
Targeting Oncogenic Mutant p53 and BCL-2 for Small Cell Lung Cancer Treatment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
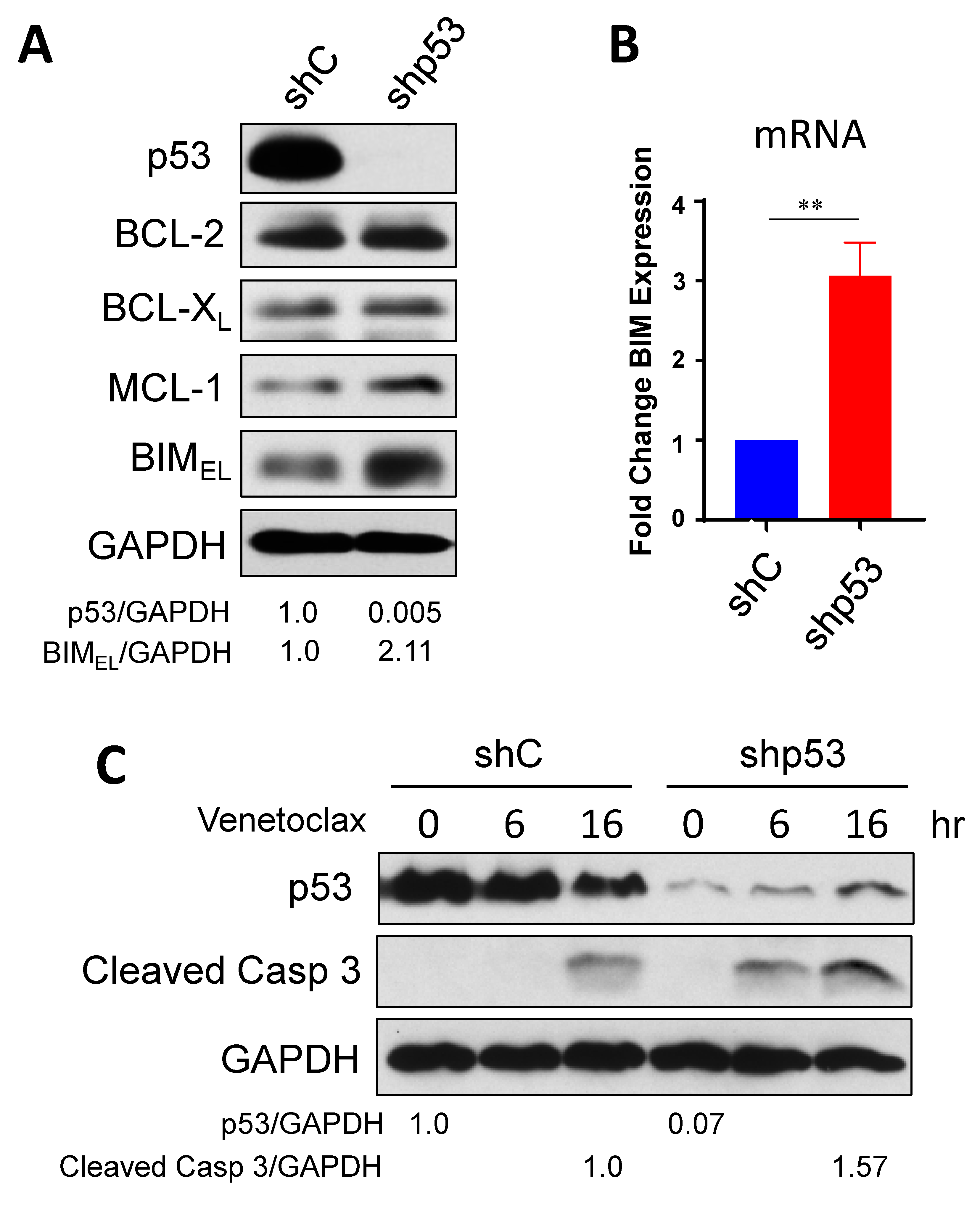
2.1. Down-Regulation of Onc-p53 Increases BIM Expression and Sensitizes to Venetoclax in SCLC-P Cells
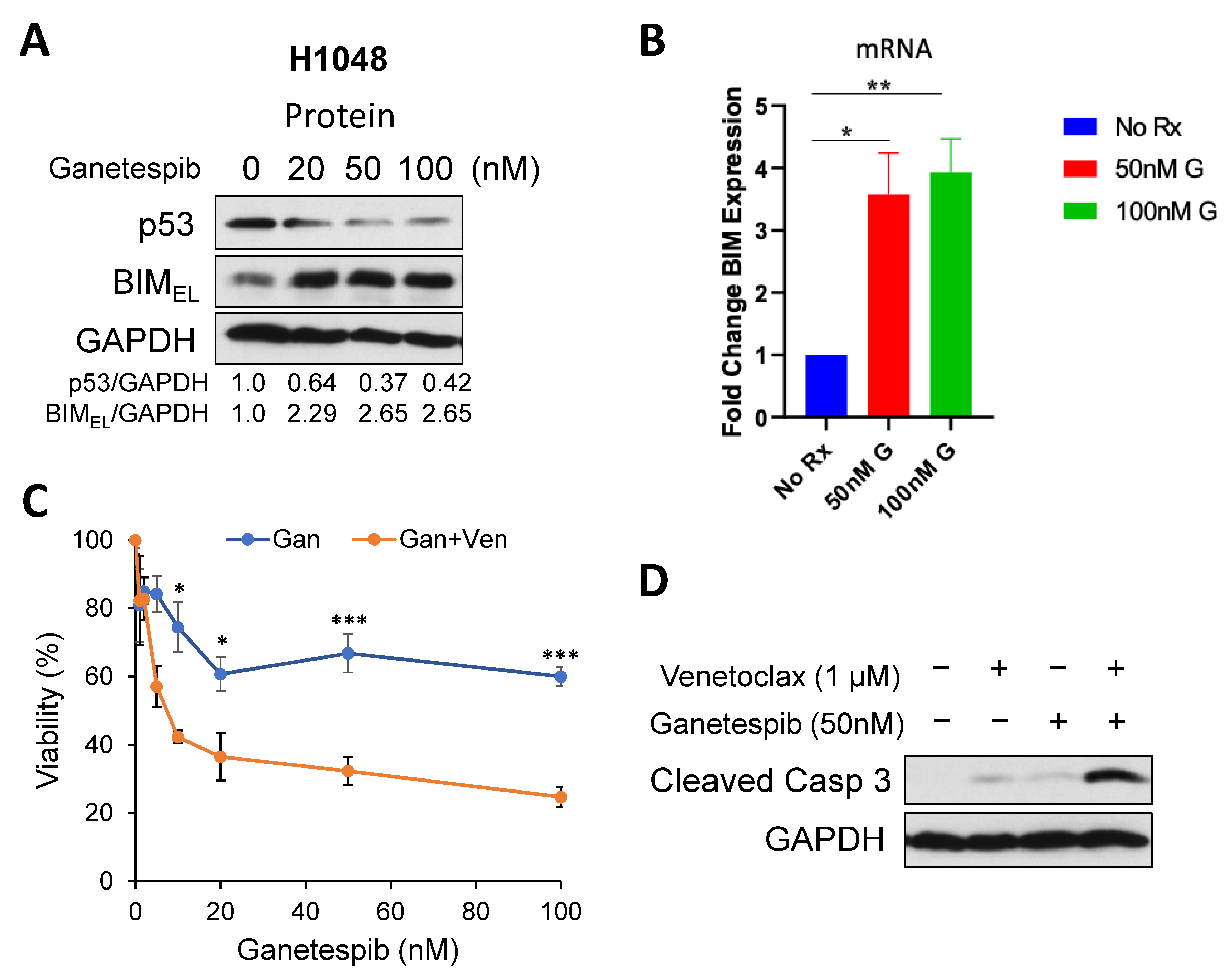
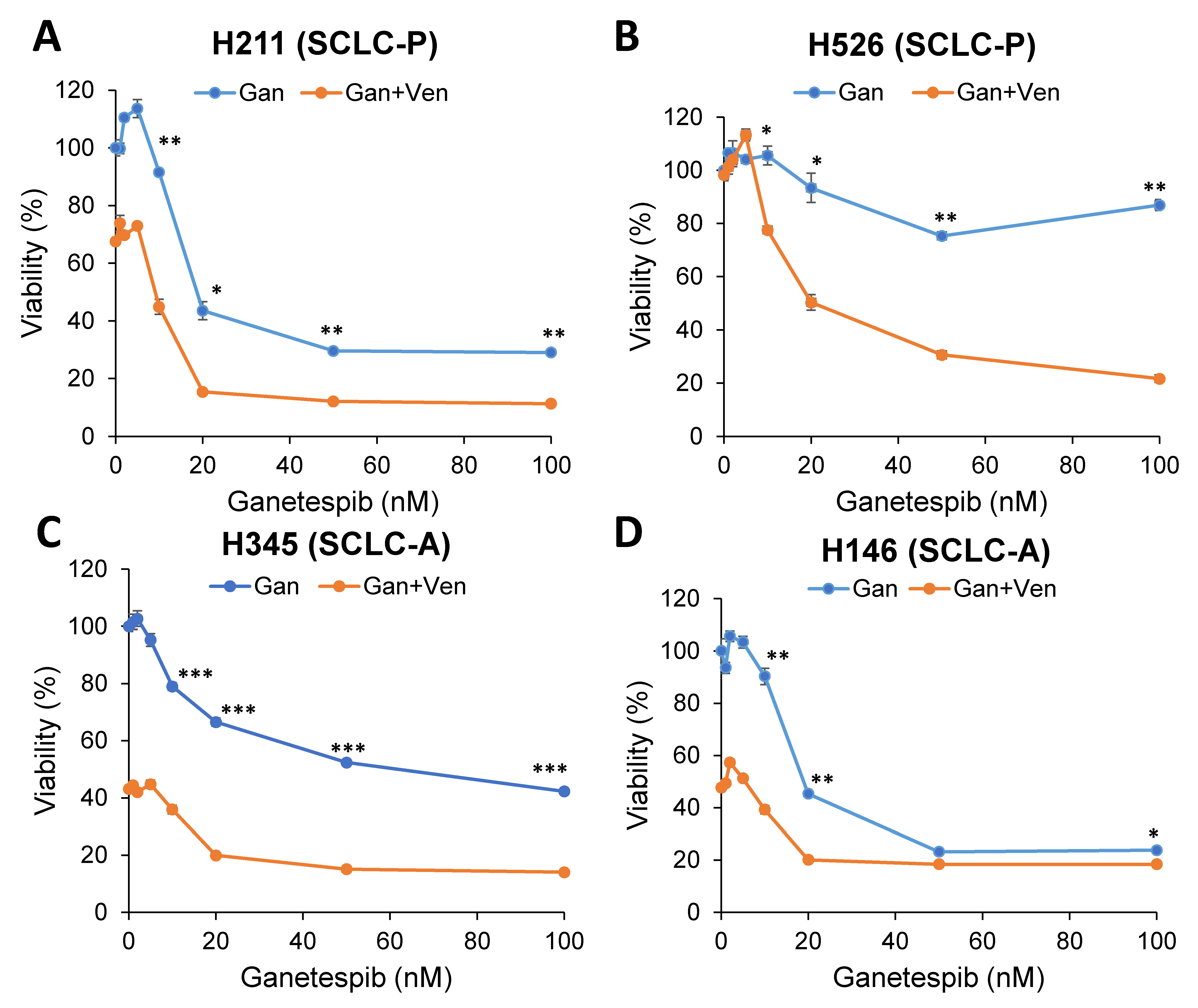
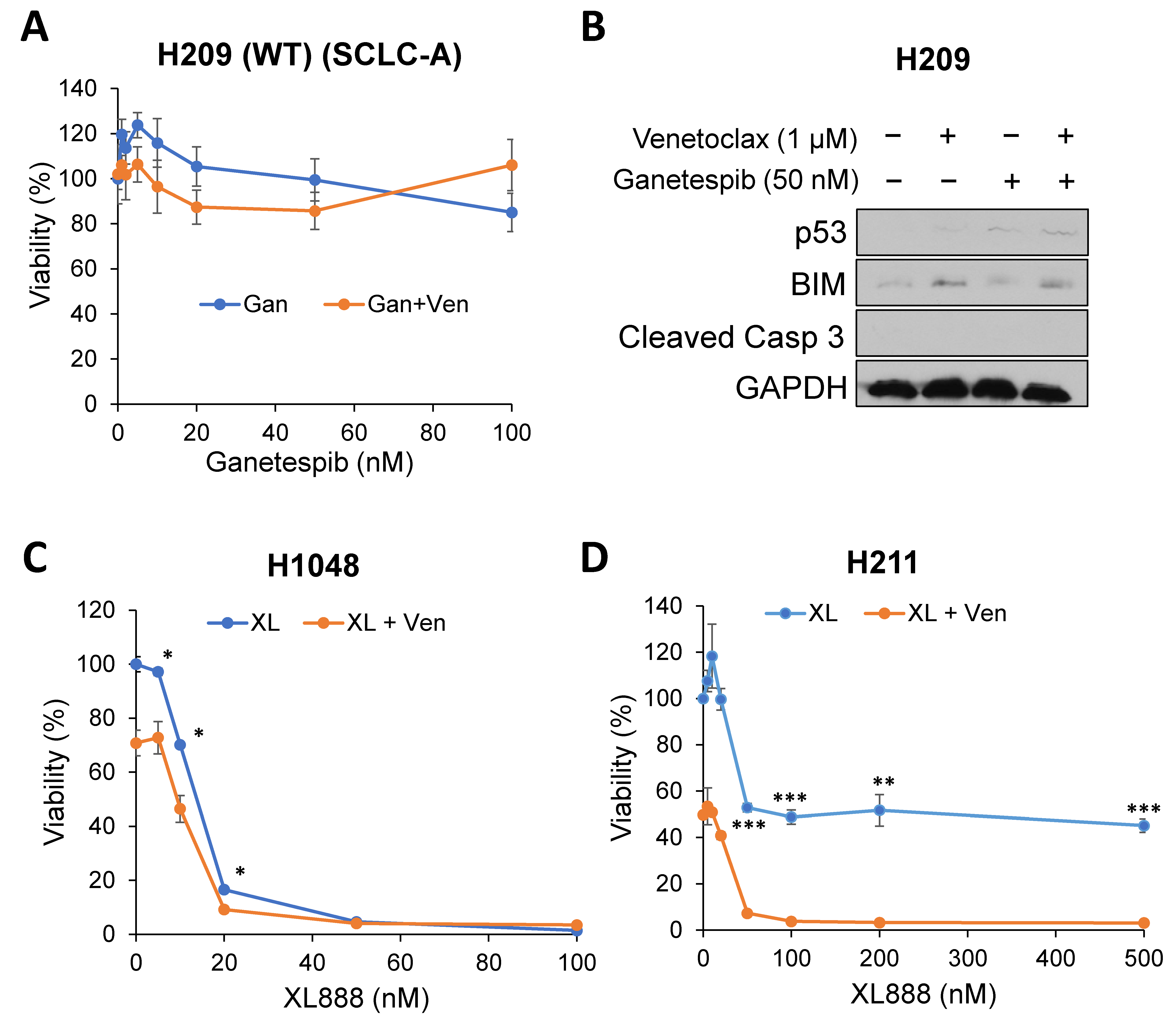
2.2. Targeting Onc-p53 by the HSP90 Inhibitor, Ganetespib, Increases BIM Expression and Sensitizes to Venetoclax in SCLC Cell Lines
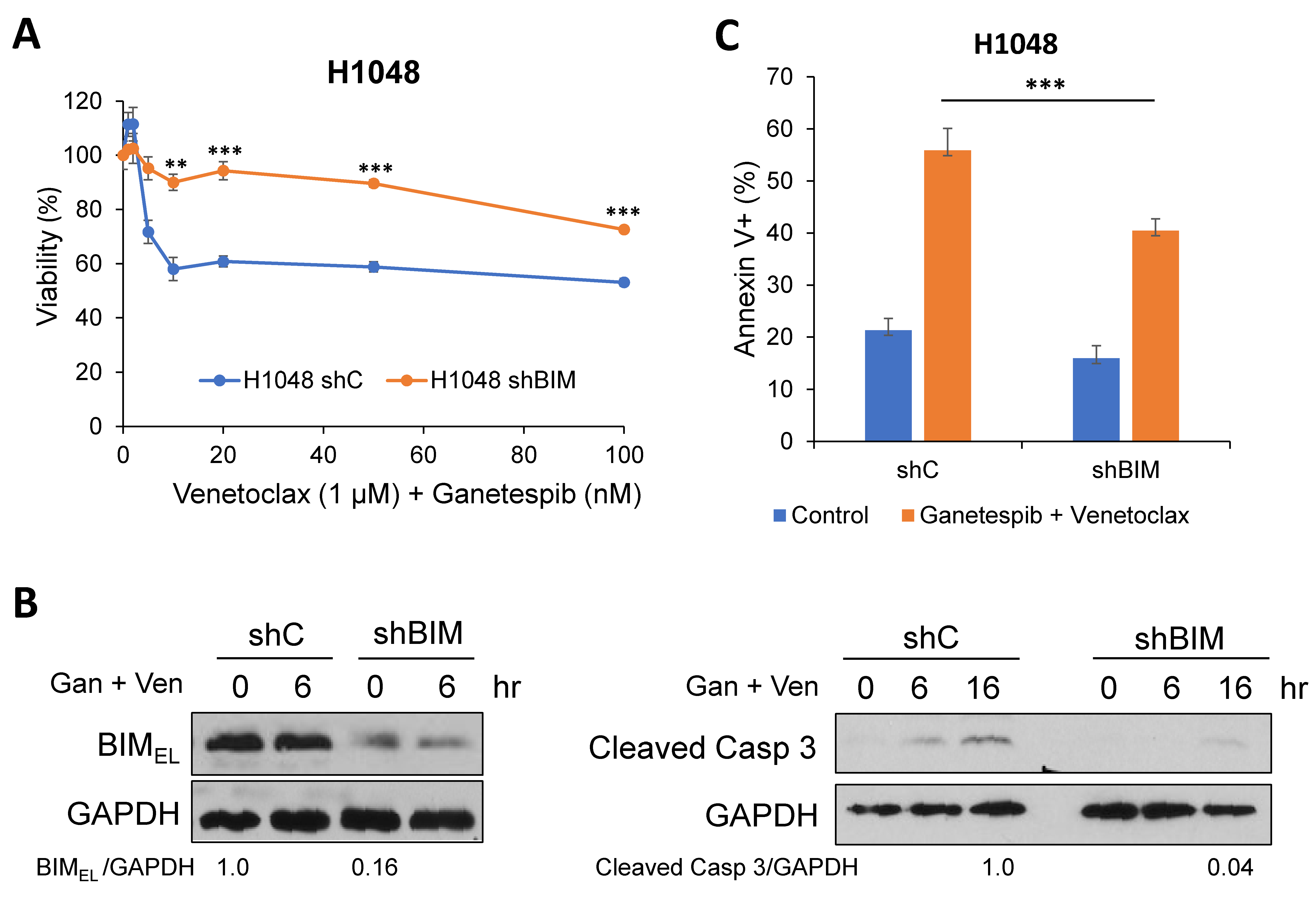
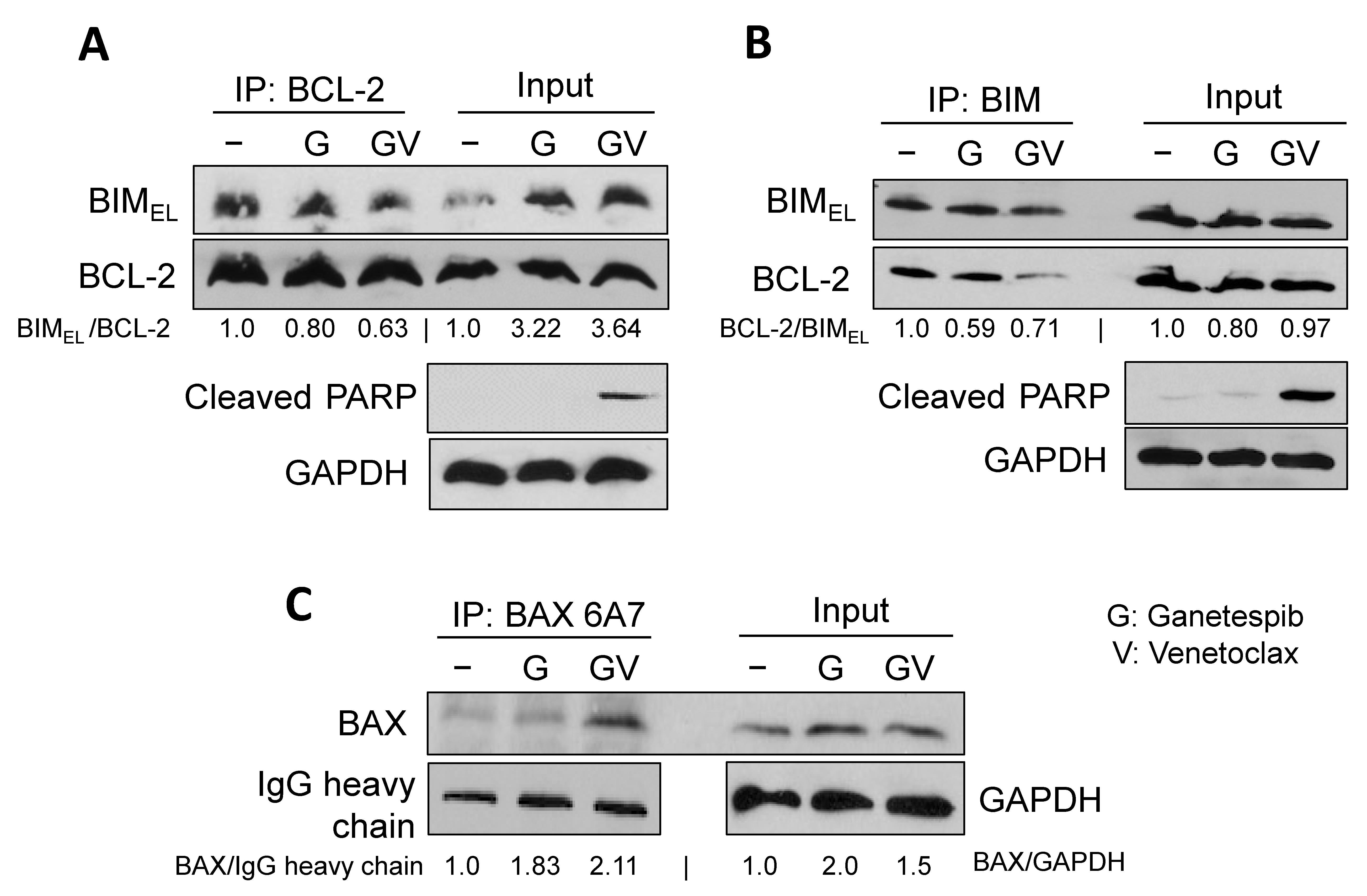
2.3. A Pro-Apoptotic BH3-Only Protein, BIM, Is Required for Cell Death in Combination of Venetoclax and Ganetespib Treatment in SCLC Cells
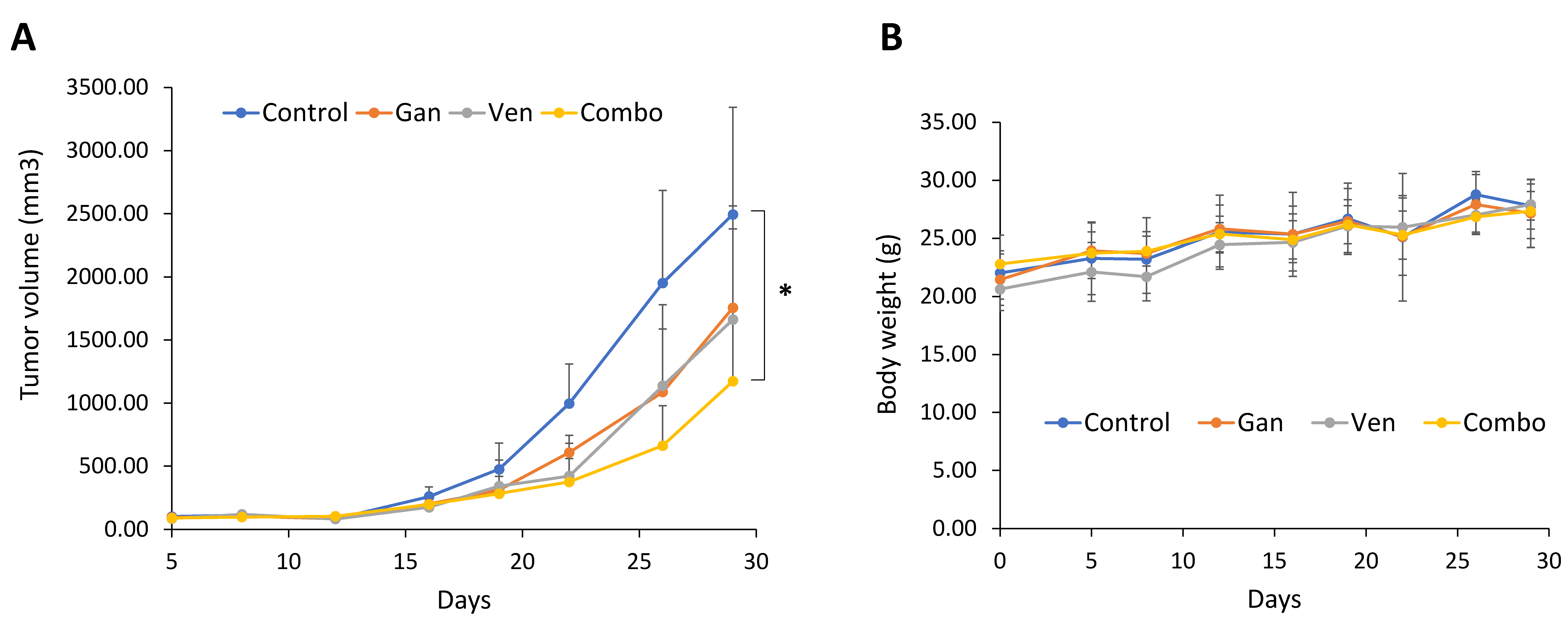
2.4. The Combination of Venetoclax and Ganetespib Treatment Delays Tumor Growth in a SCLC Xenograft Mouse Model
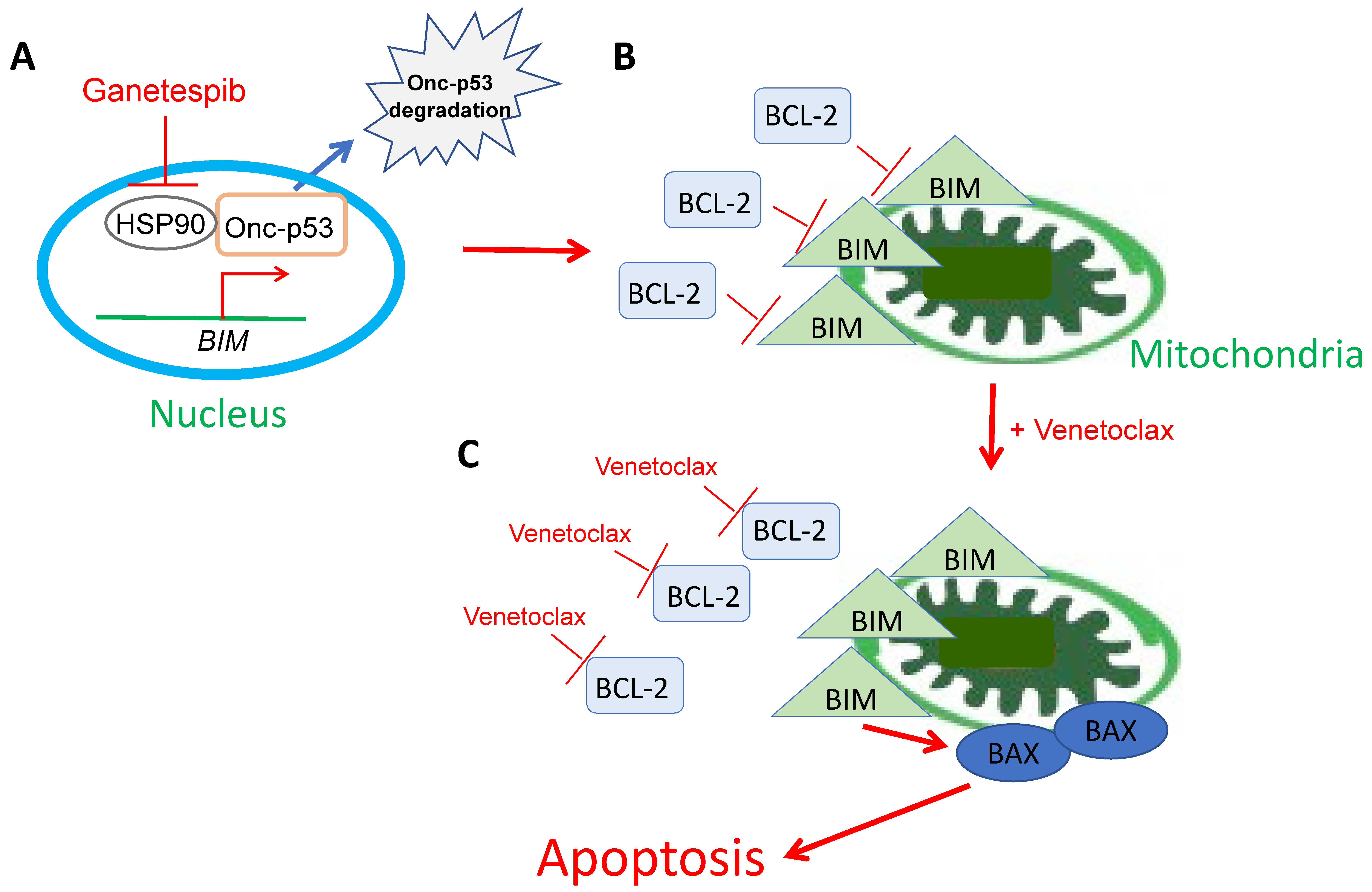
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Drugs
4.2. Lentivirus Production
4.3. Cell Viability Assays
4.4. Western Blot Analyses
4.5. Immunoprecipitation
4.6. Fluorescence-Activated Cell Sorting (FACS) Analysis
4.7. Quantitative Reverse Transcriptase Polymerase Chain Reaction (qRT-PCR)
4.8. In Vivo Studies
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rodriguez, E.; Lilenbaum, R.C. Small cell lung cancer: Past, present, and future. Curr. Oncol. Rep. 2010, 12, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Bunn, P.A., Jr.; Minna, J.D.; Augustyn, A.; Gazdar, A.F.; Ouadah, Y.; Krasnow, M.A.; Berns, A.; Brambilla, E.; Rekhtman, N.; Massion, P.P.; et al. Small Cell Lung Cancer: Can Recent Advances in Biology and Molecular Biology Be Translated into Improved Outcomes? J. Thorac. Oncol. 2016, 11, 453–474. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Fernandez-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- McFadden, D.G.; Papagiannakopoulos, T.; Taylor-Weiner, A.; Stewart, C.; Carter, S.L.; Cibulskis, K.; Bhutkar, A.; McKenna, A.; Dooley, A.; Vernon, A.; et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell 2014, 156, 1298–1311. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretic, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Ben-Ezra, J.M.; Kornstein, M.J.; Grimes, M.M.; Krystal, G. Small cell carcinomas of the lung express the Bcl-2 protein. Am. J. Pathol. 1994, 145, 1036–1040. [Google Scholar]
- Shoemaker, A.R.; Mitten, M.J.; Adickes, J.; Ackler, S.; Refici, M.; Ferguson, D.; Oleksijew, A.; O’Connor, J.M.; Wang, B.; Frost, D.J.; et al. Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small cell lung cancer xenograft models. Clin. Cancer Res. 2008, 14, 3268–3277. [Google Scholar] [CrossRef]
- Gandhi, L.; Camidge, D.R.; Ribeiro de Oliveira, M.; Bonomi, P.; Gandara, D.; Khaira, D.; Hann, C.L.; McKeegan, E.M.; Litvinovich, E.; Hemken, P.M.; et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J. Clin. Oncol. 2011, 29, 909–916. [Google Scholar] [CrossRef]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed anuclear cell death delimits platelet life span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef]
