatry 2013, 3, e244. [Google Scholar] [CrossRef]
- Strom, A.; Iaccarino, L.; Edwards, L.; Lesman-Segev, O.H.; Soleimani-Meigooni, D.N.; Pham, J.; Baker, S.L.; Landau, S.M.; Jagust, W.J.; Miller, B.L.; et al. Cortical hypometabolism reflects local atrophy and tau pathology in symptomatic Alzheimer’s disease. Brain 2022, 145, 713–728. [Google Scholar] [CrossRef]
- York, A.; Everhart, A.; Vitek, M.P.; Gottschalk, K.W.; Colton, C.A. Metabolism-Based Gene Differences in Neurons Expressing Hyperphosphorylated AT8- Positive (AT8+) Tau in Alzheimer’s Disease. ASN Neuro. 2021, 13, 17590914211019443. [Google Scholar] [CrossRef] [PubMed]
- Joly-Amado, A.; Serraneau, K.S.; Brownlow, M.; Marín de Evsikova, C.; Speakman, J.R.; Gordon, M.N.; Morgan, D. Metabolic changes over the course of aging in a mouse model of tau deposition. Neurobiol. Aging 2016, 44, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Koss, D.J.; Robinson, L.; Drever, B.D.; Plucińska, K.; Stoppelkamp, S.; Veselcic, P.; Riedel, G.; Platt, B. Mutant Tau knock-in mice display frontotemporal dementia relevant behaviour and histopathology. Neurobiol. Dis. 2016, 91, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, L.H.; Rae, C.; Ittner, L.M.; Götz, J.; Sonnewald, U. Glutamate metabolism is impaired in transgenic mice with tau hyperphosphorylation. J. Cereb. Blood Flow Metab. 2013, 33, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Traxler, L.; Herdy, J.R.; Stefanoni, D.; Eichhorner, S.; Pelucchi, S.; Szücs, A.; Santagostino, A.; Kim, Y.; Agarwal, R.K.; Schlachetzki, J.C.M.; et al. Warburg-like metabolic transformation underlies neuronal degeneration in sporadic Alzheimer’s disease. Cell Metab. 2022, 34, 1248–1263.e1246. [Google Scholar] [CrossRef]
- Tracy, T.E.; Madero-Pérez, J.; Swaney, D.L.; Chang, T.S.; Moritz, M.; Konrad, C.; Ward, M.E.; Stevenson, E.; Hüttenhain, R.; Kauwe, G.; et al. Tau interactome maps synaptic and mitochondrial processes associated with neurodegeneration. Cell 2022, 185, 712–728.e714. [Google Scholar] [CrossRef]
- Crespo, R.; Koudstaal, W.; Apetri, A. In Vitro Assay for Studying the Aggregation of Tau Protein and Drug Screening. J. Vis. Exp. 2018, 141, e58570. [Google Scholar] [CrossRef]
- Aulston, B.; Liu, Q.; Reilly, P.; Yuan, S.H. An In Vitro Model for Studying Tau Aggregation Using Lentiviral-mediated Transduction of Human Neurons. J. Vis. Exp. 2019, 147, e59433. [Google Scholar] [CrossRef]
- Kitoka, K.; Skrabana, R.; Gasparik, N.; Hritz, J.; Jaudzems, K. NMR Studies of Tau Protein in Tauopathies. Front. Mol. Biosci. 2021, 8, 761227. [Google Scholar] [CrossRef]
- Lim, S.; Haque, M.M.; Kim, D.; Kim, D.J.; Kim, Y.K. Cell-based Models To Investigate Tau Aggregation. Comput. Struct. Biotechnol. J. 2014, 12, 7–13. [Google Scholar] [CrossRef]
- Limorenko, G.; Tatli, M.; Kolla, R.; Nazarov, S.; Weil, M.-T.; Schöndorf, D.C.; Geist, D.; Reinhardt, P.; Ehrnhoefer, D.E.; Stahlberg, H.; et al. Fully co-factor-free ClearTau platform produces seeding-competent Tau fibrils for reconstructing pathological Tau aggregates. Nat. Commun. 2023, 14, 3939. [Google Scholar] [CrossRef] [PubMed]
- Manos, J.D.; Preiss, C.N.; Venkat, N.; Tamm, J.; Reinhardt, P.; Kwon, T.; Wu, J.; Winter, A.D.; Jahn, T.R.; Yanamandra, K.; et al. Uncovering specificity of endogenous TAU aggregation in a human iPSC-neuron TAU seeding model. iScience 2022, 25, 103658. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Sato, Y.; Sasaki, T.; Shimozawa, A.; Imaizumi, K.; Shindo, T.; Miyao, S.; Kiyama, K.; Kondo, T.; Shibata, S.; et al. A next-generation iPSC-derived forebrain organoid model of tauopathy with tau fibrils by AAV-mediated gene transfer. Cell Rep. Methods 2022, 2, 100289. [Google Scholar] [CrossRef]
