
Diversity Analysis and Function Prediction of Bacterial Communities in the Different Colored Pericarp of Citrus reticulata cv. ‘Shatangju’ Due to ‘Candidatus Liberibacter asiaticus’ Infection

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
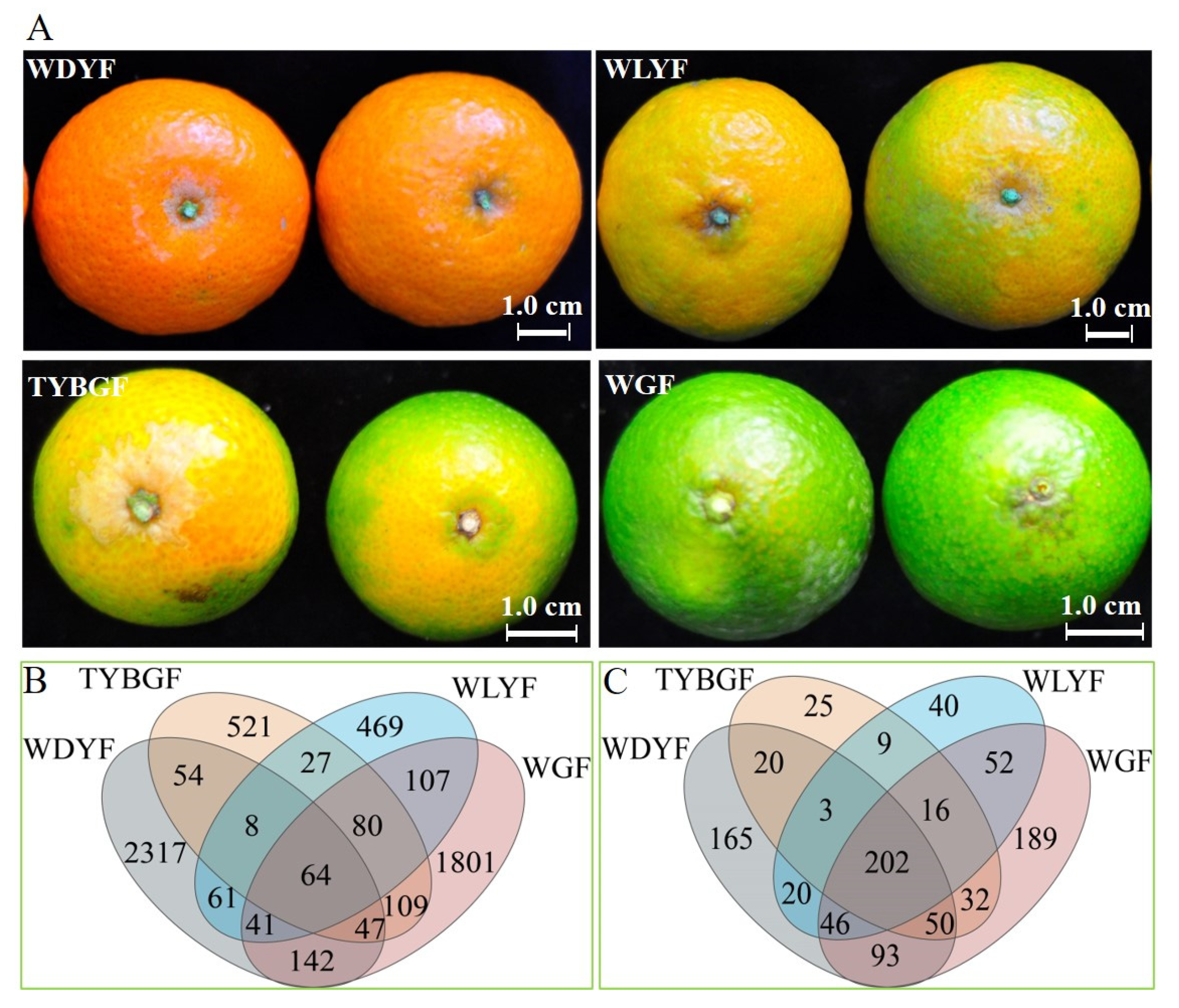
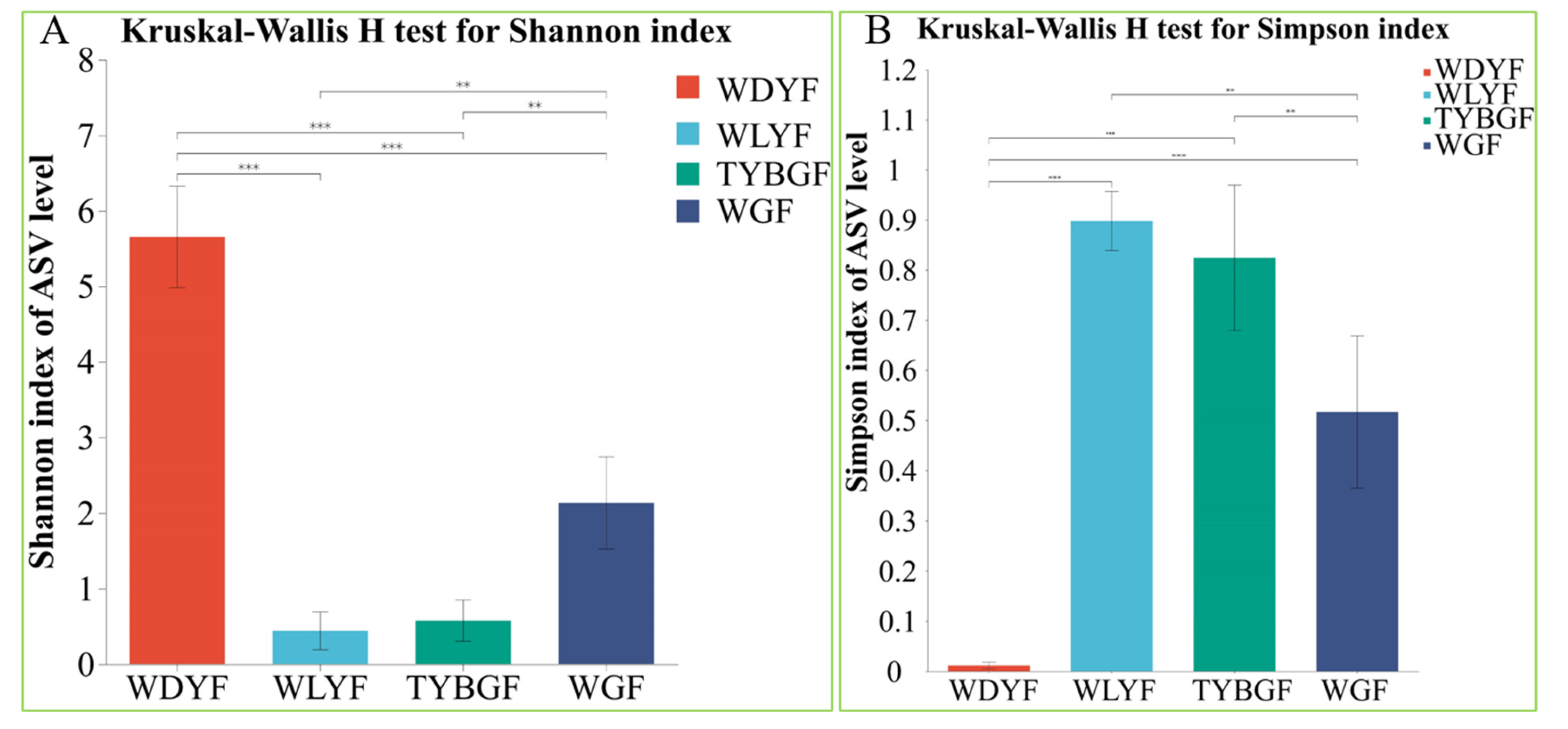
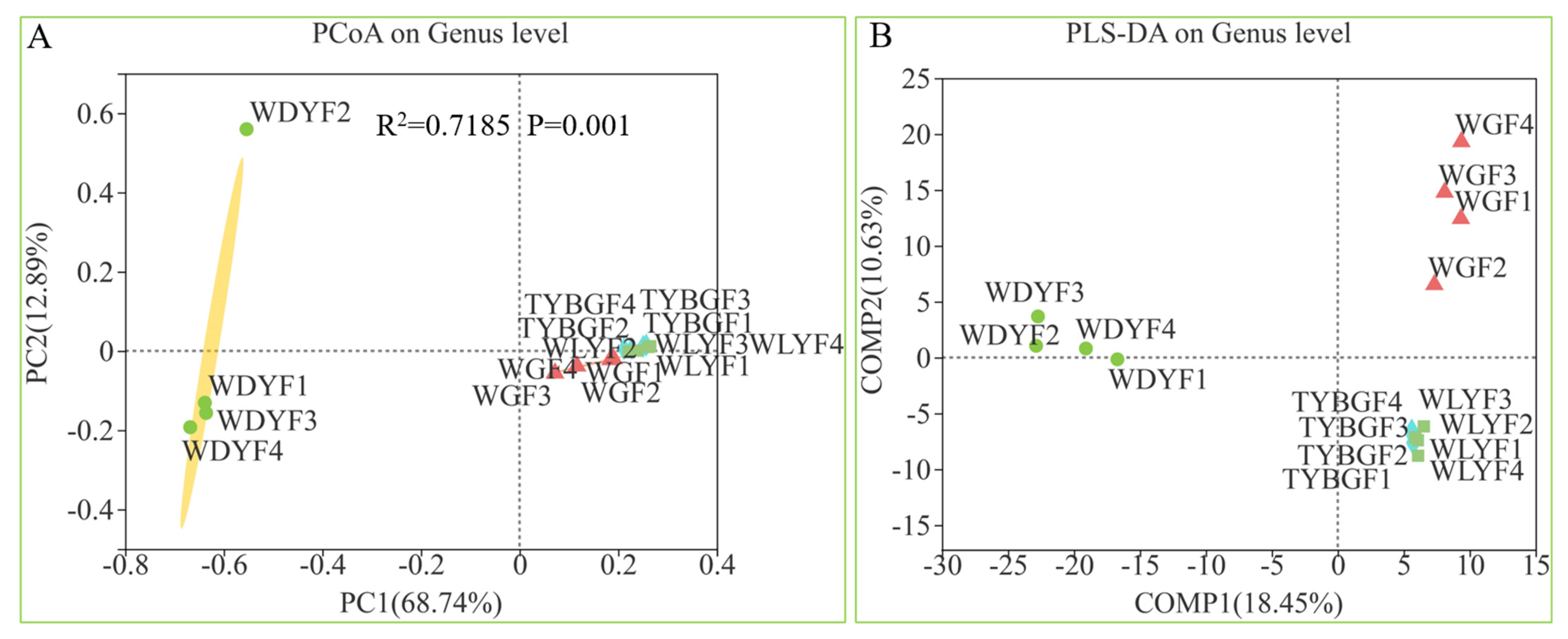
2.1. Difference in the Microbial Diversity of the Citrus Pericarp with Different Pigment
2.2. Differences in the Relative Abundance of Taxa in the Microbiome of Different Colored Pericarps
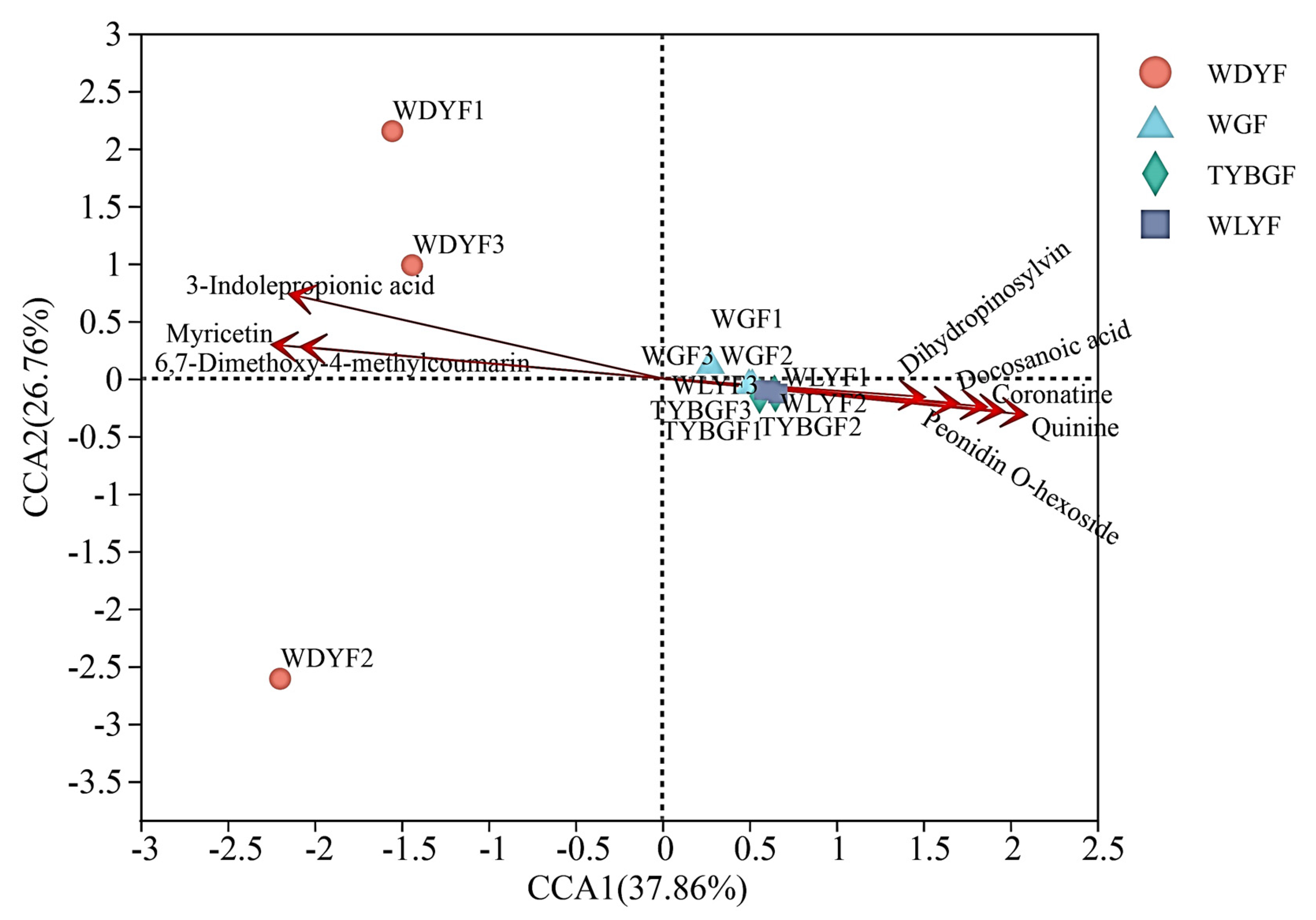
2.3. Correlation Analysis between Microbes and Metabolites of Citrus Pericarps
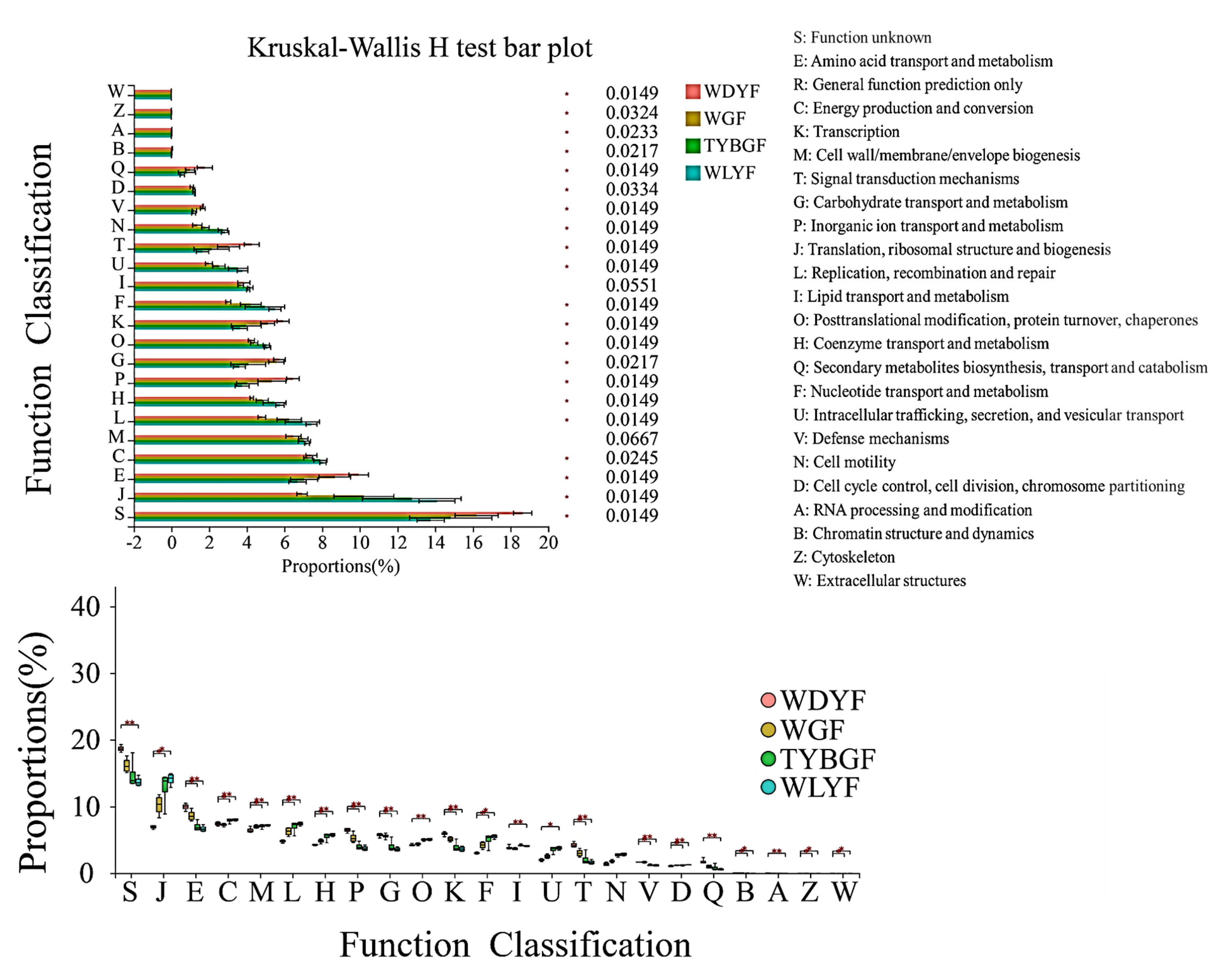
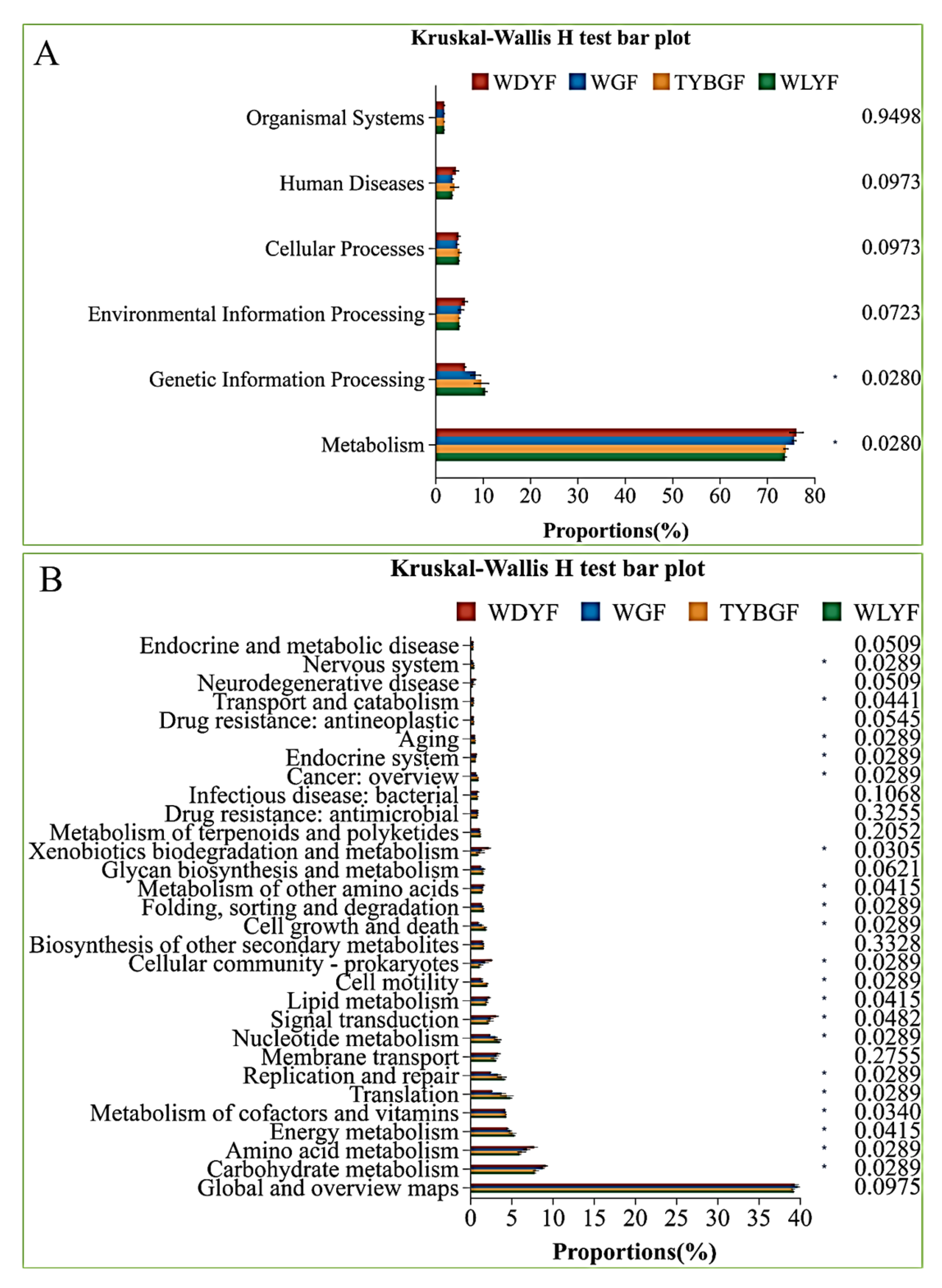
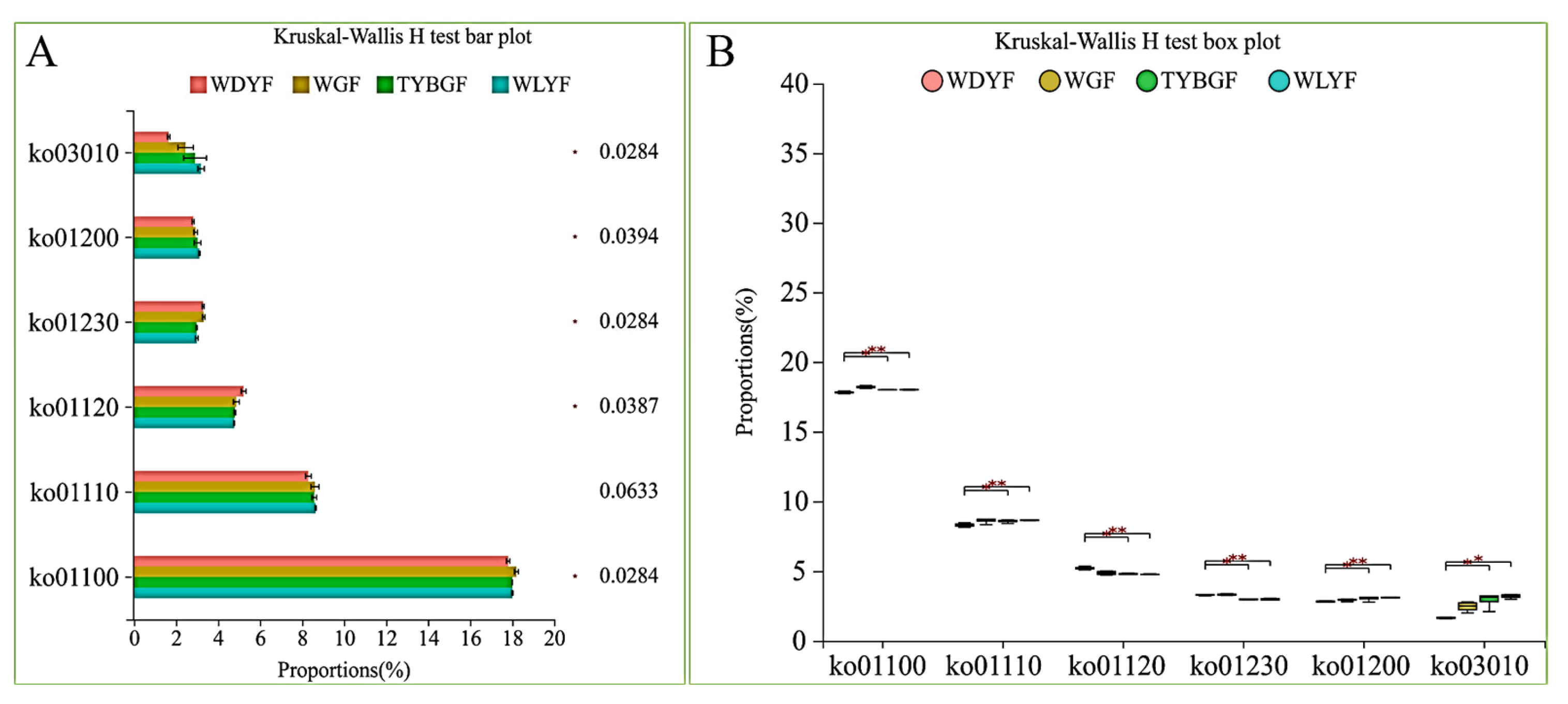
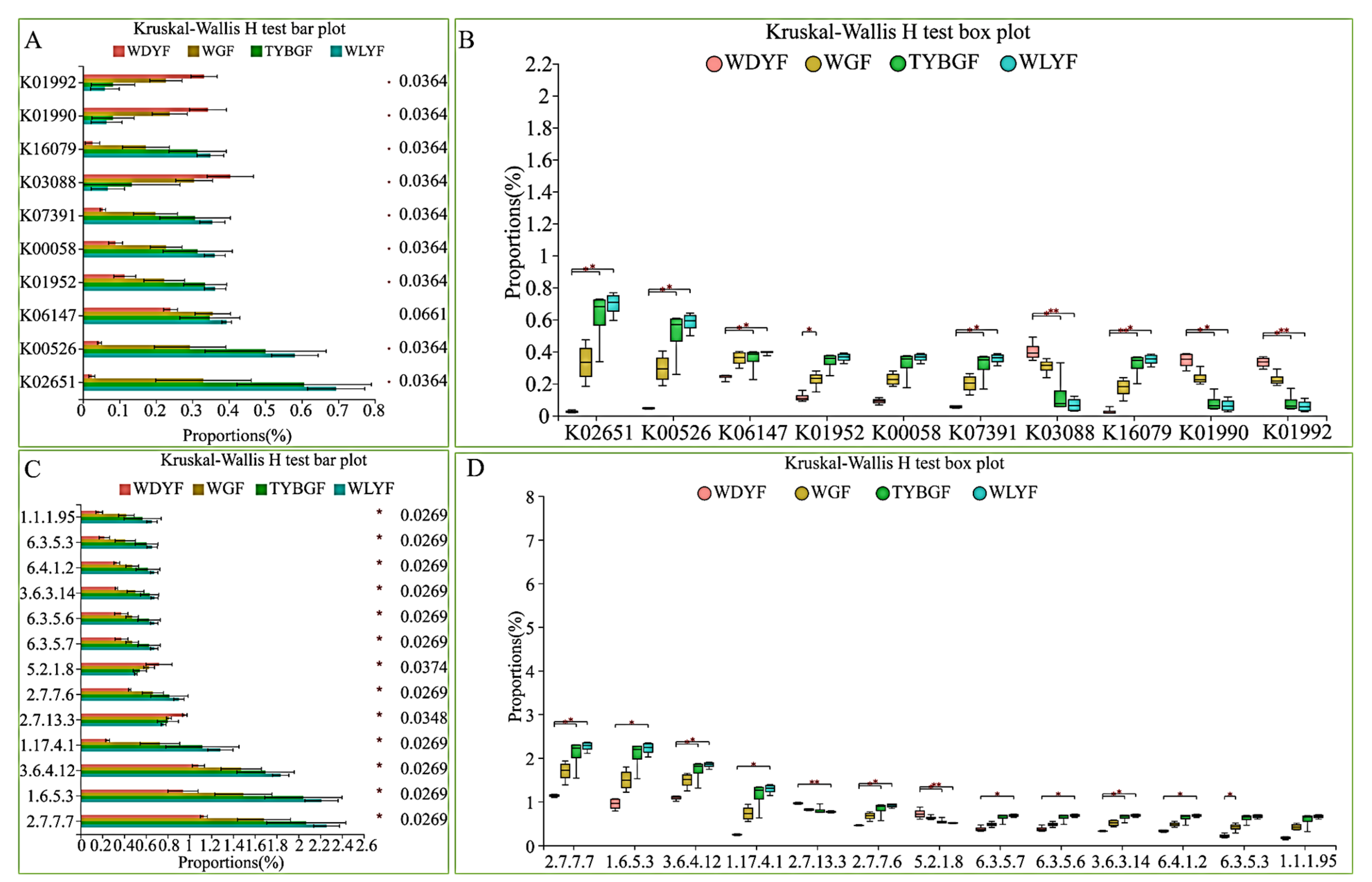
2.4. Bacterial Function Prediction in the Different Colored Citrus Pericarps
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. DNA Extraction and 16S rRNA Gene Amplification