- Rudin, C.M.; Hann, C.L.; Garon, E.B.; Ribeiro de Oliveira, M.; Bonomi, P.D.; Camidge, D.R.; Chu, Q.; Giaccone, G.; Khaira, D.; Ramalingam, S.S.; et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin. Cancer Res. 2012, 18, 3163–3169. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Lochmann, T.L.; Floros, K.V.; Naseri, M.; Powell, K.M.; Cook, W.; March, R.J.; Stein, G.T.; Greninger, P.; Maves, Y.K.; Saunders, L.R.; et al. Venetoclax Is Effective in Small-Cell Lung Cancers with High BCL-2 Expression. Clin. Cancer Res. 2018, 24, 360–369. [Google Scholar] [CrossRef]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular subtypes of small cell lung cancer: A synthesis of human and mouse model data. Nat. Rev. Cancer 2019, 19, 289–297. [Google Scholar] [CrossRef]
- Vaughan, C.; Pearsall, I.; Yeudall, A.; Deb, S.P.; Deb, S. p53: Its mutations and their impact on transcription. Subcell. Biochem. 2014, 85, 71–90. [Google Scholar] [PubMed]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-Function (GOF) Mutant p53 as Actionable Therapeutic Target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Iwakuma, T. Regulators of Oncogenic Mutant TP53 Gain of Function. Cancers 2018, 11, 4. [Google Scholar] [CrossRef]
- Blandino, G.; Levine, A.J.; Oren, M. Mutant p53 gain of function: Differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 1999, 18, 477–485. [Google Scholar] [CrossRef]
