1. Introduction
The alarming decline of biodiversity worldwide necessitates urgent conservation measures, particularly for wild, endangered, and understudied species. According to the International Union for Conservation of Nature’s (IUCN) Red List of Threatened Species, of the 5973 mammal species assessed, 1340 were classified as threatened with extinction, including 233 critically endangered taxa [
1]. This number includes wild, non-domesticated, non-model species, whose rapidly vanishing genetic resources may never be adequately explored and described. This concerns the European mink
Mustela lutreola L., 1761, a critically endangered, semiaquatic, secretive, and solitary representative of the mustelid family (Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; Mammalia; Eutheria; Laurasiatheria; Carnivora; Caniformia; Mustelidae; Mustelinae;
Mustela) [
2,
3] (
Figure 1). The species is listed in the Annex II to the Bern Convention on the Conservation of European Wildlife and Natural Habitats, the Annexes II and IV (priority species) of the Council Directive 92/43/EEC on the conservation of natural habitats and of wild fauna and flora, and in the Carpathian List of Endangered Species (critically endangered species), and considered one of the most endangered mammalian species in the world [
4,
5].
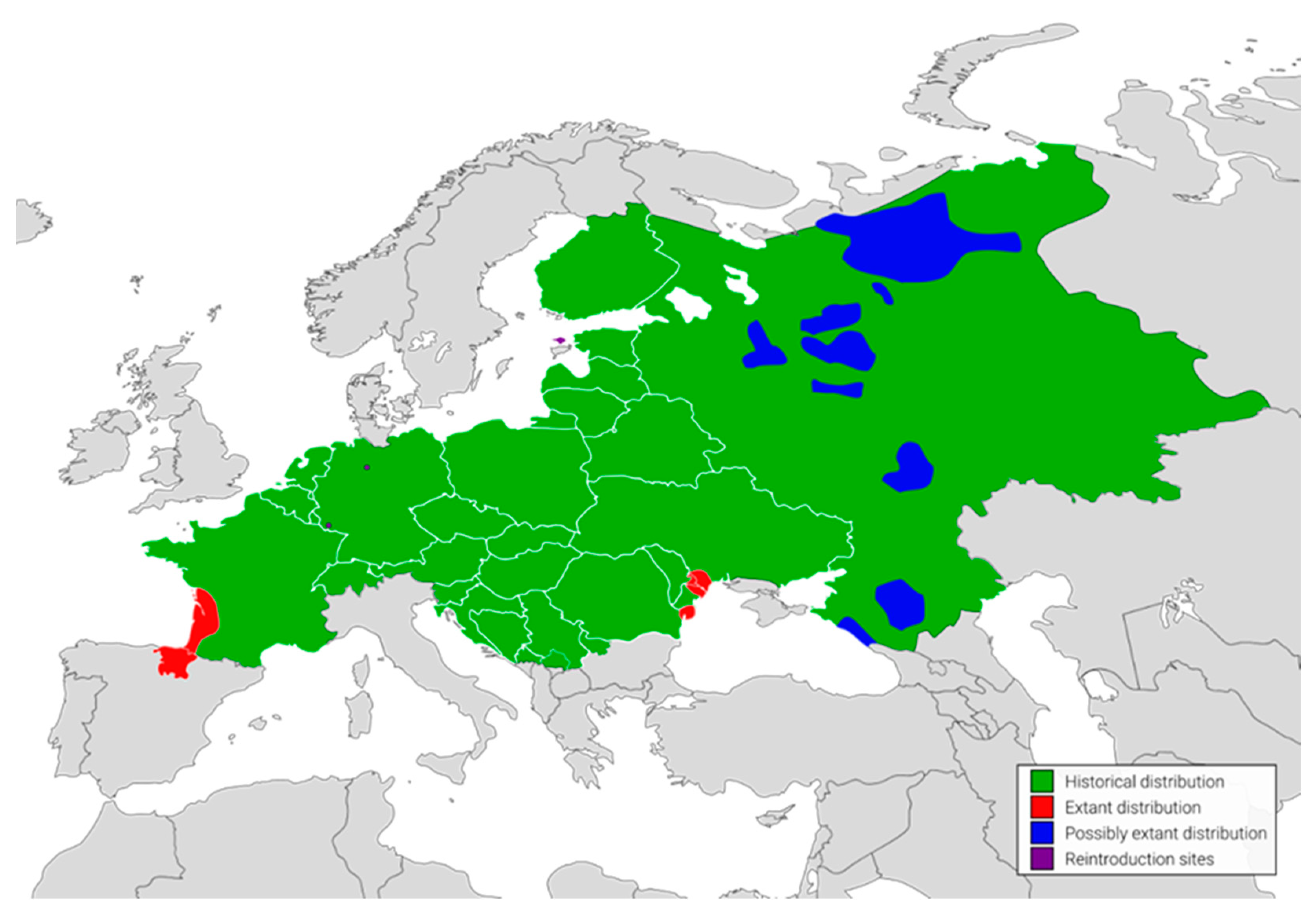
By the 19th century, European mink populations were relatively abundant and distributed across freshwater habitats throughout a large part of continental Europe [
5] (
Figure 2). Following a combination of habitat loss and fragmentation, commercial over-hunting for fur and the effects of introduced, invasive American mink
Neogale vison Schreber, 1777 lead to a dramatic depletion of the species’ populations, in terms of both shrinkage of a geographical range by 97%, and a reduction in the number of individuals persisting in the wild to about 5000 [
5,
6,
7]. What is more, an expected decline rate in number over the next three generations exceeds 80% [
5]. These days, only three isolated, declining populations, restricted to the European part of Russia, the Danube Delta, south-western France and north-eastern Spain survived [
5]. Reintroduction efforts successfully established populations in Estonia on Hiiumaa Island, as well as in Germany at two locations in Saarland and Lower Saxony [
5].
