
Molecular Mechanism of Mutational Disruption of DCLK1 Autoinhibition Provides a Rationale for Inhibitor Screening

Abstract
:1. Introduction
2. Results and Discussions
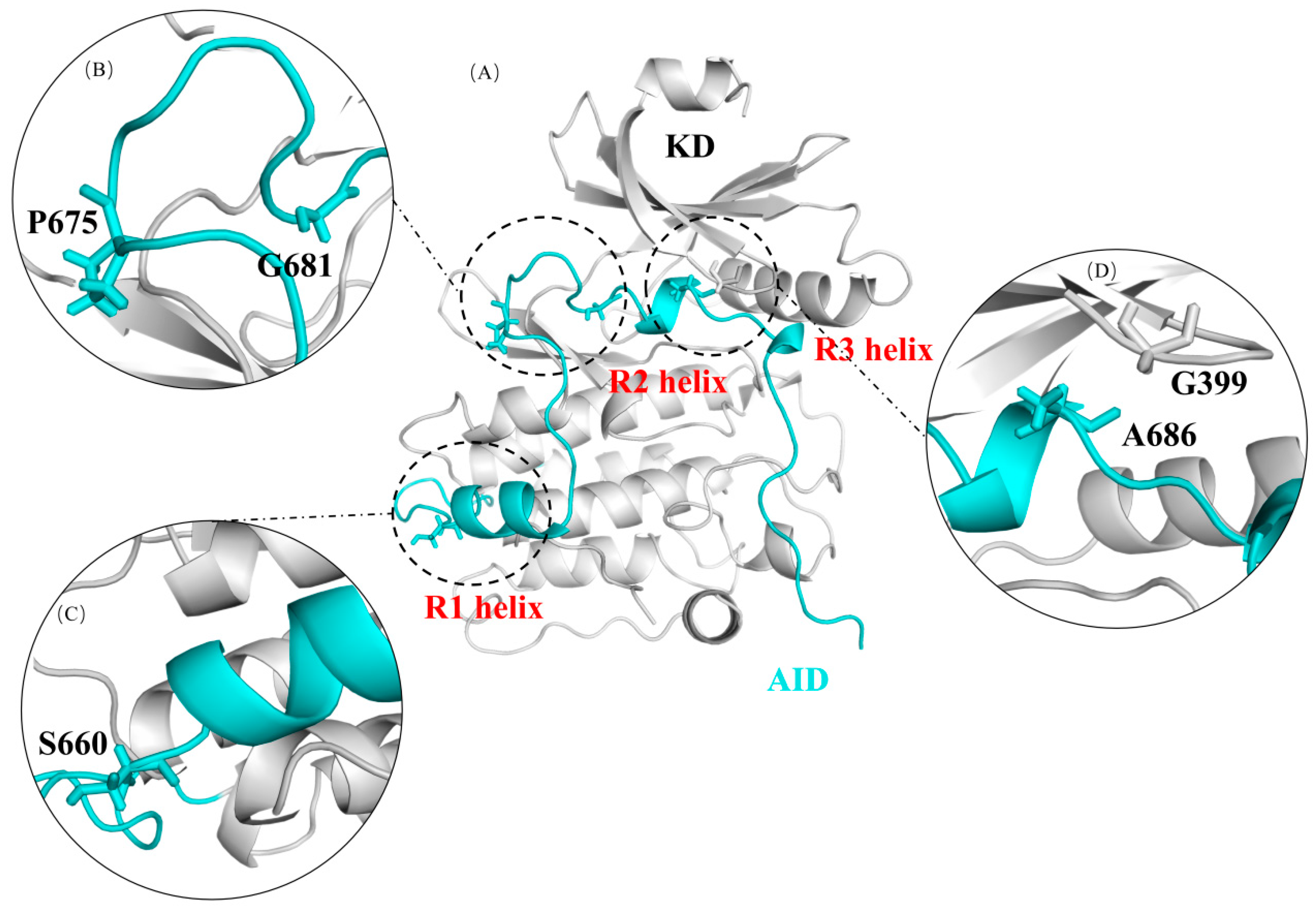
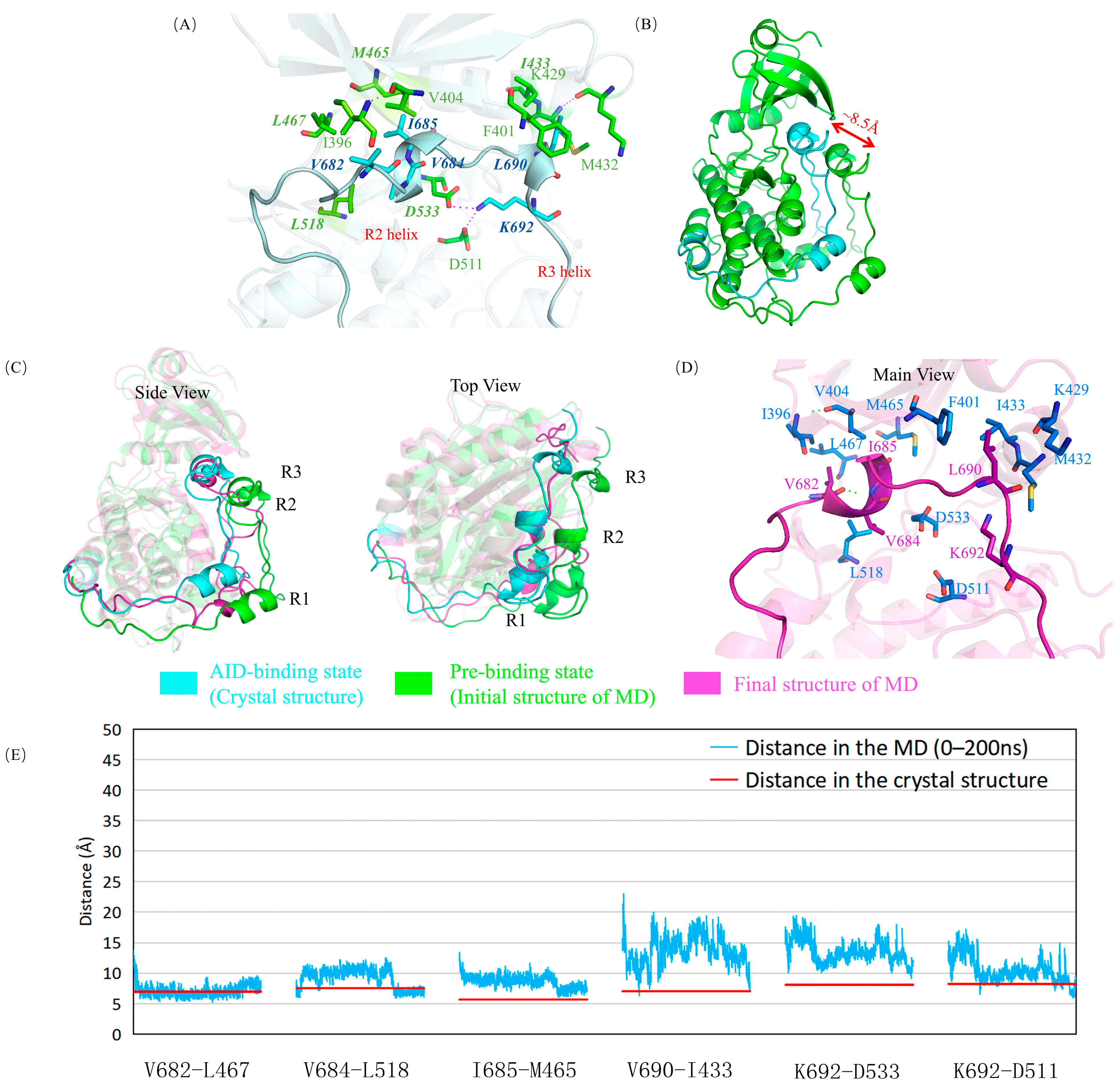
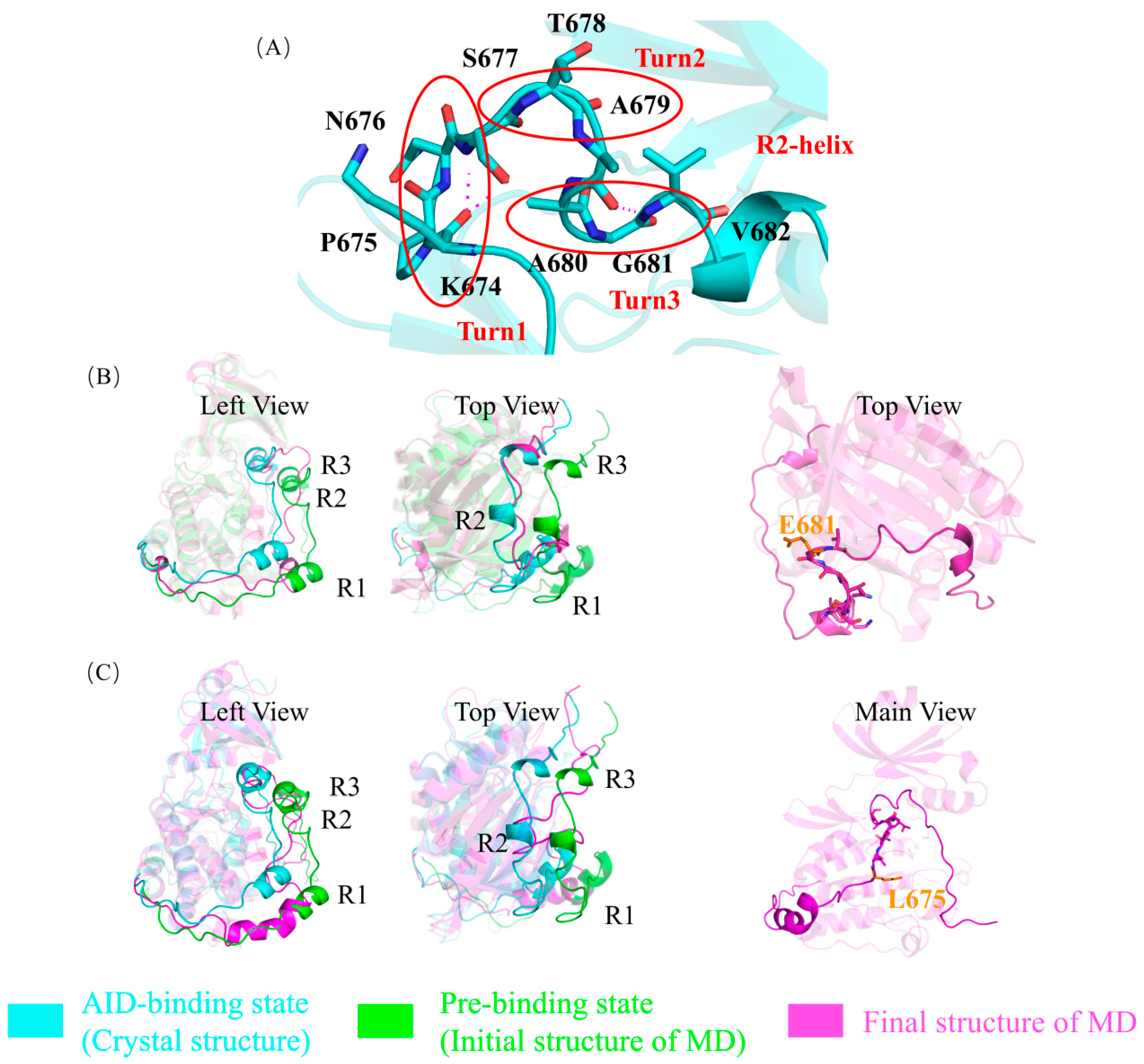
2.1. The Autoinhibition by AID Was Simulated Using Molecular Dynamics Simulation in Wild-Type DCLK1
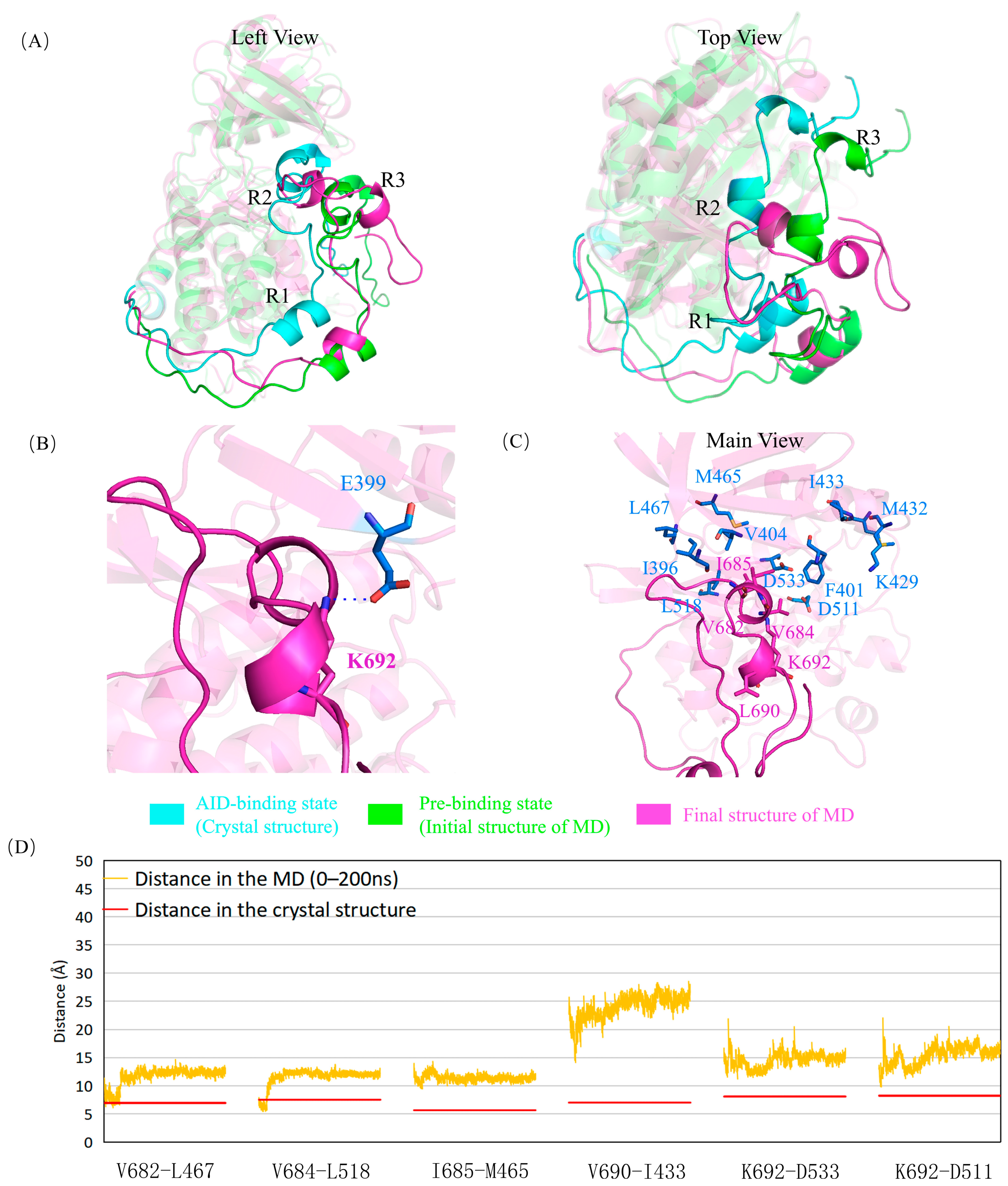
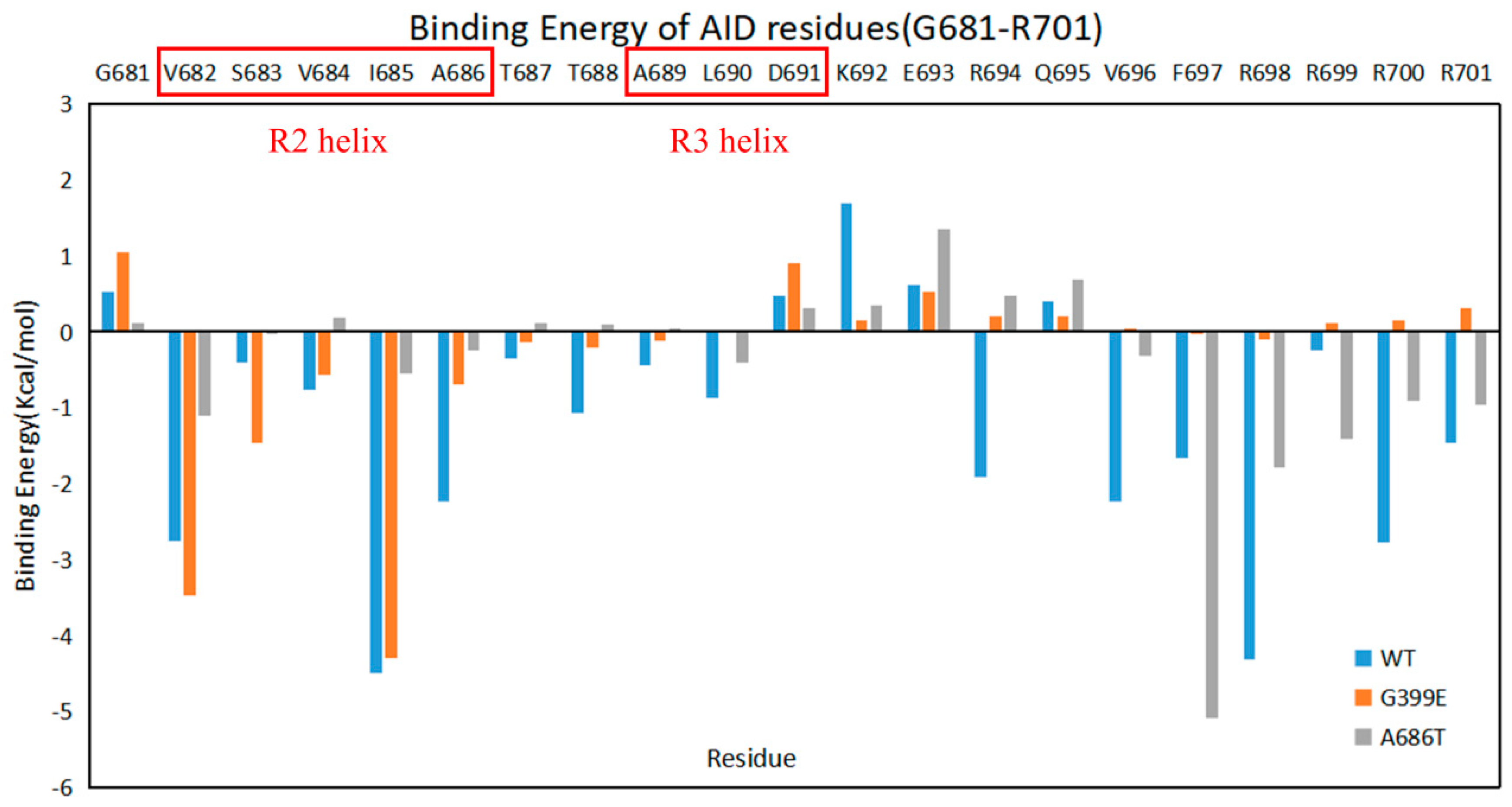
2.2. G399E and A686T Mutants Hinder the Assembly of the AID
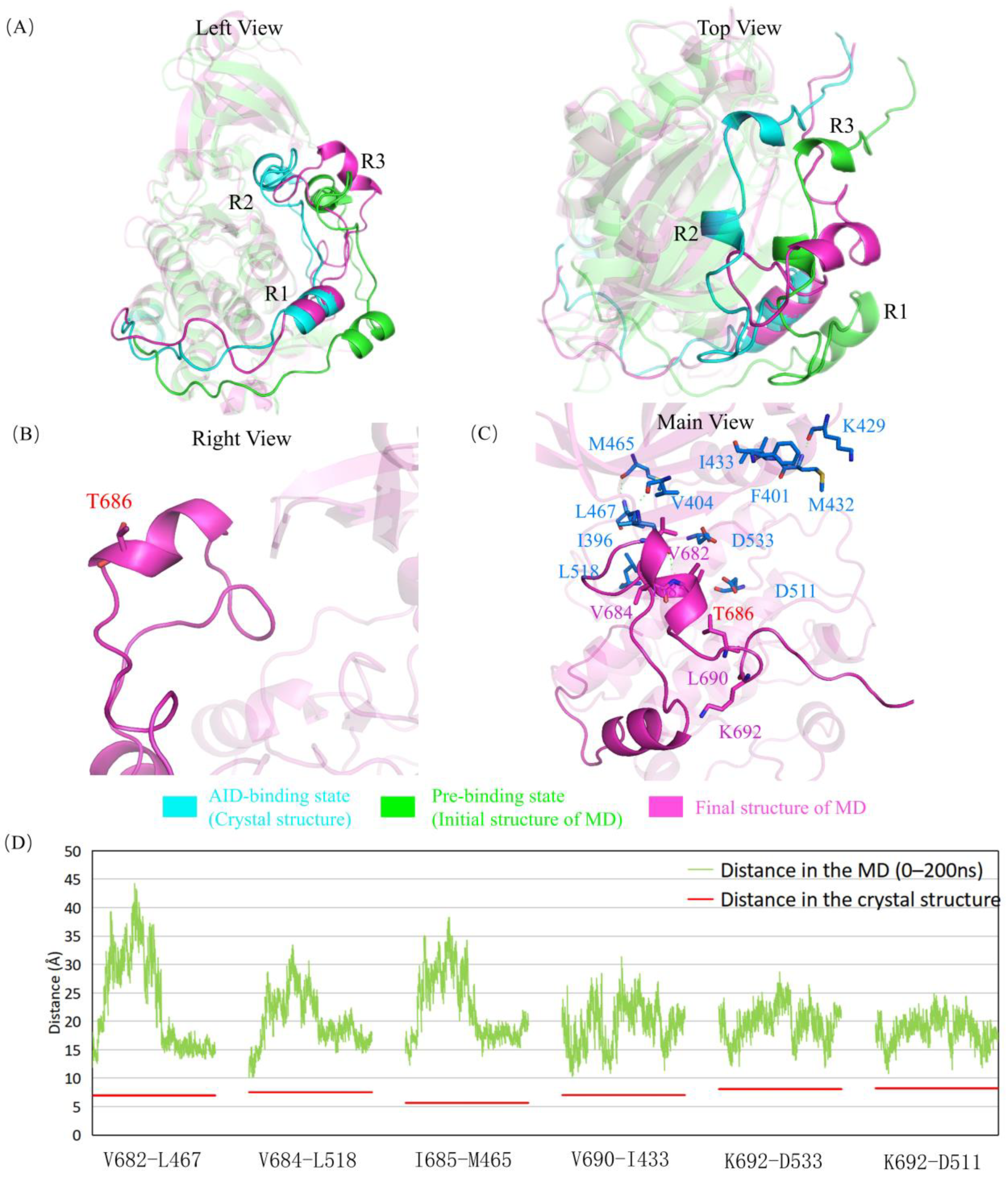
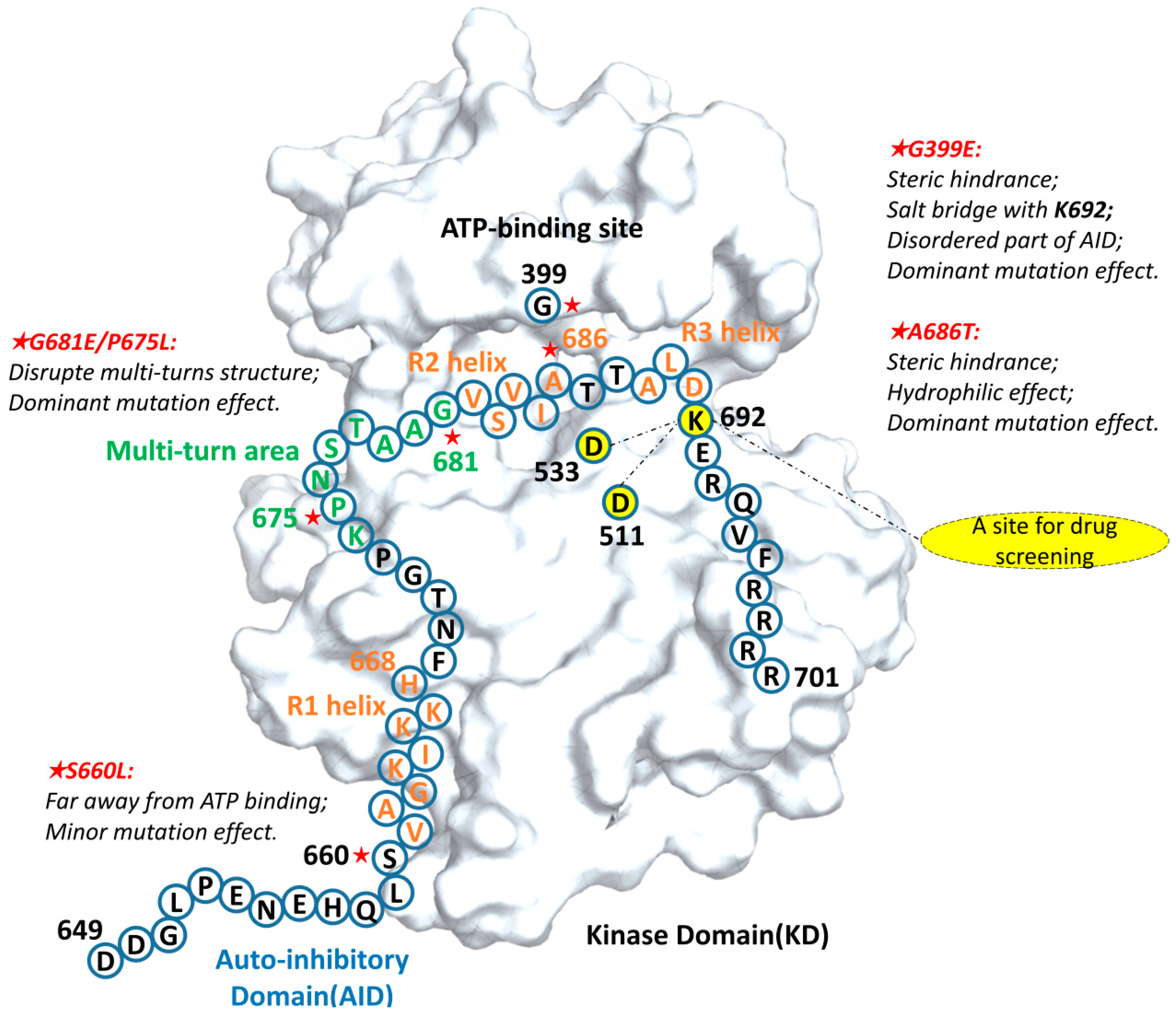
2.3. G681E and P675L Mutants Disrupt the Structural Stability of the AID
2.4. Mutations near the R1 Helix Have Relatively Minor Effects
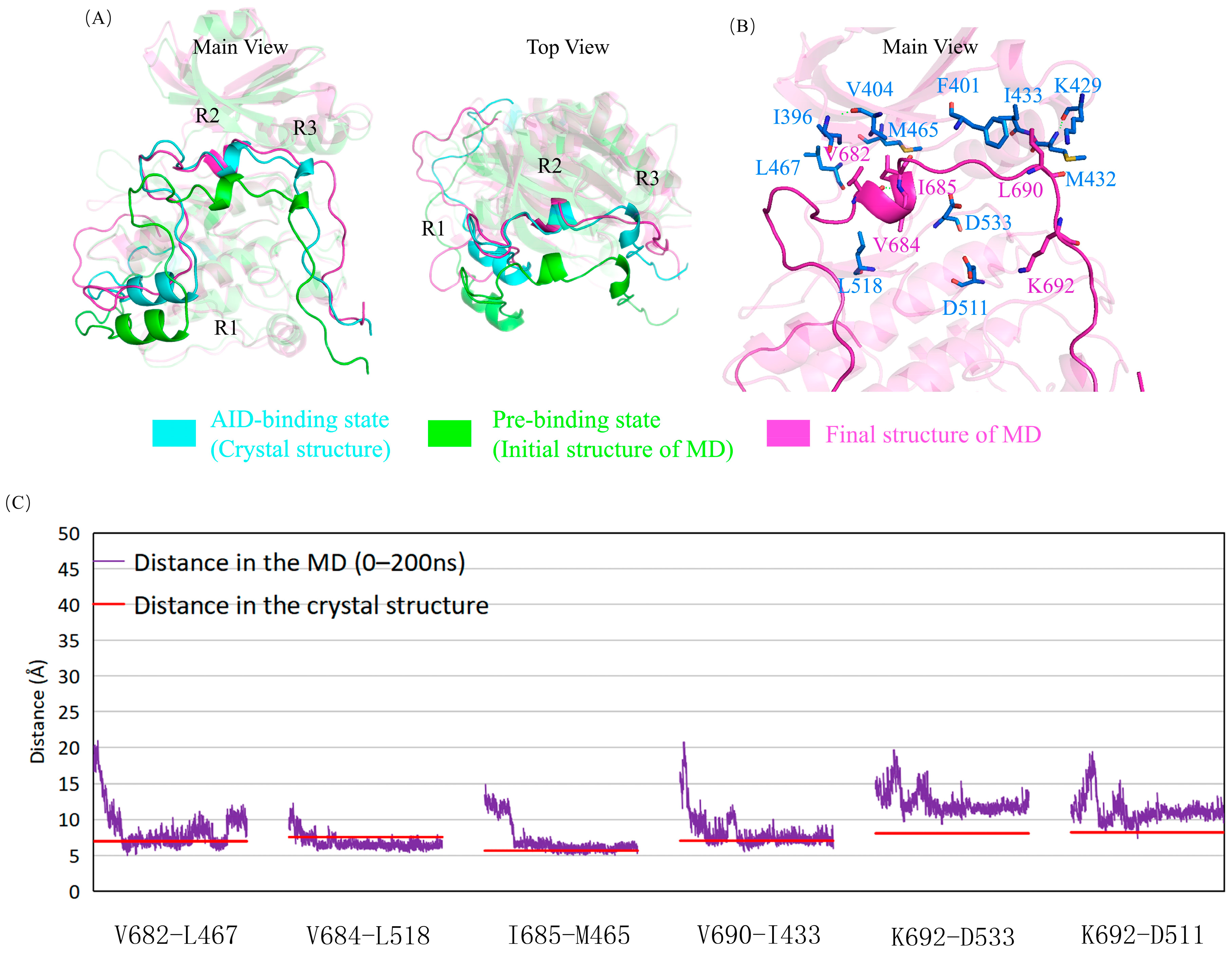
2.5. The Mutations Reduce the Binding between the AID and the KD of DCLK1 through Diverse Molecular Mechanisms
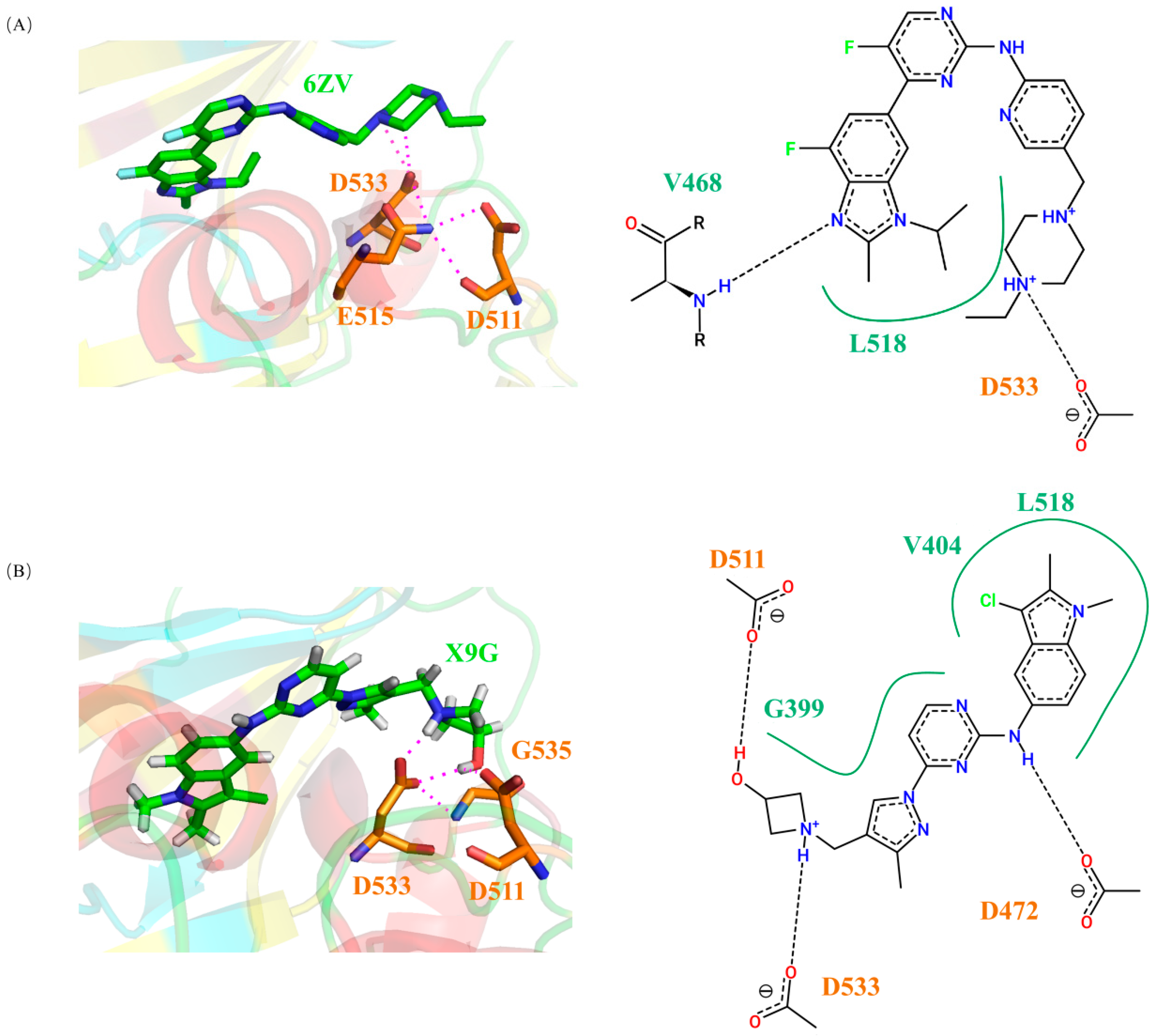
2.6. Screening and Analyzing of Possible DCLK1 Small-Molecule Inhibitors Based on the Binding Molecular Mechanisms Revealed by the Cancer Mutations
3. Materials and Methods
3.1. Construction of the Initial Models
3.2. Molecular Dynamics Simulations and Binding Free Energy Calculations on the Apoenzyme of Full-Length DCLK1
3.3. Screening, Docking, and Molecular Dynamics Simulations on the Inhibitor Complexes of the Kinase Domain of DCLK1