- Cai, Y.; Yan, X.; Zhang, G.; Zhao, W.; Jiao, S. The predictive value of ERCC1 and p53 for the effect of panobinostat and cisplatin combination treatment in NSCLC. Oncotarget 2015, 6, 18997–19005. [Google Scholar] [CrossRef]
- Fiorini, C.; Cordani, M.; Padroni, C.; Blandino, G.; Di Agostino, S.; Donadelli, M. Mutant p53 stimulates chemoresistance of pancreatic adenocarcinoma cells to gemcitabine. Biochim. Biophys. Acta 2015, 1853, 89–100. [Google Scholar] [CrossRef]
- Akeno, N.; Reece, A.L.; Callahan, M.; Miller, A.L.; Kim, R.G.; He, D.; Lane, A.; Moulton, J.S.; Wikenheiser-Brokamp, K.A. TRP53 Mutants Drive Neuroendocrine Lung Cancer Through Loss-of-Function Mechanisms with Gain-of-Function Effects on Chemotherapy Response. Mol. Cancer Ther. 2017, 16, 2913–2926. [Google Scholar] [CrossRef]
- Subramaniam, D.S.; Liu, S.V.; Crawford, J.; Kramer, J.; Thompson, J.; Wang, H.; Giaccone, G. A Phase Ib/II Study of Ganetespib With Doxorubicin in Advanced Solid Tumors Including Relapsed-Refractory Small Cell Lung Cancer. Front. Oncol. 2018, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.N.; Engelman, J.A.; Faber, A.C. The BCL2 Family: Key Mediators of the Apoptotic Response to Targeted Anticancer Therapeutics. Cancer Discov. 2015, 5, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Salem, A.H.; Agarwal, S.K.; Dunbar, M.; Enschede, S.L.; Humerickhouse, R.A.; Wong, S.L. Pharmacokinetics of Venetoclax, a Novel BCL-2 Inhibitor, in Patients With Relapsed or Refractory Chronic Lymphocytic Leukemia or Non-Hodgkin Lymphoma. J. Clin. Pharmacol. 