4.3. Illumina MiSeq Sequencing and Data Processing
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alves-Júnior, A.; Fods, A.; Tfs, A.; Uba, B.; Mgg, C. Functional and morphological analysis of isolates of phylloplane and rhizoplane endophytic bacteria interacting in different cocoa production systems in the Amazon. Curr. Res. Microb. Sci. 2021, 2, 100039. [Google Scholar] [CrossRef]
- Müller, D.B.; Vogel, C.; Bai, Y.; Vorholt, J.A. The plant microbiota: Systems-level insights and perspectives. Annu. Rev. Genet. 2016, 50, 211–234. [Google Scholar] [CrossRef] [Green Version]
- Aamir, M.; Samal, S.; Rai, A.; Kashyap, S.P.; Singh, S.K.; Ahmed, M.; Upadhyay, R.S. Plant microbiome: Diversity, distribution, and functional relevance in crop improvement and sustainable agriculture. Microbiome Stimul. Crops 2021, 417–436. [Google Scholar] [CrossRef]
- Noman, M.; Ahmed, T.; Ijaz, U.; Shahid, M.; Azizullah; Li, D.; Manzoor, I.; Song, F. Plant-microbiome crosstalk: Dawning from composition and assembly of microbial community to improvement of disease resilience in plants. Int. J. Mol. Sci. 2021, 22, 6852. [Google Scholar] [CrossRef]