- Gutknecht, M.F.; Kaku, H.; Rothstein, T.L. Microparticle immunocapture assay for quantitation of protein multimer amount and size. Cell Rep. Methods 2022, 2, 100214. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.A.; Zuniga, G.; Gonzalez, E.M.; Butler, D.; Temple, S.; Frost, B. TauLUM, an in vivo Drosophila sensor of tau multimerization, identifies neuroprotective interventions in tauopathy. Cell Rep. Methods 2022, 2, 100292. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lin, Y.; Krishnaswamy, S.; Pan, R.; Wu, Q.; Sandusky-Beltran, L.A.; Liu, M.; Kuo, M.-H.; Kong, X.-P.; Congdon, E.E.; et al. Single-domain antibody–based noninvasive in vivo imaging of α-synuclein or tau pathology. Sci. Adv. 2023, 9, eadf3775. [Google Scholar] [CrossRef]
- Zhao, J.; Jiang, L.; Matlock, A.; Xu, Y.; Zhu, J.; Zhu, H.; Tian, L.; Wolozin, B.; Cheng, J.-X. Mid-infrared chemical imaging of intracellular tau fibrils using fluorescence-guided computational photothermal microscopy. Light Sci. Appl. 2023, 12, 147. [Google Scholar] [CrossRef]
- Kimura, T.; Ono, M.; Seki, C.; Sampei, K.; Shimojo, M.; Kawamura, K.; Zhang, M.-R.; Sahara, N.; Takado, Y.; Higuchi, M. A quantitative in vivo imaging platform for tracking pathological tau depositions and resultant neuronal death in a mouse model. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 4298–4311. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Fodero-Tavoletti, M.T.; Masters, C.L.; Rowe, C.C. Tau imaging: Early progress and future directions. Lancet Neurol. 2015, 14, 114–124. [Google Scholar] [CrossRef]
- Watanabe, H.; Ono, M.; Saji, H. Novel PET/SPECT Probes for Imaging of Tau in Alzheimer’s Disease. Sci. World J. 2015, 2015, 124192. [Google Scholar] [CrossRef]
- Chen, C.; Kumbhar, R.R.; Wang, H.; Yang, X.; Gadhave, K.; Rastegar, C.; Kimura, Y.; Behensky, A.; Katakam, S.; Jeong, D. Pathological Tau transmission initiated by binding lymphocyte-activation gene 3. bioRxiv 2023. [Google Scholar] [CrossRef]
- Zhu, R.; Makwana, K.M.; Zhang, Y.; Rajewski, B.; Del Valle, J.; Wang, Y. Blocking Tau Transmission by Biomimetic Graphene Nanoparticles. J. Mater. Chem. B 2023, 11, 7378–7388. [Google Scholar] [CrossRef] [PubMed]
- Puangmalai, N.; Bhatt, N.; Montalbano, M.; Sengupta, U.; Gaikwad, S.; Ventura, F.; McAllen, S.; Ellsworth, A.; Garcia, S.; Kayed, R. Internalization mechanisms of brain-derived tau oligomers from patients with Alzheimer’s disease, progressive supranuclear palsy and dementia with Lewy bodies. Cell Death Dis. 2020, 11, 314. [Google Scholar] [CrossRef] [PubMed]
- Mothes, T.; Portal, B.; Konstantinidis, E.; Eltom, K.; Libard, S.; Streubel-Gallasch, L.; Ingelsson, M.; Rostami, J.; Lindskog, M.; Erlandsson, A. Astrocytic uptake of neuronal corpses promotes cell-to-cell spreading of tau pathology. Acta Neuropathol. Commun. 2023, 11, 97. [Google Scholar] [CrossRef]
- Batenburg, K.L.; Sestito, C.; Cornelissen-Steijger, P.; van Weering, J.R.; Price, L.S.; Heine, V.M.; Scheper, W. A 3D human co-culture to model neuron-astrocyte interactions in tauopathies. Biol. Proced. Online 2023, 25, 4. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, S.; Changolkar, L.; Riddle, D.M.; Kats, A.; Stieber, A.; Weitzman, S.A.; Zhang, B.; Li, Z.; Roberson, E.D.; Trojanowski, J.Q. Human tau pathology transmits glial tau aggregates in the absence of neuronal tau. J. Exp. Med. 2020, 217, e20190783. [Google Scholar] [CrossRef] [PubMed]