2017, 57, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Stilgenbauer, S.; Seymour, J.F.; Huang, D.C.S. Venetoclax in Patients with Previously Treated Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2017, 23, 4527–4533. [Google Scholar] [CrossRef]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef]
- Eroglu, Z.; Chen, Y.A.; Gibney, G.T.; Weber, J.S.; Kudchadkar, R.R.; Khushalani, N.I.; Markowitz, J.; Brohl, A.S.; Tetteh, L.F.; Ramadan, H.; et al. Combined BRAF and HSP90 Inhibition in Patients with Unresectable BRAF (V600E)-Mutant Melanoma. Clin. Cancer Res. 2018, 24, 5516–5524. [Google Scholar] [CrossRef]
- Nakajima, W.; Hicks, M.A.; Tanaka, N.; Krystal, G.W.; Harada, H. Noxa determines localization and stability of MCL-1 and consequently ABT-737 sensitivity in small cell lung cancer. Cell Death Dis. 2014, 5, e1052. [Google Scholar] [CrossRef]
- Augustyn, A.; Borromeo, M.; Wang, T.; Fujimoto, J.; Shao, C.; Dospoy, P.D.; Lee, V.; Tan, C.; Sullivan, J.P.; Larsen, J.E.; et al. ASCL1 is a lineage oncogene providing therapeutic targets for high-grade neuroendocrine lung cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 14788–14793. [Google Scholar] [CrossRef]
- Seymour, J.F.; Ma, S.; Brander, D.M.; Choi, M.Y.; Barrientos, J.; Davids, M.S.; Anderson, M.A.; Beaven, A.W.; Rosen, S.T.; Tam, C.S.; et al. Venetoclax plus rituximab in relapsed or refractory chronic lymphocytic leukaemia: A phase 1b study. Lancet Oncol. 2017, 18, 230–240. [Google Scholar] [CrossRef]