- Cordovez, V.; Dini-Andreote, F.; Carrión, V.J.; Raaijmakers, J.M. Ecology and evolution of plant microbiomes. Annu. Rev. Microbiol. 2019, 73, 69–88. [Google Scholar] [CrossRef]
- Bashir, I.; War, A.; Rafiq, I.; Reshi, Z.; Rashid, I.; Shouche, Y. Phyllosphere microbiome: Diversity and functions. Microbiol. Res. 2022, 254, 126888. [Google Scholar] [CrossRef] [PubMed]
- Ginnan, N.A.; Dang, T.; Bodaghi, S.; Ruegger, P.M.; Mccollum, G.; England, G. Disease-induced microbial shifts in citrus indicate microbiome derived responses to Huanglongbing across the disease severity spectrum. Phytobiomes J. 2022, 4, 375–387. [Google Scholar] [CrossRef]
- Yan, H.; Zhou, B.; Jiang, B.; Lv, Y.; Moniruzzaman, M.; Zhong, G. Comparative analysis of bacterial and fungal endophytes responses to Candidatus Liberibacter asiaticus infection in leaf midribs of Citrus reticulata cv. Shatangju. Physiol. Mol. Plant Pathol. 2021, 113, 101590. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, J.; Riera, N.; Jin, T.; Li, J.; Wang, N. Huanglongbing impairs the rhizosphere-to-rhizoplane enrichment process of the citrus root-associated microbiome. Microbiome 2017, 5, 97. [Google Scholar] [CrossRef]
- Wang, N.; Trivedi, P. Citrus Huanglongbing: A newly relevant disease presents unprecedented challenges. Phytopathology 2013, 103, 652–665. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.G.; Wu, J.; Bright, D.B.; Graham, J.H. Association of “Candidatus Liberibacter asiaticus” root infection, but not phloem plugging with root loss on huanglongbing-affected trees prior to appearance of foliar symptoms. Plant Pathol. 2014, 63, 290–298. [Google Scholar] [CrossRef]
- Manjunath, K.L.; Halbert, S.E.; Ramadugu, C.; Webb, S.; Lee, R.F. Detection of ‘Candidatus Liberibacter asiaticus’ in Diaphorina citri and its importance in the management of citrus Huanglongbing in Florida. Phytopathology 2008, 98, 387–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Ding, Y.; Nan, J.; Yang, X.; Sun, L.; Zhao, X. Transcriptome