In response to the critical status of
M. lutreola, conservation efforts have been initiated in the late 20th century, focusing on captive breeding programmes (i.e., the European Association of Zoos and Aquaria (EAZA) Ex-situ Programme (EEP) for European mink, established in 1992, regional captive breeding programme initiated in Spain in 2004, and the European Mink Breeding Centre of the Ilmen Nature Reserve in Russia, operating since 2010), habitat restoration, local reintroduction initiatives (launched in Estonia, France, Germany, Russia and Spain), reintroduced populations monitoring, and public awareness campaigns [
8]. Of key importance for the effectiveness and efficiency of these measures are genomic research, informing targeted and evidence-based conservation strategies [
9,
10]. By studying the genomic makeup of the European mink, valuable insights into its population structure, genetic diversity, and evolutionary history can be gained. This knowledge is essential for understanding the unique adaptations and vulnerabilities of this species, as well as identifying and quantifying inbreeding, hybridization, and introgression [
11,
12,
13,
14,
15]. Such information is vital for developing targeted conservation efforts, including captive breeding programmes, reintroduction and translocation strategies, and genetic management plans, to halt ongoing depletion of genetic diversity of
M. lutreola sparse, local populations, and preserve their genetic integrity and long-term viability, ultimately aiding in their survival and recovery in the wild [
8].
Comprehensive research of the genetic resources of a critically endangered species is of paramount importance from both cognitive and practical perspectives. Not only do we gain new knowledge, enable a deeper understanding of its biology and conservation needs, but we can also provide critical information for informed conservation decision-making. This knowledge is invaluable for collecting and preserving irreplaceable genetic heritage that is on the verge of being lost forever and unnoticed, particularly in the case of a non-charismatic, rare, and elusive species. Meanwhile, despite the alarming situation of the European mink, the number of studies on its genetics and genomics is limited and urgently need to be completed [
8]. To date, there has not been a reference genome for the species, its mitochondrial genome was only sequenced in 2022, and no data were stored in the Sequence Read Archive (SRA) in the GenBank [
8,
16].
To address this problem, we present for the first time a platinum-standard reference-quality genome, i.e., high confidence, contiguous, de novo assembly of the haplotype-resolved diploid genome with full chromosome scaffold, for the European mink. By sharing this valuable resource, we aim to shed light on the species genomics, facilitate future research on its evolutionary history and present population structure, dynamics and adaptation, and thus catalyze global efforts towards the conservation and management of M. lutreola and other threatened mustelids, by offering novel perspectives on conservation genomics of this taxa.