3.4. Surface Plasmon Resonance (SPR) Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zou, W.; Greenblatt, M.B.; Brady, N.; Lotinun, S.; Zhai, B.; de Rivera, H.; Singh, A.; Sun, J.; Gygi, S.P.; Baron, R.; et al. The microtubule-associated protein DCAMKL1 regulates osteoblast function via repression of Runx2. J. Exp. Med. 2013, 210, 1793–1806. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhao, Y.; Luo, W.; Zhu, W.; Jin, L.; Wang, M.; Ye, L.; Wang, Y.; Liang, G. Macrophage DCLK1 promotes obesity-induced cardiomyopathy via activating RIP2/TAK1 signaling pathway. Cell Death Dis. 2023, 14, 419. [Google Scholar] [CrossRef] [PubMed]
- Westphalen, C.B.; Quante, M.; Wang, T.C. Benedikt Functional implication of Dclk1 and Dclk1-expressing cells in cancer. Small GTPases 2017, 8, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.T.; Gleeson, J.G.; Corbo, J.C.; Flanagan, L.; Walsh, C.A. DCAMKL1 encodes a protein kinase with homology to doublecortin that regulates microtubule polymerization. J. Neurosci. 2000, 20, 9152–9161. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Cierpicki, T.; Derewenda, U.; Krowarsch, D.; Feng, Y.; Devedjiev, Y.; Dauter, Z.; Walsh, C.A.; Otlewski, J.; Bushweller, J.H.; et al. The DCX-domain tandems of doublecortin and doublecortin-like kinase. Nat. Struct. Biol. 2003, 10, 324–333. [Google Scholar] [CrossRef]