- Bouillet, T.; Ciba, M.; Alves, C.L.; Rodrigues, F.A.; Thielemann, C.; Colin, M.; Buée, L.; Halliez, S. Revisiting the involvement of tau in complex neural network remodeling: Analysis of the extracellular neuronal activity in organotypic brain slice co-cultures. J. Neural Eng. 2022, 19, 066026. [Google Scholar] [CrossRef]
- Mewes, A.; Franke, H.; Singer, D. Organotypic brain slice cultures of adult transgenic P301S mice—A model for tauopathy studies. PLoS ONE 2012, 7, e45017. [Google Scholar] [CrossRef]
- Croft, C.L.; Wade, M.A.; Kurbatskaya, K.; Mastrandreas, P.; Hughes, M.M.; Phillips, E.C.; Pooler, A.M.; Perkinton, M.S.; Hanger, D.P.; Noble, W. Membrane association and release of wild-type and pathological tau from organotypic brain slice cultures. Cell Death Dis. 2017, 8, e2671. [Google Scholar] [CrossRef]
- Katsikoudi, A.; Ficulle, E.; Cavallini, A.; Sharman, G.; Guyot, A.; Zagnoni, M.; Eastwood, B.J.; Hutton, M.; Bose, S. Quantitative propagation of assembled human Tau from Alzheimer’s disease brain in microfluidic neuronal cultures. J. Biol. Chem. 2020, 295, 13079–13093. [Google Scholar] [CrossRef]
- Calafate, S.; Buist, A.; Miskiewicz, K.; Vijayan, V.; Daneels, G.; De Strooper, B.; de Wit, J.; Verstreken, P.; Moechars, D. Synaptic contacts enhance cell-to-cell tau pathology propagation. Cell Rep. 2015, 11, 1176–1183. [Google Scholar] [CrossRef]
- Usenovic, M.; Niroomand, S.; Drolet, R.E.; Yao, L.; Gaspar, R.C.; Hatcher, N.G.; Schachter, J.; Renger, J.J.; Parmentier-Batteur, S. Internalized tau oligomers cause neurodegeneration by inducing accumulation of pathogenic tau in human neurons derived from induced pluripotent stem cells. J. Neurosci. 2015, 35, 14234–14250. [Google Scholar] [CrossRef] [PubMed]
- Winston, C.N.; Aulston, B.; Rockenstein, E.M.; Adame, A.; Prikhodko, O.; Dave, K.N.; Mishra, P.; Rissman, R.A.; Yuan, S.H. Neuronal exosome-derived human tau is toxic to recipient mouse neurons in vivo. J. Alzheimers Dis. 2019, 67, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Peeraer, E.; Bottelbergs, A.; Van Kolen, K.; Stancu, I.-C.; Vasconcelos, B.; Mahieu, M.; Duytschaever, H.; Ver Donck, L.; Torremans, A.; Sluydts, E. Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol. Dis. 2015, 73, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Darricau, M.; Katsinelos, T.; Raschella, F.; Milekovic, T.; Crochemore, L.; Li, Q.; Courtine, G.; McEwan, W.A.; Dehay, B.; Bezard, E. Tau seeds from patients induce progressive supranuclear palsy pathology and symptoms in primates. Brain 2023, 146, 2524–2534. [Google Scholar] [CrossRef]
- Nguyen, A.P.T.; Daniel, G.; Valdés, P.; Islam, M.S.; Schneider, B.L.; Moore, D.J. G2019S LRRK2 enhances the neuronal transmission of tau in the mouse brain. Hum. Mol. Genet. 2018, 27, 120–134. [Google Scholar] [CrossRef]
- Kim, Y.; Park, H.; Kim, Y.; Kim, S.-H.; Lee, J.H.; Yang, H.; Kim, S.J.; Li, C.M.; Lee, H.; Na, D.-H. Pathogenic role of RAGE in tau transmission and memory deficits. Biol. Psychiatry 2023, 93, 829–841. [Google Scholar] [CrossRef]
- He, Z.; McBride, J.D.; Xu, H.; Changolkar, L.; Kim, S.-j.; Zhang, B.; Narasimhan, S.; Gibbons, G.S.; Guo, J.L.; Kozak, M. Transmission of tauopathy strains is independent of their isoform composition. Nat. Commun. 2020, 11, 7. [Google Scholar] [CrossRef]