2. Results
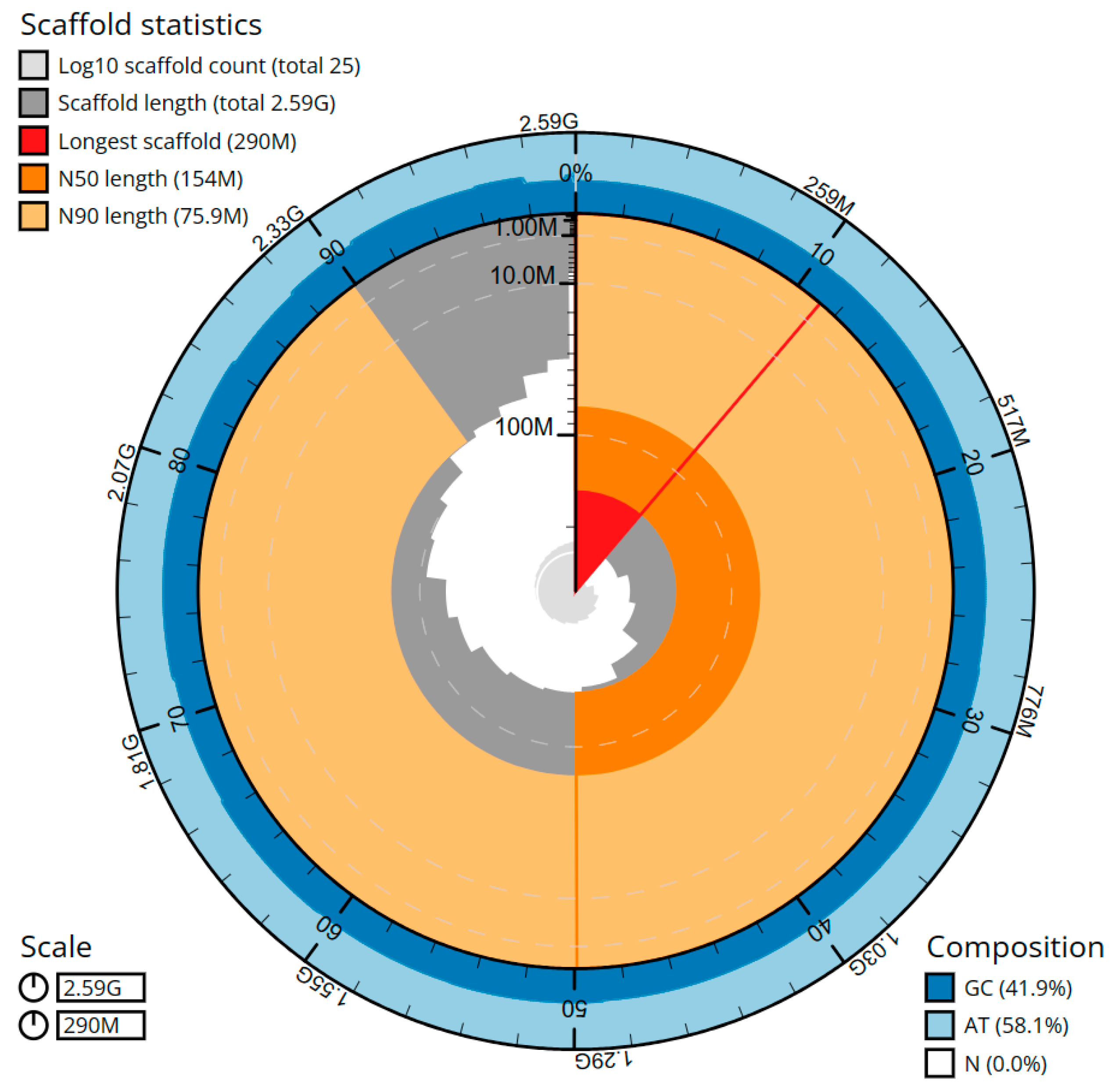
The cumulative length of the final assembly amounts to 2586.27 Mbp, including 39 gaps and encompassing 64 sequence contigs and 25 scaffolds, with a contig N50 (LG = 11) and a scaffold N50 (LG = 7) of 83.36 Mbp and 154.08 Mbp, respectively (
Figure 3,
Table 1). The presence of the 11 largest contigs containing 50% of the genomic sequences demonstrates significant contiguity within the assembly. On average, 36.15× coverage for the PacBio sequencing and 66.85× for the Hi-C sequencing was achieved. The genome assembly exhibited an average contig length of 40.41 Mbp, representing the mean size of the individual DNA fragments prior to scaffolding. The average scaffold length was 103.45 Mbp, indicating the typical size of the contiguous DNA sequences generated. The total gap length in the scaffolds amounted to 7.8 kbp, with an average gap length of 200 bp. Through manual assembly curation, a total of 27 missing or missed joins were rectified, and no sequence removed as haplotypic duplication, resulting in a 26.5% drop in the scaffold number. The scaffold N50 value remained unaltered, while a minor increase of 0.25% in the N90 length was observed as a result of the curation process.