- Liu, J.S.; Schubert, C.R.; Fu, X.; Fourniol, F.J.; Jaiswal, J.K.; Houdusse, A.; Stultz, C.M.; Moores, C.A.; Walsh, C.A. Molecular basis for specific regulation of neuronal kinesin-3 motors by doublecortin family proteins. Mol. Cell 2012, 47, 707–721. [Google Scholar] [CrossRef]
- Cheng, L.; Yang, Z.; Guo, W.; Wu, C.; Liang, S.; Tong, A.; Cao, Z.; Thorne, R.F.; Yang, S.-Y.; Yu, Y.; et al. DCLK1 autoinhibition and activation in tumorigenesis. Innovation 2022, 3, 100191. [Google Scholar] [CrossRef]
- Silverman, M.A.; Benard, O.; Jaaro, H.; Rattner, A.; Citri, Y.; Seger, R. PG16, a novel protein serine/threonine kinase downstream of cAMP-dependent protein kinase. J. Biol. Chem. 1999, 274, 2631–2636. [Google Scholar] [CrossRef]
- Shang, L.; Kwon, Y.-G.; Nandy, S.; Lawrence, D.S.; Edelman, A.M. Catalytic and regulatory domains of doublecortin kinase-1. Biochemistry 2003, 42, 2185–2194. [Google Scholar] [CrossRef]
- Mirzaei, A.; Tavoosidana, G.; Rad, A.A.; Rezaei, F.; Kadijani, A.A.; Khalili, E.; Madjd, Z. A new insight into cancer stem cell markers: Could local and circulating cancer stem cell markers correlate in colorectal cancer? Tumor Biol. 2016, 37, 2405–2414. [Google Scholar] [CrossRef]
