
Structural Insights into M1 Muscarinic Acetylcholine Receptor Signaling Bias between Gαq and β-Arrestin through BRET Assays and Molecular Docking

Abstract
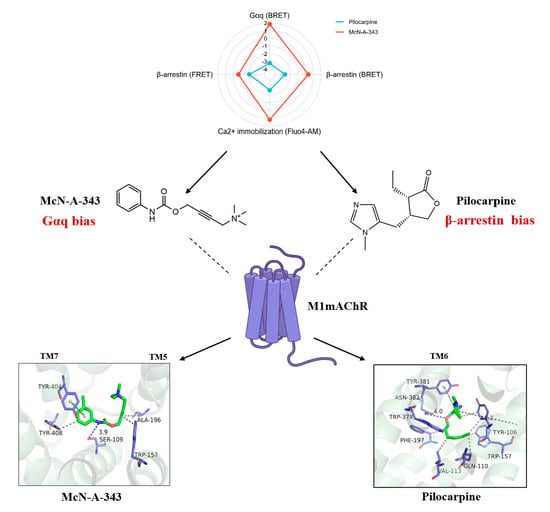
:
1. Introduction
2. Results
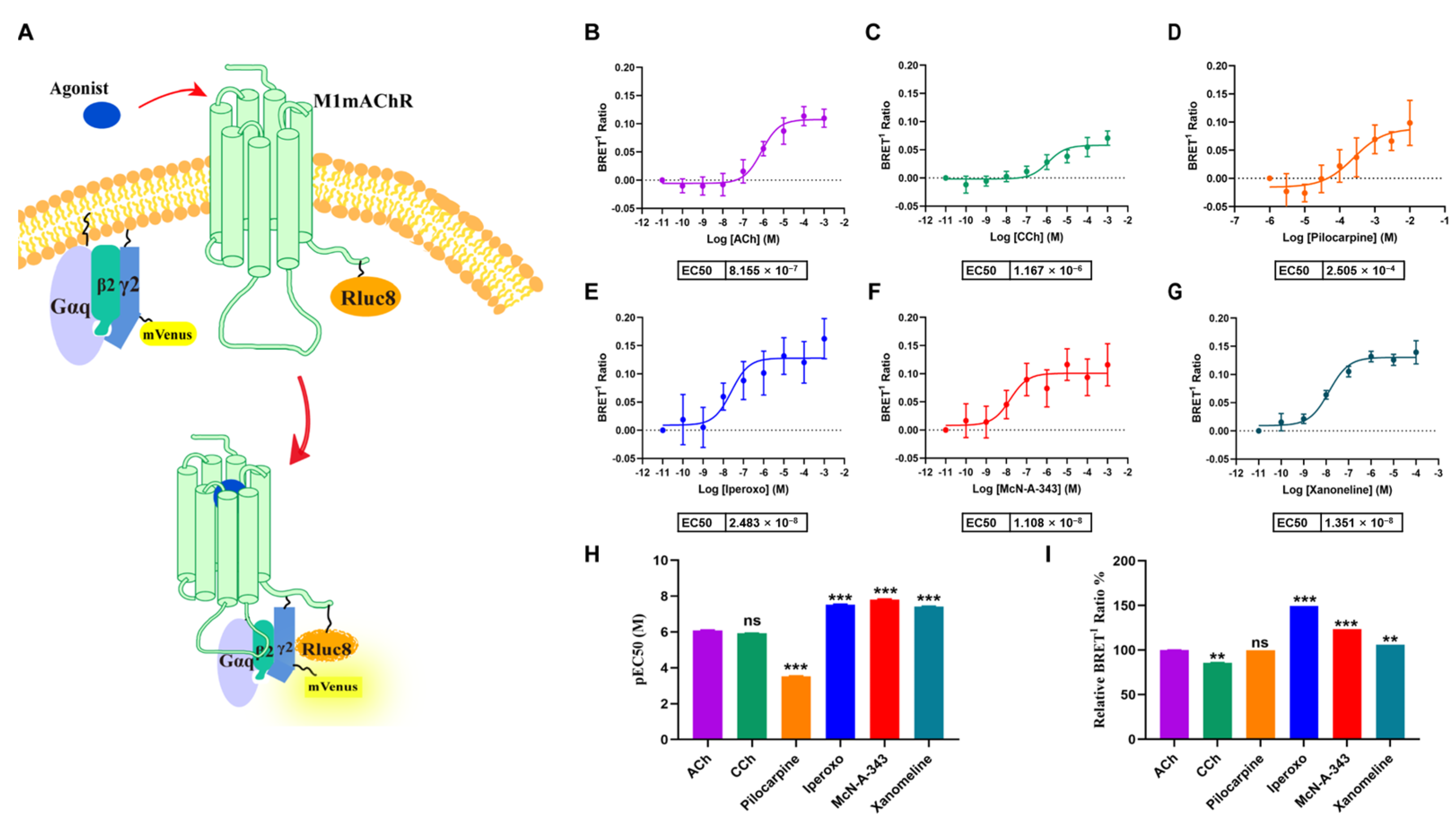
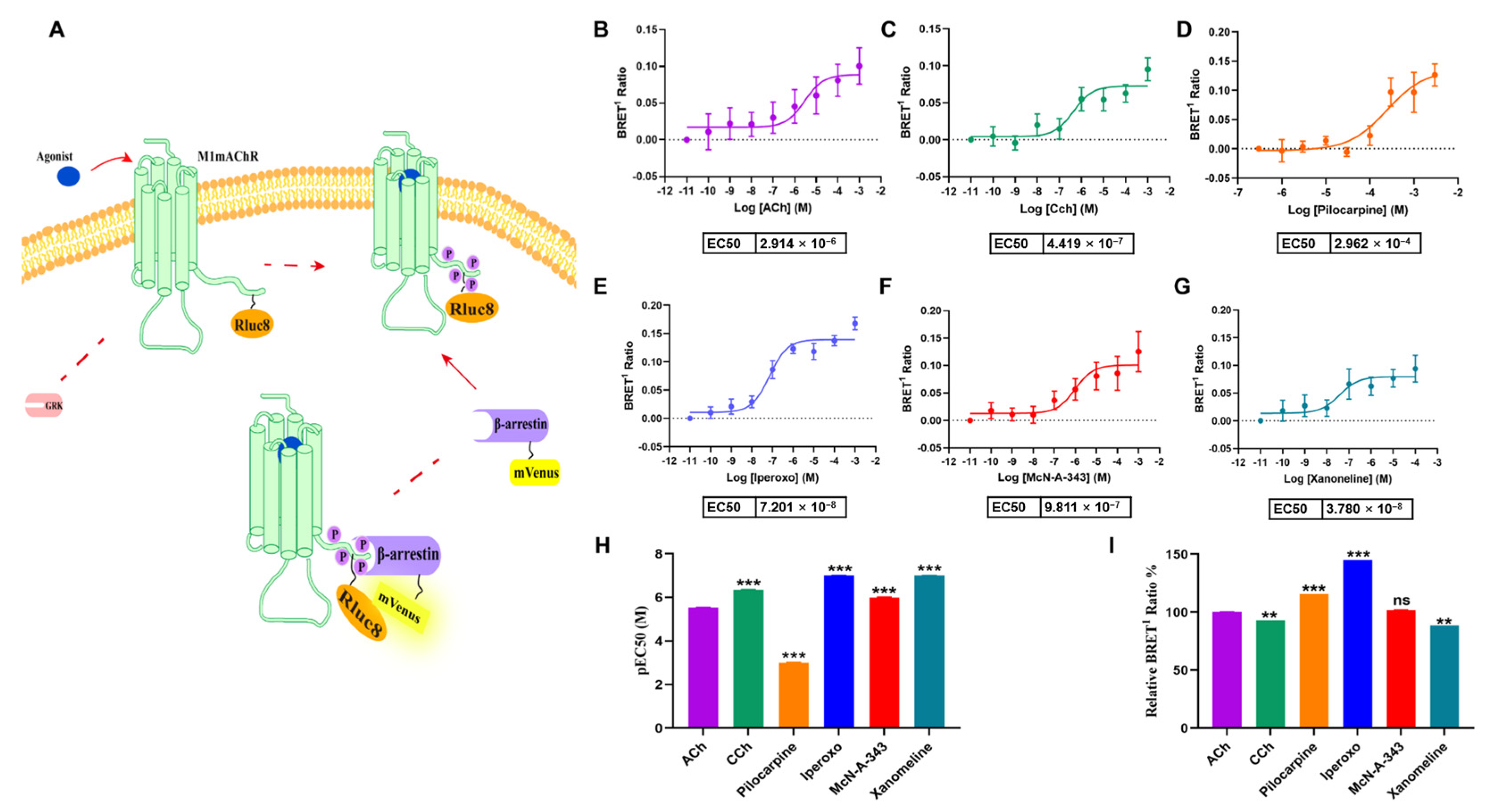
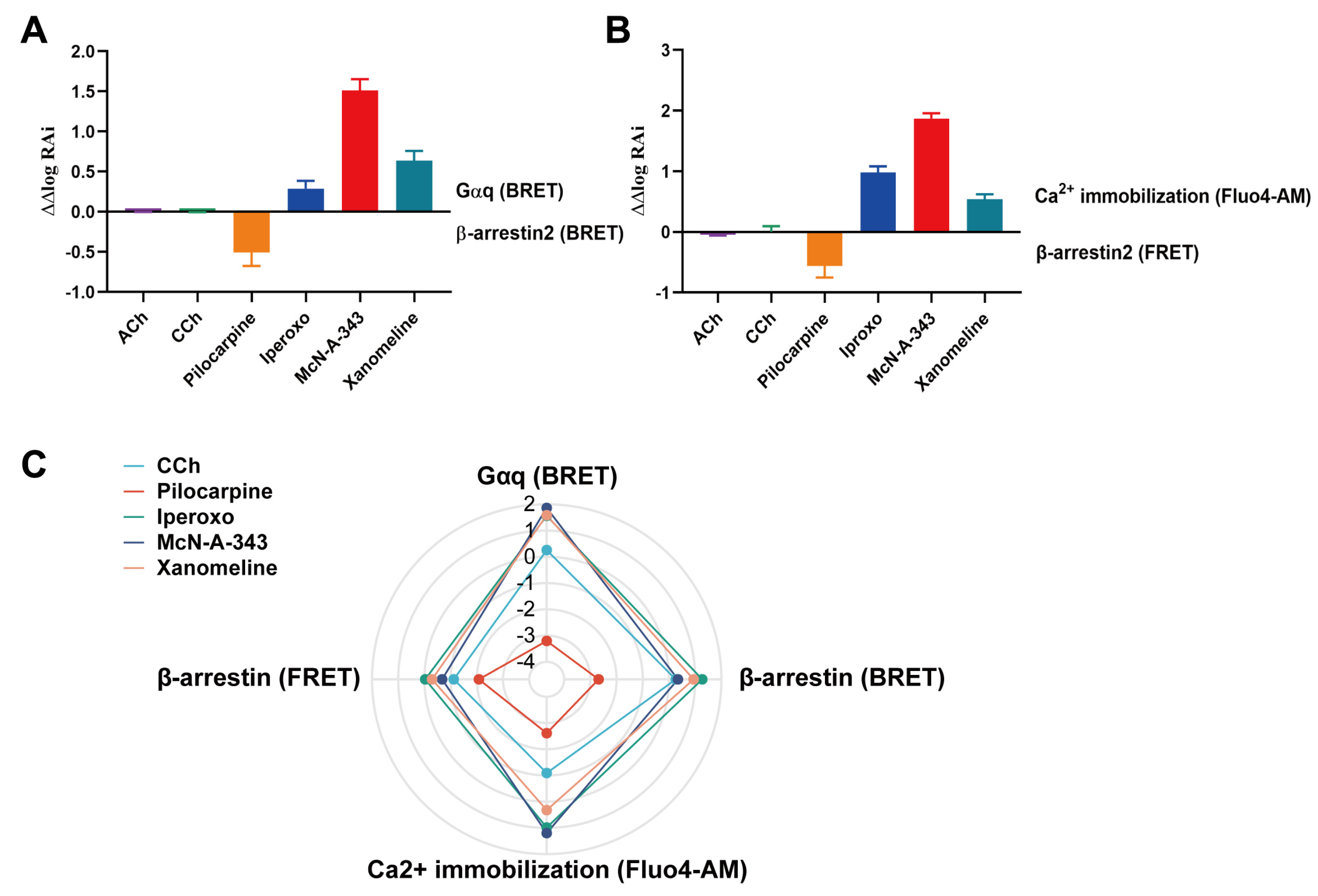
2.1. Efficacy of M1mAChR Agonists in Gαq-Binding Revealed by BRET Assays
2.2. Kinetics of Gαq-M1mAChR Coupling Assessed by BRET Assays
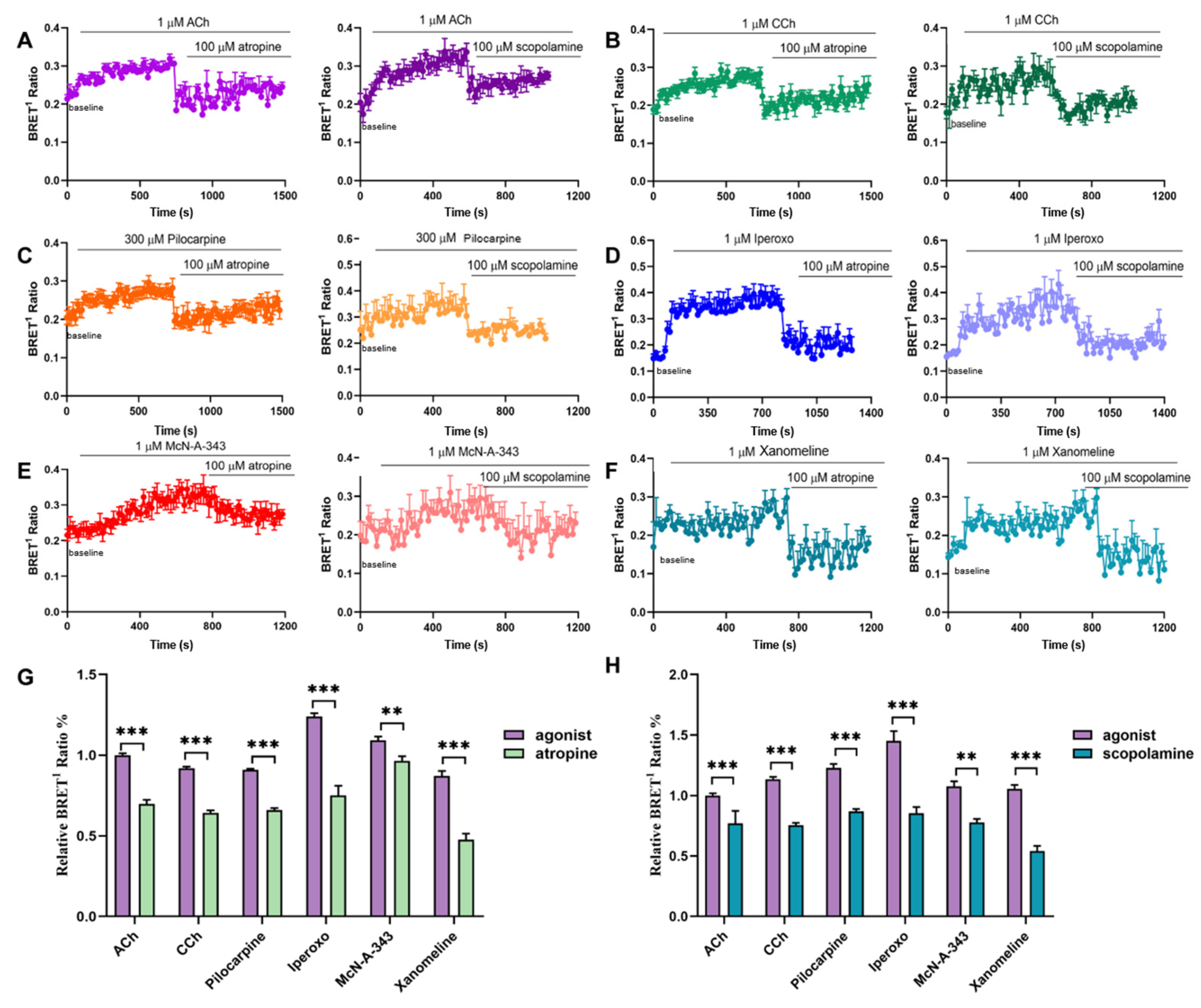
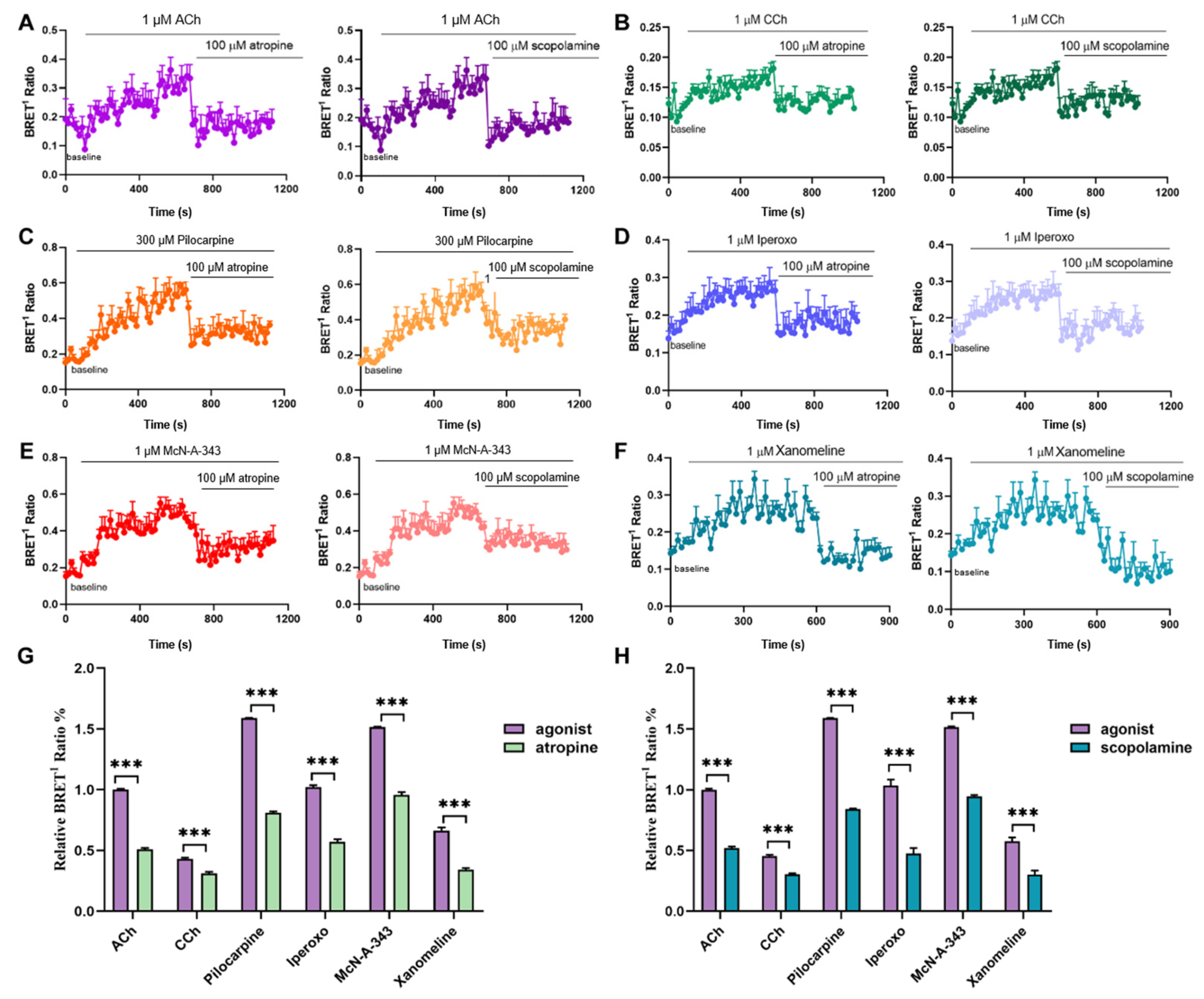
2.3. Efficacy of Agonists in β-Arrestin2-Binding to M1mAChR Revealed by BRET Assays
2.4. Kinetics of β-Arrestin2-M1mAChR Coupling Assessed by BRET Assays
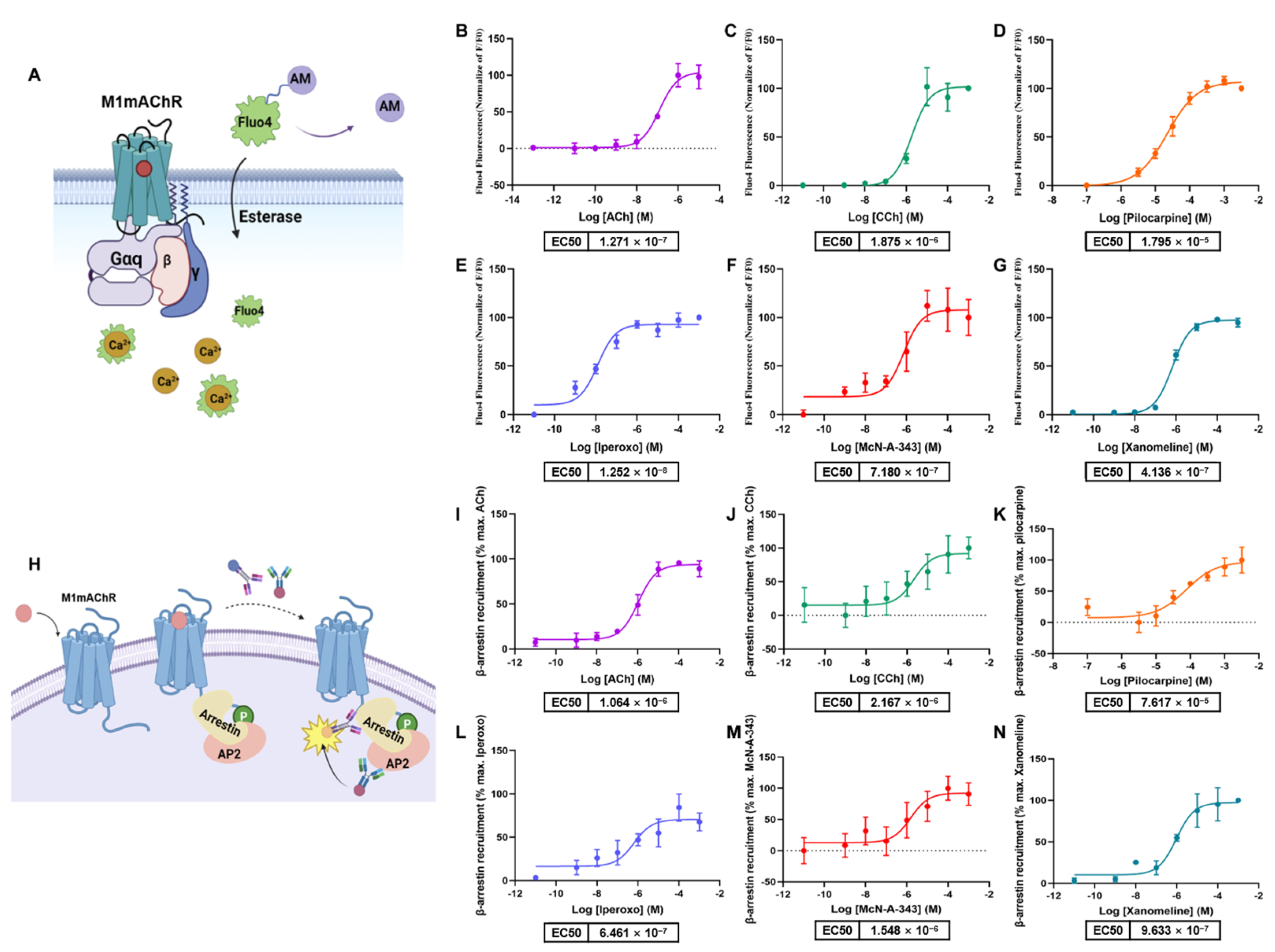
2.5. Determination of Gαq and β-Arrestin2 Pathway Activation by Commercial Methods
2.6. Evaluation of the Signaling Bias of M1mAChR Agonists
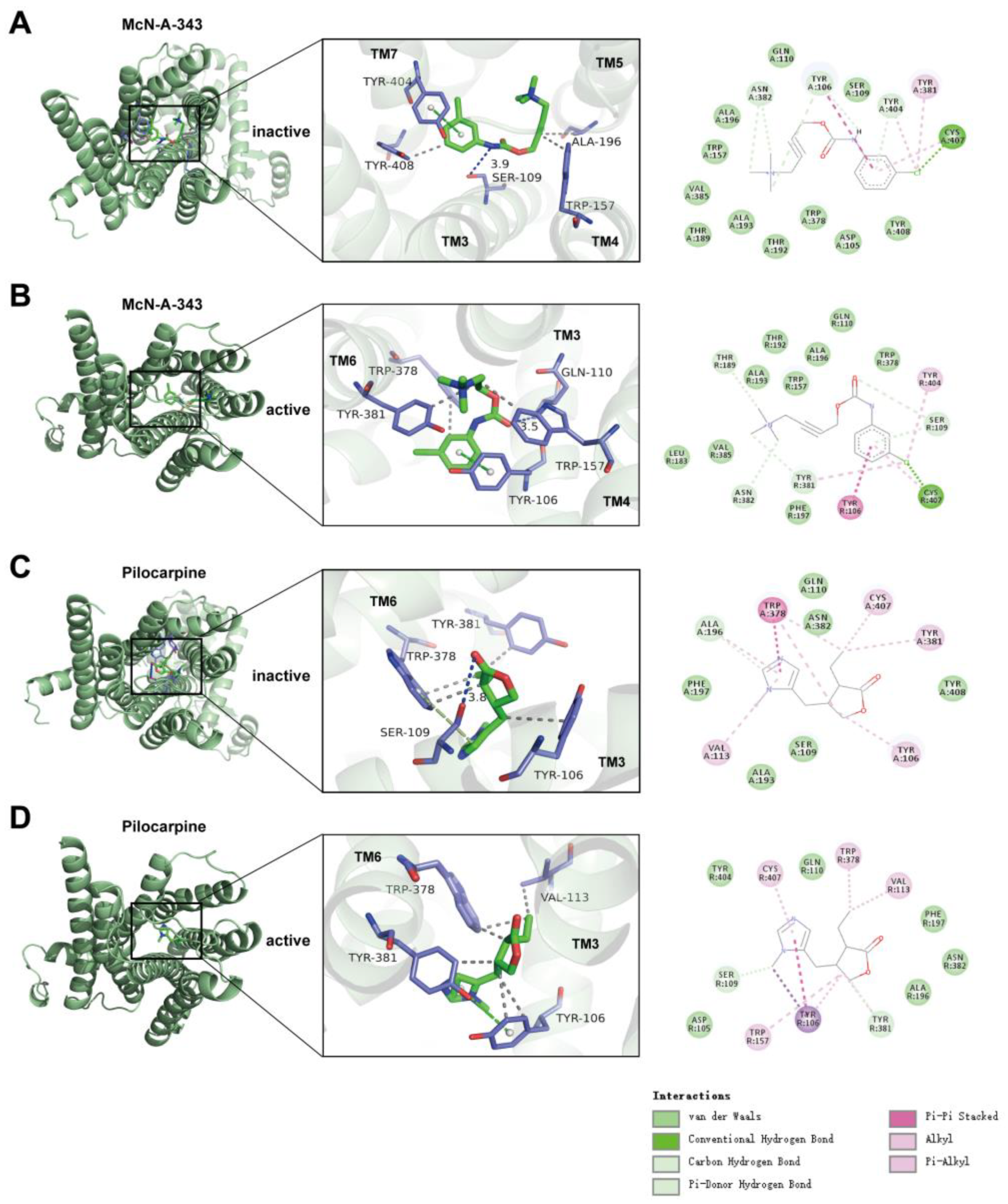
2.7. The Interaction Underlying the Signaling Bias Observed in Molecular Docking