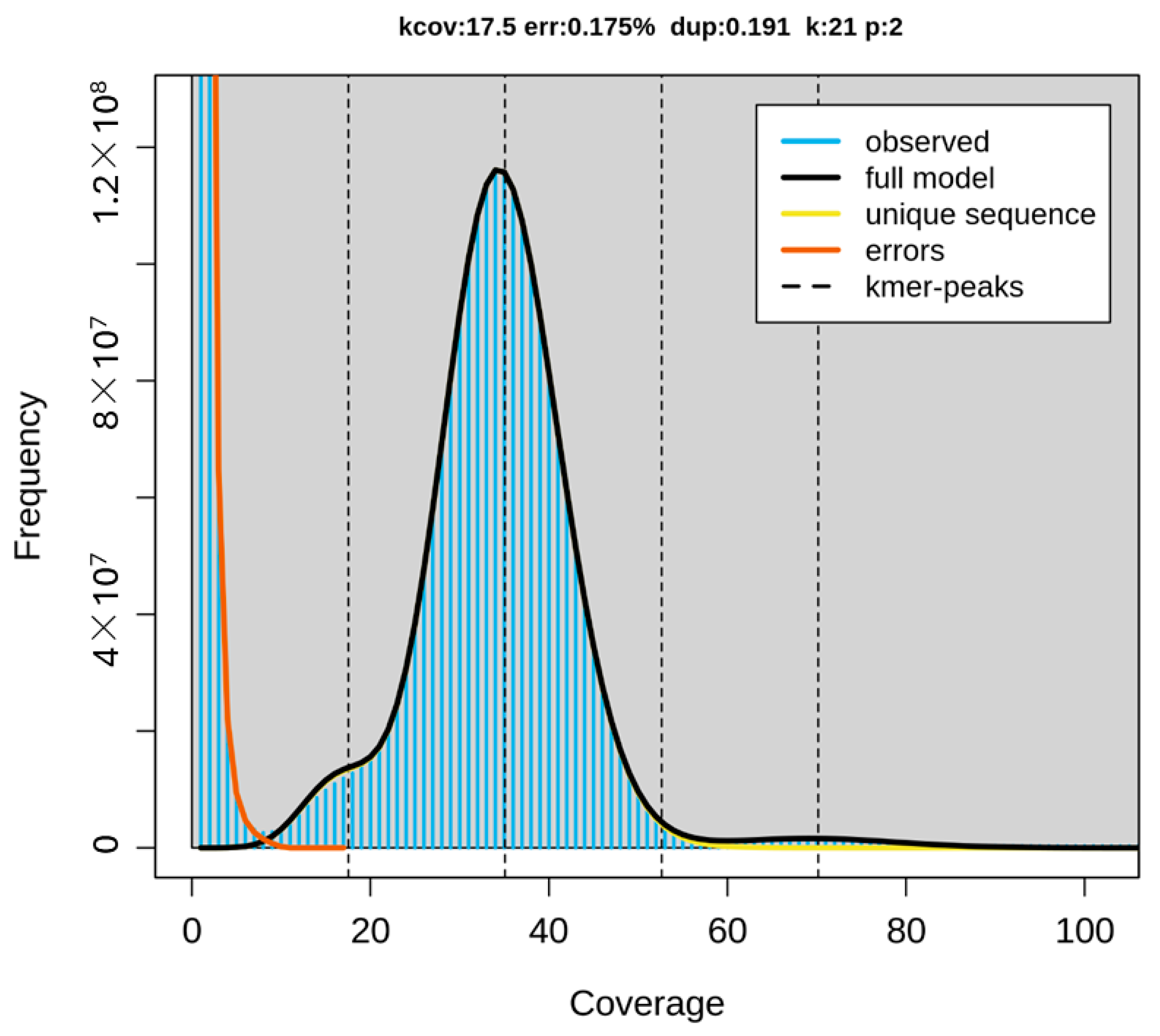
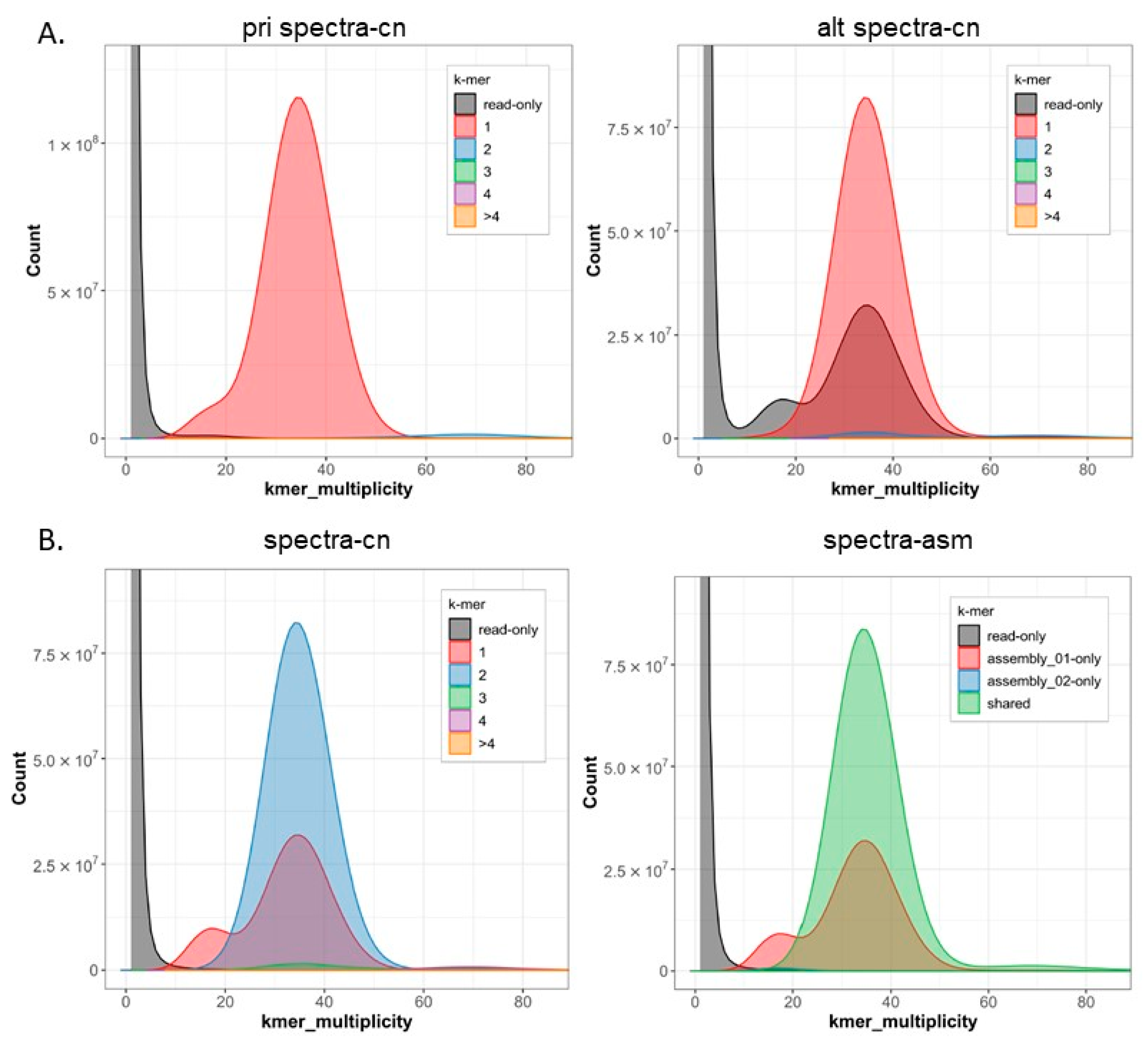
The newly sequenced genome displayed a GC base content of 41.85%, with adenine, cytosine, guanine, and thymine nucleotides accounting for 29.08%, 20.94%, 20.91%, and 29.07%, respectively. Repeat sequences constitute approximately 24.37% of the genome. The genome assembly analysis revealed that 99.83% of the regions were homozygous, while the remaining 0.17% were heterozygous (
Figure 4 and
Figure 5).
The estimated Quality Value (QV) of the Hifiasm assembly was 64.34, with k-mer completeness of 99.17%, and error rate of 3.6826 × 10
−7. The final, purged assembly had a BUSCO completeness score (C) of 98.15%. The score breakdown indicates that 95.92% of the expected complete single-copy genes (S) were identified as complete sequences, while 2.24% of the single-copy genes were found as duplicates or fragmented sequences (D). The fragmented fraction (F) score was 0.86%, denoting a proportion of expected complete duplicated genes found in the assembly. The missing (M) score, indicating a rate of missing expected complete genes, was 0.98%. Regarding false duplications, the BUSCO results showed promising outcomes, and the primary-only spectra plot from Merqury displayed a clean pattern. The purged assembly demonstrated completeness of 99.13%, a QV of 64.44, and an error rate of 3.6005 × 10
−7. Detailed characteristics of pre- and curated genome assemblies of a
M. lutreola are presented in
Table 2.