- Ito, H.; Tanaka, S.; Akiyama, Y.; Shimada, S.; Adikrisna, R.; Matsumura, S.; Aihara, A.; Mitsunori, Y.; Ban, D.; Ochiai, T.; et al. Dominant expression of DCLK1 in human pancreatic cancer stem cells accelerates tumor invasion and metastasis. PLoS ONE 2016, 11, e0146564. [Google Scholar] [CrossRef]
- Westphalen, C.B.; Takemoto, Y.; Tanaka, T.; Houchen, C.W.; Olive, K.P.; Wang, T.C. Dclk1 defifines quiescent pancreatic progenitors that promote injury-induced regeneration and tumorigenesis. Cell Stem Cell 2016, 18, 441–455. [Google Scholar] [CrossRef]
- Lu, Q.; Feng, H.; Chen, H.; Weygant, N.; Du, J.; Yan, Z.; Cao, Z. Role of DCLK1 in oncogenic signaling (Review). Int. J. Oncol. 2022, 61, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, E.; Razmi, M.; Tajik, F.; Asadi-Lari, M.; Ghods, R.; Madjd, Z. Oncogenic functions and clinical significances of DCLK1 isoforms in colorectal cancer: A systematic review and meta-analysis. Cancer Cell Int. 2022, 22, 217. [Google Scholar] [CrossRef] [PubMed]
- Qu, D.; Weygant, N.; Yao, J.; Chandrakesan, P.; Berry, W.L.; May, R.; Pitts, K.; Husain, S.; Lightfoot, S.; Li, M.; et al. Overexpression of DCLK1-AL Increases Tumor Cell Invasion, Drug Resistance, and KRAS Activation and Can Be Targeted to Inhibit Tumorigenesis in Pancreatic Cancer. J. Oncol. 2019, 2019, 6402925. [Google Scholar] [CrossRef] [PubMed]