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. cDNA Constructs
4.3. BRET Assays for Gαq and β-Arrestin Coupling
4.4. Fluo-4-AM
4.5. β-Arrestin Recruitment
4.6. Molecular Docking
4.7. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GPCRs | G-protein-coupled receptors |
| M1mAChR | M1 muscarinic acetylcholine receptor |
| BRET | Bioluminescence resonance energy transfer |
| FRET | Fluorescence resonance energy transfer |
| ACh | Acetylcholine |
| CCh | Carbachol |
| RAi | Relative intrinsic activity |
References
- Masuho, I.; Skamangas, N.K.; Muntean, B.S.; Martemyanov, K.A. Diversity of the Gbetagamma complexes defines spatial and temporal bias of GPCR signaling. Cell Syst. 2021, 12, 324–337.e5. [Google Scholar] [CrossRef] [PubMed]
- Werder, R.B.; Ullah, M.A.; Rahman, M.M.; Simpson, J.; Lynch, J.P.; Collinson, N.; Rittchen, S.; Rashid, R.B.; Sikder, M.A.A.; Handoko, H.Y.; et al. Targeting the P2Y(13) Receptor Suppresses IL-33 and HMGB1 Release and Ameliorates Experimental Asthma. Am. J. Respir. Crit. Care Med. 2022, 205, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, G.; Austin, C.; Anderson, J.; Pawlyk, A.; Colvis, C.; Margolis, R.; Baker, J. Glimmers in illuminating the druggable genome. Nat. Rev. Drug Discov. 2018, 17, 301–302. [Google Scholar] [CrossRef]
- Wang, W.; Qiao, Y.; Li, Z. New Insights into Modes of GPCR Activation. Trends Pharmacol. Sci. 2018, 39, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Dixon, A.D.; Inoue, A.; Robson, S.A.; Culhane, K.J.; Trinidad, J.C.; Sivaramakrishnan, S.; Bumbak, F.; Ziarek, J.J. The Effect of Ligands and Transducers on the Neurotensin Receptor 1 (NTS1) Conformational Ensemble. J. Am. Chem. Soc. 2022, 144, 10241–10250. [Google Scholar] [CrossRef]
- Ilter, M.; Mansoor, S.; Sensoy, O. Utilization of Biased G Protein-Coupled ReceptorSignaling towards Development of Safer andPersonalized Therapeutics. Molecules 2019, 24, 2052. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Yanagawa, M.; Hiratsuka, S.; Yoshida, M.; Ono, Y.; Hiroshima, M.; Ueda, M.; Aoki, J.; Sako, Y.; Inoue, A. Heterotrimeric Gq proteins act as a switch for GRK5/6 selectivity underlying beta-arrestin transducer bias. Nat. Commun. 2022, 13, 487. [Google Scholar] [CrossRef]
- Yohn, S.E.; Weiden, P.J.; Felder, C.C.; Stahl, S.M. Muscarinic acetylcholine receptors for psychotic disorders: Bench-side to clinic. Trends Pharmacol. Sci. 2022, 43, 1098–1112. [Google Scholar] [CrossRef]
- Van der Westhuizen, E.T.; Choy, K.H.C.; Valant, C.; McKenzie-Nickson, S.; Bradley, S.J.; Tobin, A.B.; Sexton, P.M.; Christopoulos, A. Fine Tuning Muscarinic Acetylcholine Receptor Signaling Through Allostery and Bias. Front. Pharmacol. 2020, 11, 606656. [Google Scholar] [CrossRef]
- Bradley, S.J.; Molloy, C.; Valuskova, P.; Dwomoh, L.; Scarpa, M.; Rossi, M.; Finlayson, L.; Svensson, K.A.; Chernet, E.; Barth, V.N.; et al. Biased M1-muscarinic-receptor-mutant mice inform the design of next-generation drugs. Nat. Chem. Biol. 2020, 16, 240–249. [Google Scholar] [CrossRef]