sequencing and ITRAQ reveal the detoxification mechanism of Bacillus GJ1, a potential biocontrol agent for Huanglongbing. PLoS ONE 2018, 13, e0200427. [Google Scholar] [CrossRef] [PubMed]
- Munir, S.; Li, Y.; He, P.; Ahmed, A.; Wu, Y. Unraveling the metabolite signature of citrus showing defense response towards Candidatus Liberibacter asiaticus after application of endophyte Bacillus subtilis L1-21. Microbiol. Res. 2020, 234, 126425. [Google Scholar] [CrossRef]
- Hopkins, D.; Wall, K. Biological control of citrus Huanglongbing with EB92-1, a benign strain of Xylella fastidiosa. Plant Dis. 2021, 105, 2914–2918. [Google Scholar] [CrossRef]
- Blaustein, R.A.; Lorca, G.L.; Meyer, J.L.; Gonzalez, C.F.; Teplitski, M. Defining the core citrus leaf-and root-associated microbiota: Factors associated with community structure and implications for managing Huanglongbing (citrus greening) disease. Appl. Environ. Microbiol. 2017, 83, e00210–e00217. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.Y.; Wu, Y.L.; Wu, W.; Huang, Y.J.; Zhu, C.Y.; Zhang, R.M.; Chen, J.; Zeng, J. Integrative analysis of metabolome and transcriptome profiles provides insight into the fruit pericarp pigmentation disorder caused by ‘Candidatus Liberibacter asiaticus’ infection. BMC Plant Biol. 2021, 21, 397. [Google Scholar] [CrossRef]
- Trivedi, P.; He, Z.L.; Van-Nostrand, J.D.; Albrigo, G.; Zhou, J.Z.; Wang, N. Huanglongbing alters the structure and functional diversity of microbial communities associated with citrus rhizosphere. ISME J. 2012, 6, 363–383. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Gomba, A.; Chidamba, L.; Korsten, L. Effect of postharvest practices including degreening on citrus carpoplane microbial biomes. J. Appl. Microbiol. 2017, 122, 1057–1070. [Google Scholar] [CrossRef] [Green Version]
- Kong, H.G.; Ham, H.; Lee, M.H.; Dong, S.P.; Yong, H.L. Microbial community dysbiosis and functional gene content changes in apple flowers due to fire blight. Plant Pathol. J. 2021, 37, 404–412. [Google Scholar] [CrossRef]