- Chandrakesan, P.; Yao, J.; Qu, D.; May, R.; Weygant, N.; Ge, Y.; Ali, N.; Sureban, S.M.; Gude, M.; Vega, K.; et al. Dclk1, a tumor stem cell marker, regulates pro-survival signaling and self-renewal of intestinal tumor cells. Mol. Cancer 2017, 16, 30. [Google Scholar] [CrossRef]
- Chandrakesan, P.; Weygant, N.; May, R.; Qu, D.; Chinthalapally, H.R.; Sureban, S.M.; Ali, N.; Lightfoot, S.A.; Umar, S.; Houchen, C.W. DCLK1 facilitates intestinal tumor growth via enhancing pluripotency and epithelial mesenchymal transition. Oncotarget 2014, 5, 9269–9280. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hom, M.E.; Bearrood, T.E.; Rosenthal, Z.C.; Fernández, D.; Ondrus, A.E.; Gu, Y.; McCormick, A.K.; Tomaske, M.G.; Marshall, C.R.; et al. Targeting colorectal cancer with small-molecule inhibitors of ALDH1B1. Nat. Chem. Biol. 2022, 18, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, Y.; Seno, H.; Fukuoka, A.; Ueo, T.; Yamaga, Y.; Maruno, T.; Nakanishi, N.; Kanda, K.; Komekado, H.; Kawada, M.; et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat. Genet. 2013, 45, 98–103. [Google Scholar] [CrossRef]
- Chen, Y. Research progress of small molecule inhibitors of doublecortin-like kinase 1. Yaoxue Xuebao 2022, 57, 2914–2920. [Google Scholar]
- Berger, M.F.; Hodis, E.; Heffernan, T.P.; Deribe, Y.L.; Lawrence, M.S.; Protopopov, A.; Ivanova, E.; Watson, I.R.; Nickerson, E.; Ghosh, P.; et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 2012, 485, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.N.