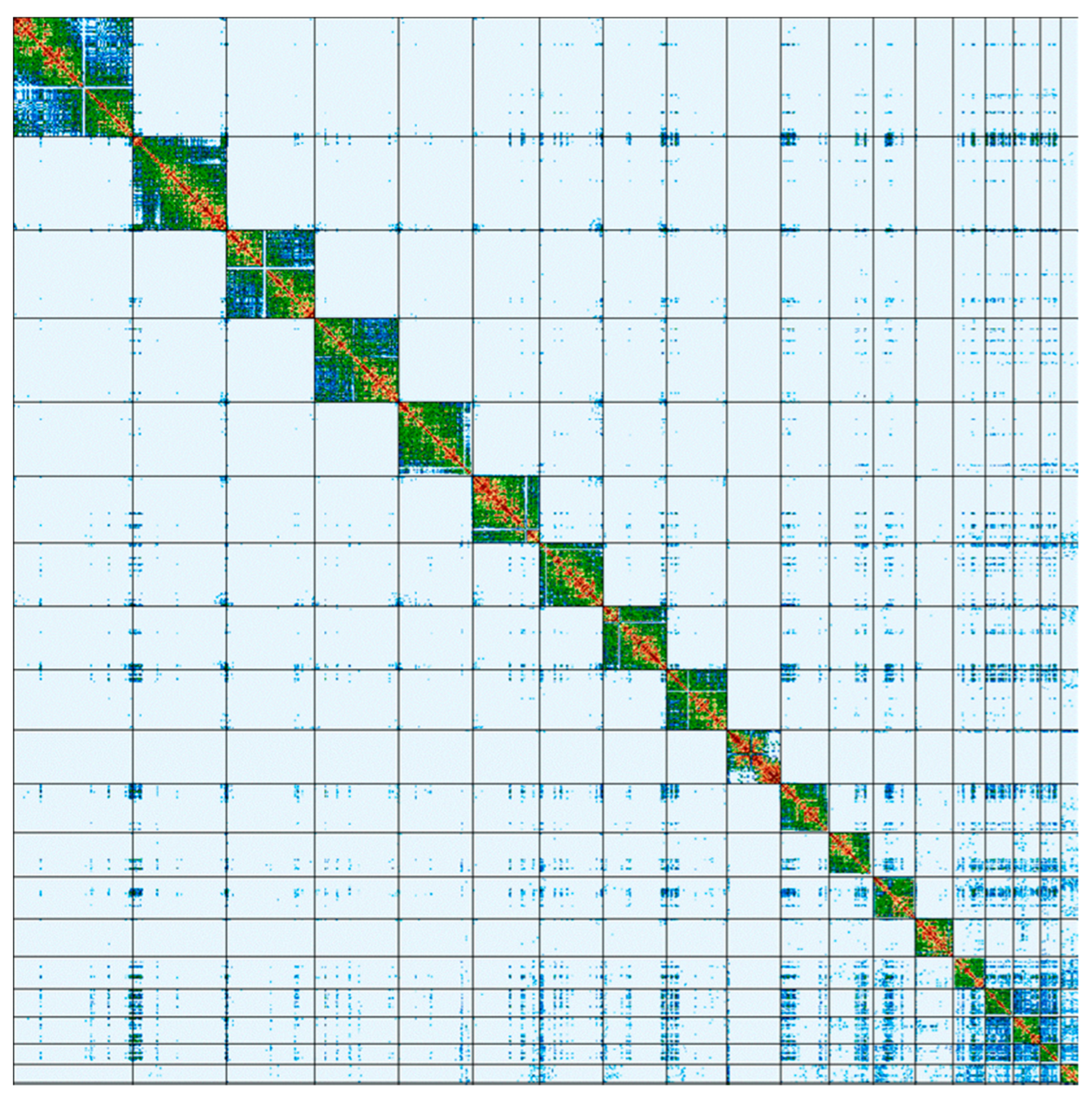
The vast majority (99.9985%) of the genome assembly was assigned to 20 C-scaffolds (pseudomolecules) [
17], comprising 18 autosomes, and the X and Y sex chromosomes (
Figure 6). Additionally, one unplaced (without a chromosome assignment) and four unlocalized (not localized to a specific position in the chromosome) scaffolds were identified in the assembly. Chromosome-scale scaffolds, confirmed by the Hi-C data, are named in order of size and characterized in
Table 3.
The complete mitochondrial genome assembly is 16,552 bp in length, and displayed a level of identity ranging from 99.62% to 99.94% with previously sequenced mitochondrial genomes of
M. lutreola, deposited in the GenBank [
16].
3. Discussion
The genome sequencing of various mustelids has provided valuable insights into their genetic composition and evolutionary history. Out of the 67 species in the Mustelidae family, 17 have reference genome assemblies ready and deposited in GenBank, while only in the case of seven species has a chromosome-level assembly been achieved (
Table 4) [
18,
19]. European mink stands as the only critically endangered species within this group that has now attained a reference genome of a platinum quality. A platinum genome is defined as a high-quality, near error-free and gapless, chromosome-level, haplotype-phased, reference genome assembly [
20,
21]. Additionally, the following standards were drawn for the Vertebrate Genomes Project of the Genome 10K consortium: N50 size of at least 1 Mbp for contigs and 10 Mbp for scaffolds, sequence error frequency of up to 1 in 10,000 bp, structural variants confirmed by multiple technologies, at least 90% of the sequence assigned to chromosomes and haplotype-phased [
21,
22]. Furthermore, a standard VGP reference genome involves an automated workflow that combines long-read sequencing, linked-read sequencing, optical mapping, and Hi-C data, with a final manual curation step to enhance the genome assembly and minimize errors [
20,
23]. The genome sequence reported in this article was assembled following the VGP quality requirements and meets the abovementioned conditions.