- Luo, Q.; Pan, W.; Zhou, S.; Wang, G.; Yi, H.; Zhang, L.; Yan, X.; Yuan, L.; Liu, Z.; Wang, J.; et al. A Novel BCL-2 Inhibitor APG-2575 Exerts Synthetic Lethality with BTK or MDM2-p53 Inhibitor in Diffuse Large B-Cell Lymphoma. Oncol. Res. 2020, 28, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.D.; Zhu, H.; Tang, Q.; Wang, G.; Min, P.; Wang, Q.; Li, N.; Yang, D.; Zhai, Y. FLT3 inhibition by olverembatinib (HQP1351) downregulates MCL-1 and synergizes with BCL-2 inhibitor lisaftoclax (APG-2575) in preclinical models of FLT3-ITD mutant acute myeloid leukemia. Transl. Oncol. 2022, 15, 101244. [Google Scholar] [CrossRef]
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.; Xiong, H.; et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012, 30, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Huemer, F.; Melchardt, T.; Jansko, B.; Wahida, A.; Jilg, S.; Jost, P.J.; Klieser, E.; Steiger, K.; Magnes, T.; Pleyer, L.; et al. Durable remissions with venetoclax monotherapy in secondary AML refractory to hypomethylating agents and high expression of BCL-2 and/or BIM. Eur. J. Haematol. 2019, 102, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Ham, J.; Costa, C.; Sano, R.; Lochmann, T.L.; Sennott, E.M.; Patel, N.U.; Dastur, A.; Gomez-Caraballo, M.; Krytska, K.; Hata, A.N.; et al. Exploitation of the Apoptosis-Primed State of