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Stawiski, E.W.; Pavía-Jiménez, A.; Modrusan, Z.; Kapur, P.; Jaiswal, B.S.; Zhang, N.; Toffessi-Tcheuyap, V.; Nguyen, T.T.; Pahuja, K.B.; et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat. Genet. 2015, 47, 13–21. [Google Scholar] [CrossRef]
- Giannakis, M.; Hodis, E.; Mu, X.J.; Yamauchi, M.; Rosenbluh, J.; Cibulskis, K.; Saksena, G.; Lawrence, M.S.; Qian, Z.R.; Nishihara, R.; et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat. Genet. 2014, 46, 1264–1266. [Google Scholar] [CrossRef]
- Marios, G.; Mu, X.J.; Shukla, S.A.; Fuchs, C.S.; Ogino, S.; Garraway, L.A. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 17, 1206. [Google Scholar]
- Barbieri, C.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.-P.; White, T.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef]
- Sanborn, J.Z.; Chung, J.; Purdom, E.; Cho, R.J. Zachary Phylogenetic analyses of melanoma reveal complex patterns of metastatic dissemination. Proc. Natl. Acad. Sci. USA 2015, 112, 10995–11000. [Google Scholar] [CrossRef] [PubMed]
- Patel, O.; Dai, W.; Mentzel, M.; Griffin, M.D.; Serindoux, J.; Gay, Y.; Fischer, S.; Sterle, S.; Kropp, A.; Burns, C.J.; et al. Biochemical and Structural Insights into Doublecortin-like Kinase Domain 1. Structure 2016, 24, 1550–1561. [Google Scholar] [CrossRef]