The size of the reported genome of a
M. lutreola fits well with previously sequenced genomes of other representatives of the Mustela genus, being the biggest among the so-called ferret group, clustering the European polecat, the steppe polecat, the black-footed ferret, and the European mink [
16,
24,
25]. The European mink genome assembly was found to be noticeably close in size and the GC-content to the earlier estimates, which predicted the genome size of the species to be around 2.411–2.474 Gbp and the GC content to be approximately 42%, based on the sequenced genomes of the ferret (MusPutFur1.0, GenBank assembly accession: GCF_000215625.1) and the European polecat (polecat_10x_lmp_bionano, GenBank assembly accession: GCA_902207235.1) [
16].
The number of whole-chromosome pseudomolecules assembled in the European mink’s reference genome (18 autosomes and two heterosomes) is consistent with a diploid chromosome number reported for this species (2
n = 38) [
26]. Such a diploid number of chromosomes is typical of the family Mustelidae, occurring in over 60% of its representatives [
27].
The high-quality reference genome of European mink represents the fulfilment of earlier calls for whole-genome sequencing of this critically endangered species [
8,
28], marking a significant step forward in advancing our understanding of the species’ genetic composition and promising to enhance conservation efforts. A platinum quality reference genome serves as a highly accurate and reliable resource for various research applications, including evolutionary studies, population genomics, comparative genomics and functional genomics [
13,
29]. Such reference genomes are particularly valuable for species of conservation concern, where accurate genomic information is crucial for effective conservation efforts and understanding the species’ biology [
10,
13]. The significant application potential of genomics in addressing conservation problems is well-documented for various carnivorans, e.g., African wild dog
Lycaon pictus, Eastern wolf
Canis lupus lycaon, puma
Puma concolor, Iberian lynx
Lynx pardinus, and wolverine
Gulo gulo [
11,
30,
31,
32].
The reference genome of the European mink can play a pivotal role in addressing the conservation problems faced by this species, enabling the revision of management units to effectively manage genetic diversity within populations and minimize the outbreeding risk associated with inter-population translocations, providing comprehensive insights into the impacts of captive breeding, resolving phylogeographic questions, and facilitating the evaluation of conservation program efficiency and effectiveness [
8,
33]. It is important to recognize that the application of genomics to conservation often encounters a significant challenge—the high costs associated with molecular analyses. However, this is where advanced genomic tools come into play. Through techniques like whole-genome re-sequencing and reduced-representation approaches, conservation genomics offers a potential solution to mitigate these cost constraints, as these methodologies can reduce the number of markers required for analysis and monitoring projects in the field of conservation activities [
11,
12,
13,
14,
15,
33].
Comprehensive population genomics studies are crucial, as the limited data on interpopulation genetic diversity could significantly impede the effectiveness of captive breeding, reintroduction programs, and potential translocations for persisting wild populations of
M. lutreola [
28,
34,
35,
36,
37]. By analysing the genetic variation, relatedness between populations, and identifying adaptive traits loci, it aids in the establishment of appropriate management units for targeted conservation. One prominent example of the application of population genomics is the ongoing debate surrounding the potential inclusion of the Spanish conservation breeding initiative in the European Endangered Species Programme (EEP) for European mink [
28]. Additionally, plans to obtain new founders from the wild Romanian population emphasize the importance of understanding historical population dynamics and connectivity between existing European mink populations [
14,
28].
Compared to traditional genetic approaches, the reference genome provides advanced genomic tools to investigate evolutionary relationships among different European mink populations. It helps in determining historical patterns of migration and divergence, shedding light on the species’ phylogenomic and phylogeographic history [
38,
39]. Such understanding is essential for re-evaluating Evolutionarily Significant Units (ESUs) and Management Units (MUs) for
M. lutreola [
34,
40,
41,
42]. These units guide conservation actions based on distinct evolutionary lineages and aim to preserve the genetic diversity of the species [
40,
41]. Moreover, the reference genome of the European mink serves as a valuable tool in resolving uncertainties about its past distribution over continental Europe [
43,
44]. It helps identify regions of high genetic diversity, indicating historical refugia or areas of long-term stability for the species, as well as regions of genetic variation associated with adaptation to specific environments, habitat use, disease resistance, responses to changing conditions, or other crucial ecological factors [
10,
29]. Furthermore, it can provide insights into the evolutionary history of the species and its relationship with other mustelids.