- Ardano, P.A.; Häggman, H.; Kozyrovska, N.; Pirttilä, A.M. Methylobacterium-induced endophyte community changes correspond with protection of plants against pathogen attack. PLoS ONE 2012, 7, e46802. [Google Scholar] [CrossRef]
- Luo, L.F.; Wang, L.T.; Deng, L.M.; Mei, X.Y.; Liu, Y.X.; Huang, H.C.; Du, F.; Zhu, S.; Yang, M. Enrichment of Burkholderia in the rhizosphere by autotoxic ginsenosides to alleviate negative plant-soil feedback. Microbiol. Spectr. 2021, 9, e01400-21. [Google Scholar] [CrossRef]
- Liu, C.; Zhao, D.; Ma, W.; Guo, Y.; Wang, A.; Wang, Q. Denitrifying sulfide removal process on high-salinity wastewaters in the presence of Halomonas sp. Appl. Microbiol. Biot. 2016, 100, 1421–1426. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. FASTP: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.; Holmes, S.P. DADA2: High-resolution sample inference from illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Zhu, C.; Zhang, R.; Huang, Y.; Wu, W.; Chen, J.; Zeng, J. Diversity Analysis and Function Prediction of Bacterial Communities in the Different Colored Pericarp of Citrus reticulata cv. ‘Shatangju’ Due to ‘Candidatus Liberibacter asiaticus’ Infection. Int. J. Mol. Sci. 2023, 24, 11472. https://doi.org/10.3390/ijms241411472
Wang F, Zhu C, Zhang R, Huang Y, Wu W, Chen J, Zeng J. Diversity Analysis and Function Prediction of Bacterial Communities in the Different Colored Pericarp of Citrus reticulata cv. ‘Shatangju’ Due to ‘Candidatus Liberibacter asiaticus’ Infection. International Journal of Molecular Sciences. 2023; 24(14):11472. https://doi.org/10.3390/ijms241411472
Chicago/Turabian StyleWang, Feiyan, Congyi Zhu, Ruimin Zhang, Yongjing Huang, Wen Wu, Jiezhong Chen, and Jiwu Zeng. 2023. "Diversity Analysis and Function Prediction of Bacterial Communities in the Different Colored Pericarp of Citrus reticulata cv. ‘Shatangju’ Due to ‘Candidatus Liberibacter asiaticus’ Infection" International Journal of Molecular Sciences 24, no. 14: 11472. https://doi.org/10.3390/ijms241411472