- Jang, D.M.; Lim, H.J.; Hahn, H.; Lee, Y.; Kim, H.K.; Kim, H.S. Man Structural Basis of Inhibition of DCLK1 by Ruxolitinib. Int. J. Mol. Sci. 2021, 22, 8488. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, F.M.; Nabet, B.; Raghavan, S.; Liu, Y.; Leggett, A.L.; Luljanin, M.; Kalekar, R.L.; Yang, A.; He, S.; Wang, J. Discovery of a selective inhibitor of doublecortin like kinase 1. Nat. Chem. Biol. 2020, 16, 635–643. [Google Scholar] [CrossRef]
- Kooistra, A.J.; Kanev, G.K.; van Linden, O.P.J.; Leurs, R.; de Esch, I.J.P.; de Graaf, C. KLIFS: A structural kinase-ligand interaction database. Nucleic Acids Res. 2016, 44, D365–D371. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation Site | Region | COSMIC |
|---|---|---|
| S660L | AID-R1 | COSP28834 [21]/COSP35781 [22] |
| I665M | AID-R1 | COSP37842 [23] |
| P675L | AID | COSP37678 [24]/COSP44021 [25] |
| G681E | AID | COSP28835 [26] |
| A686T | AID-R2 | COSU419 |
| G399E | AID Binding site | COSP40391 [27] |
| Mutant | Average Binding Energy (Kcal/mol) |
|---|---|
| Wild type | −90.8021 |
| G399E | −38.4060 |
| A686T | −60.9727 |
| Small Molecules | Original Kinase Receptor | Main Experimental Methods | IC50 (nM) | Hydrogen Bond with D533 |
|---|---|---|---|---|
| NVP-TAE684 [28] | ALK | TSA | 1084 | Yes |