- Wegmann, S.; Maury, E.A.; Kirk, M.J.; Saqran, L.; Roe, A.; DeVos, S.L.; Nicholls, S.; Fan, Z.; Takeda, S.; Cagsal-Getkin, O. Removing endogenous tau does not prevent tau propagation yet reduces its neurotoxicity. EMBO J. 2015, 34, 3028–3041. [Google Scholar] [CrossRef]
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 2016, 19, 1085–1092. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tau Isoforms | Tauopathies | Neuropathological Hallmarks | References |
|---|---|---|---|
| 3R Tau | Pick’s disease (PiD) | Ballooned neurons, gliosis, Pick bodies | [10,11] |
| 3R + 4R Tau | Primary age-related tauopathy (PART) | Neurofibrillary tangles (NFTs), absence of amyloid (Aβ) plaques | [12] |
| Alzheimer’s disease (AD) | NFTs, Aβ plaques, dystrophic neurites Plaques, pretangle neurons, tangle neurons | [11] | |
| Familial British dementia (FBD) | Cerebral amyloid angiopathy (CAA), parenchymal Aβ plaques, NFTs | [13] | |
| Familial Danish dementia (FDD) | CAA, NFTs, Aβ plaques, Danish amyloid | [13] | |
| Chronic traumatic encephalopathy (CTE) | NFTs, astrocytic tangles | [14] | |
| 4R Tau | Progressive supranuclear paralysis (PSP) | NFTs, tufted tau-positive astrocytes, coiled bodies | [15] |
| Globular glial tauopathy (GGT) | Globular oligodendrocytic inclusions, globular astrocytic inclusions | [16] | |
| Corticobasal degeneration (CBD) | Astrocytic plaques, preganglionic neurons, coiled bodies, argyrophilic threads | [17,18] | |
| Argyrophilic grain disease (AGD) | Argyrophilic grains, small spindle-shaped lesions, pretangle neurons, oligodendroglial coiled bodies | [19,20] | |
| Aging-related tau astrogliopathy (ARTAG) | Thorny or granular/fuzzy astrocytic tau | [21] |
| Tau Species | Molecular Weight (kDa) | Features | Isoforms or Compositions | Folding Types | References |
|---|---|---|---|---|---|
| Monomer | 55–74 | soluble | tau0N3R, tau1N3R, tau2N3R, tau0N4R, tau1N4R, and tau2N4R | [56] | |
| Dimer | 180 | cysteine-dependent and reducible | 2 Tau monomers | [45] | |
| 130 | cysteine-independent and unreducible | [57] | |||
| Small oligomers | 300–500 | soluble | 6–8 Tau monomers | [43] | |
| Granular oligomers | 1800 | insoluble, granule | 40 Tau monomers | ||
| Filaments | two-layered | 3R | Narrow Pick filaments | [51] | |
| Wide Pick filaments | |||||
| three-layered | 4R | PSP fold | |||
| GPT fold | |||||
| GGT type 1, type 2, and type 3 folds | |||||
| four-layered | AGD type 1, type 2 folds | ||||
| CBD fold | |||||
| two-layered | 3R + 4R | AD fold (including FBD and FDD folds) | |||
| CTE fold |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, J.; Sha, W.; Yuan, S.; Wu, J.; Huang, Y. Aggregation, Transmission, and Toxicity of the Microtubule-Associated Protein Tau: A Complex Comprehension. Int. J. Mol. Sci. 2023, 24, 15023. https://doi.org/10.3390/ijms241915023
Hu J, Sha W, Yuan S, Wu J, Huang Y. Aggregation, Transmission, and Toxicity of the Microtubule-Associated Protein Tau: A Complex Comprehension. International Journal of Molecular Sciences. 2023; 24(19):15023. https://doi.org/10.3390/ijms241915023
Chicago/Turabian StyleHu, Jiaxin, Wenchi Sha, Shuangshuang Yuan, Jiarui Wu, and Yunpeng Huang. 2023. "Aggregation, Transmission, and Toxicity of the Microtubule-Associated Protein Tau: A Complex Comprehension" International Journal of Molecular Sciences 24, no. 19: 15023. https://doi.org/10.3390/ijms241915023