Another pressing concern in the European mink conservation revolves around the assessment of the impact of the conservation breeding process on the development of traits essential for survival in the wild, specifically focusing on the adaptation to captivity [
8]. Farquharson et al. [
45] reported strong effects of inbreeding on the European mink offspring fitness in the EEP captive breeding program, highlighting the importance of addressing genetic management. One of the factors that reduces reproductive success in captivity and reconstituted (reintroduced) populations is aggressive behaviour exhibited by males toward females, which can lead to their exclusion from mating [
46,
47]. However, without a clear understanding of the heritability of these personality traits, assessing the risk of reducing genetic variation in reintroduced populations due to the release of individuals with specific personality types becomes challenging [
46,
47,
48]. Furthermore, the reference genome can help to identify regions of the genome that are associated with local adaptation or specific traits relevant to survival in the wild [
10,
29]. This information can guide breeding strategies to ensure that valuable adaptive and survival traits are retained in captive populations, even when introducing new individuals to counteract inbreeding. By avoiding excessive inbreeding, the fitness and resilience of an offspring can be improved, increasing their chances of survival in both captive and future reintroduction settings.
Genomic information is essential to examine the impact of reintroduced individuals on shaping the gene pools of wild populations to address potential issues of outbreeding and the risk of losing unique adaptations [
40,
43]. In this regard, genomic-scale analyses serve as a valuable tool in evaluating potential fitness losses, thus facilitating more informed decisions and enhancing the success of reintroduction and translocation efforts [
10,
13]. The reference genome can support conservation breeding strategies by assisting in the selection of founders for captive breeding programs. It also helps optimize breeding pairs and prevents over-representation of certain lineages, ensuring that individuals chosen for reproduction possess optimal genetic diversity and reduce the risk of inbreeding depression [
10,
13].
With the reference genome, it becomes feasible to conduct genome-wide monitoring of the European mink local populations. This allows researchers to track changes in genetic diversity, detect potential threats to specific populations, and evaluate the effectiveness of conservation interventions over time, across multiple generations. Understanding the genetic variation within and between populations can help identify populations with low genetic diversity, which may be at greater risk of inbreeding depression and reduced adaptive potential, as well as genetically unique and vulnerable populations. Conservation efforts can then prioritize these populations for targeted management and genetic supplementation if necessary (genetic rescue) [
49]. Genome-scale monitoring provides real-time information on the genetic health and status of endangered species populations, enabling adaptive management strategies to ensure their long-term survival, recovery, and genetic health [
50]. Adaptive management is a dynamic and flexible approach characterized by continuous revisions of conservation strategies based on genetic data and observed fitness outcomes, allowing for timely adjustments and improvements to enhance conservation effectiveness.
In conclusion, embracing reference genome offers a powerful and comprehensive approach to address the conservation issues faced by
M. lutreola, enhancing the prospects for its preservation and sustainable recovery. Future research directions in the European mink conservation genomics, based on the reported genome assembly, may involve building a reference pangenome to enable detailed population genomic studies, monitoring interpopulation genetic diversity patterns using restriction-site associated DNA sequencing (RADseq, ddRAD), and conducting a genome-wide scan for runs of homozygosity (ROH) to detect signatures of selection and estimate inbreeding [
15,
29,
51]. Many of the conservation issues observed in this species could have been prevented if decisions regarding the necessary actions were not made before obtaining knowledge about its genome, but resulted from it.
The reported genome assembly also provides perspectives for planning, implementing, monitoring, and evaluating conservation interventions for other closely related taxa. The reference genome of the European mink can serve as a valuable reference for conducting reference-based assembly or designing primers for targeted sequencing of specific genomic locations in mustelids, whose genomes are yet to be fully revealed (e.g., Mustela eversmanii, Mustela sibirica, Mustela itatsi).
In the context of large genome sequencing programs, the prioritization of species sequencing order becomes crucial, as it enables the optimization of funding allocation, research interest, and workload alignment, ensuring that endangered species receive the necessary resources and attention commensurate with their conservation urgency. By prioritizing the comprehensive research of genomic resources in critically endangered species like the European mink, we not only gain valuable knowledge for conservation decision-making, but also preserve the irreplaceable genetic heritage that is on the brink of being lost forever.
The relatively low research interest in the European mink is incongruent with its critical situation. This highlights the significance of promotional and informational campaigns aimed at drawing attention and raising awareness in society about its alarming threat of extinction, ultimately ensuring adequate focus and attention of the scientific community towards understanding and conserving this fascinating species, deserving effective protection.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}