| DCLK-IN-1 [30] | N/A | chemoproteomic profiling | 2346 | No |
| Ruxolitinib [29] | JAK | DSF | 1645 (1230–2207) | No |
| Ligand HET-Code/Name | IFP Similarity (with AMPPN) | Binding Energy with DCLK1 | Binding Energy with Original Kinase |
|---|---|---|---|
| SGV | 0.76 | −35.58 | −37.72 [GRK5] |
| 6ZV(Abemaciclib) | 0.75 | −50.94 | −55.37 [CDK6] |
| X9G | 0.82 | −51.13 | −48.23 [SYK] |
| IM9 | 0.79 | −42.53 | −43.90 [CDK2] |
| NVP-TAE684 | −54.54 | ||
| DCLK-IN−1 | −49.33 | ||
| Ruxolitinib | −41.31 | ||
| AMPPN | −34.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.; Liu, R.; Yu, Y.; Wei, D.; Chen, Q.; Xu, Q. Molecular Mechanism of Mutational Disruption of DCLK1 Autoinhibition Provides a Rationale for Inhibitor Screening. Int. J. Mol. Sci. 2023, 24, 14020. https://doi.org/10.3390/ijms241814020
Chen W, Liu R, Yu Y, Wei D, Chen Q, Xu Q. Molecular Mechanism of Mutational Disruption of DCLK1 Autoinhibition Provides a Rationale for Inhibitor Screening. International Journal of Molecular Sciences. 2023; 24(18):14020. https://doi.org/10.3390/ijms241814020
Chicago/Turabian StyleChen, Weizhi, Rui Liu, Yamei Yu, Dongqing Wei, Qiang Chen, and Qin Xu. 2023. "Molecular Mechanism of Mutational Disruption of DCLK1 Autoinhibition Provides a Rationale for Inhibitor Screening" International Journal of Molecular Sciences 24, no. 18: 14020. https://doi.org/10.3390/ijms241814020