- Scarpa, M.; Molloy, C.; Jenkins, L.; Strellis, B.; Budgett, R.F.; Hesse, S.; Dwomoh, L.; Marsango, S.; Tejeda, G.S.; Rossi, M.; et al. Biased M1 muscarinic receptor mutant mice show accelerated progression of prion neurodegenerative disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2107389118. [Google Scholar] [CrossRef] [PubMed]
- Powers, A.S.; Pham, V.; Burger, W.A.C.; Thompson, G.; Laloudakis, Y.; Sexton, P.M.; Paul, S.M.; Christopoulos, A.; Thal, D.M.; Felder, C.C.; et al. Structural basis of efficacy-driven ligand selectivity at GPCRs. Nat. Chem. Biol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Flöser, A.; Becker, K.; Kostenis, E.; König, G.; Krasel, C.; Kolb, P.; Bünemann, M. Disentangling bias between Gq, GRK2, and arrestin3 recruitment to the M3 muscarinic acetylcholine receptor. eLife 2021, 10, e58442. [Google Scholar] [CrossRef] [PubMed]
- Pfeil, E.M.; Brands, J.; Merten, N.; Vogtle, T.; Vescovo, M.; Rick, U.; Albrecht, I.M.; Heycke, N.; Kawakami, K.; Ono, Y.; et al. Heterotrimeric G Protein Subunit Galphaq Is a Master Switch for Gbetagamma-Mediated Calcium Mobilization by Gi-Coupled GPCRs. Mol. Cell 2020, 80, 940–954.e6. [Google Scholar] [CrossRef]
- Clark, W.A.; Jian, X.; Chen, L.; Northup, J.K. Independent and synergistic interaction of retinal G-protein subunits with bovine rhodopsin measured by surface plasmon resonance. Biochem. J. 2001, 358, 389–397. [Google Scholar] [CrossRef]
- Nagai, T.; Ibata, K.; Park, E.S.; Kubota, M.; Mikoshiba, K.; Miyawaki, A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002, 20, 87–90. [Google Scholar] [CrossRef]
- Lohse, M.J.; Nuber, S.; Hoffmann, C. Fluorescence/bioluminescence resonance energy transfer techniques to study G-protein-coupled receptor activation and signaling. Pharmacol. Rev. 2012, 64, 299–336. [Google Scholar] [CrossRef]
- Wu, P.; Brand, L. Resonance energy transfer: Methods and applications. Anal. Biochem. 1994, 218, 1–13. [Google Scholar] [CrossRef]