MYCN-Amplified Neuroblastoma to Develop a Potent and Specific Targeted Therapy Combination. Cancer Cell 2016, 29, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Bate-Eya, L.T.; den Hartog, I.J.; van der Ploeg, I.; Schild, L.; Koster, J.; Santo, E.E.; Westerhout, E.M.; Versteeg, R.; Caron, H.N.; Molenaar, J.J.; et al. High efficacy of the BCL-2 inhibitor ABT199 (venetoclax) in BCL-2 high-expressing neuroblastoma cell lines and xenografts and rational for combination with MCL-1 inhibition. Oncotarget 2016, 7, 27946–27958. [Google Scholar] [CrossRef]
- Workman, P. Combinatorial attack on multistep oncogenesis by inhibiting the Hsp90 molecular chaperone. Cancer Lett. 2004, 206, 149–157. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Sanchez, J.; Carter, T.R.; Cohen, M.S.; Blagg, B.S.J. Old and New Approaches to Target the Hsp90 Chaperone. Curr. Cancer Drug Targets 2020, 20, 253–270. [Google Scholar] [CrossRef]
- Li, Z.N.; Luo, Y. HSP90 inhibitors and cancer: Prospects for use in targeted therapies (Review). Oncol. Rep. 2023, 49, 6. [Google Scholar] [CrossRef]
- Jhaveri, K.; Taldone, T.; Modi, S.; Chiosis, G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim. Biophys. Acta 2012, 1823, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Ferraldeschi, R.; Armstrong, H.K.; Centenera, M.M.; Workman, P. Maximizing the Therapeutic Potential of HSP90 Inhibitors. Mol. Cancer Res. 2015, 13, 1445–1451. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Ingallina, E.; Sorrentino, G.; Bertolio, R.; Lisek, K.; Zannini, A.; Azzolin, L.; Severino, L.U.; Scaini, D.; Mano, M.; Mantovani, F.; et al. Mechanical cues control mutant p53 stability through a mevalonate-RhoA axis. Nat. Cell Biol. 2018, 20, 28–35. [Google Scholar] [CrossRef]
- Perdrix, A.; Najem, A.; Saussez, S.; Awada, A.; Journe, F.; Ghanem, G.; Krayem, M. PRIMA-1 and PRIMA-1(Met) (APR-246): From Mutant/Wild Type p53 Reactivation to Unexpected Mechanisms Underlying Their Potent Anti-Tumor Effect in Combinatorial Therapies. Cancers 2017, 9, 172. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neely, V.; Manchikalapudi, A.; Nguyen, K.; Dalton, K.; Hu, B.; Koblinski, J.E.; Faber, A.C.; Deb, S.; Harada, H. Targeting Oncogenic Mutant p53 and BCL-2 for Small Cell Lung Cancer Treatment. Int. J. Mol. Sci. 2023, 24, 13082. https://doi.org/10.3390/ijms241713082
Neely V, Manchikalapudi A, Nguyen K, Dalton K, Hu B, Koblinski JE, Faber AC, Deb S, Harada H. Targeting Oncogenic Mutant p53 and BCL-2 for Small Cell Lung Cancer Treatment. International Journal of Molecular Sciences. 2023; 24(17):13082. https://doi.org/10.3390/ijms241713082
Chicago/Turabian StyleNeely, Victoria, Alekhya Manchikalapudi, Khanh Nguyen, Krista Dalton, Bin Hu, Jennifer E. Koblinski, Anthony C. Faber, Sumitra Deb, and Hisashi Harada. 2023. "Targeting Oncogenic Mutant p53 and BCL-2 for Small Cell Lung Cancer Treatment" International Journal of Molecular Sciences 24, no. 17: 13082. https://doi.org/10.3390/ijms241713082