- Pfleger, K.; Eidne, K.A. New technologies: Bioluminescence resonance energy transfer (BRET) for the detection of real time interactions involving G-protein coupled receptors. Pituitary 2003, 6, 141–151. [Google Scholar] [CrossRef]
- Pfleger, K.D.; Eidne, K.A. Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat. Methods 2006, 3, 165–174. [Google Scholar] [CrossRef]
- Felder, C.C.; Kanterman, R.Y.; Ma, A.L.; Axelrod, J. A transfected m1 muscarinic acetylcholine receptor stimulates adenylate cyclase via phosphatidylinositol hydrolysis. J. Biol. Chem. 1989, 264, 20356–20362. [Google Scholar] [CrossRef] [PubMed]
- Premont, R.T.; Gainetdinov, R.R. Physiological roles of G protein-coupled receptor kinases and arrestins. Annu. Rev. Physiol. 2007, 69, 511–534. [Google Scholar] [CrossRef]
- Maeda, S.; Qu, Q.; Robertson, M.J.; Skiniotis, G.; Kobilka, B.K. Structures of the M1 and M2 muscarinic acetylcholine receptor/G-protein complexes. Science 2019, 364, 552–557. [Google Scholar] [CrossRef]
- Chen, M.W.; Zhu, H.; Xiong, C.H.; Li, J.B.; Zhao, L.X.; Chen, H.Z.; Qiu, Y. PKC and Ras are Involved in M1 Muscarinic Receptor-Mediated Modulation of AMPA Receptor GluA1 Subunit. Cell Mol. Neurobiol. 2020, 40, 547–554. [Google Scholar] [CrossRef]
- Wingler, L.M.; Elgeti, M.; Hilger, D.; Latorraca, N.R.; Lerch, M.T.; Staus, D.P.; Dror, R.O.; Kobilka, B.K.; Hubbell, W.L.; Lefkowitz, R.J. Angiotensin Analogs with Divergent Bias Stabilize Distinct Receptor Conformations. Cell 2019, 176, 468–478.e11. [Google Scholar] [CrossRef] [PubMed]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. Beta-arrestins and cell signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef]
- Latorraca, N.R.; Wang, J.K.; Bauer, B.; Townshend, R.J.L.; Hollingsworth, S.A.; Olivieri, J.E.; Xu, H.E.; Sommer, M.E.; Dror, R.O. Molecular mechanism of GPCR-mediated arrestin activation. Nature 2018, 557, 452–456. [Google Scholar] [CrossRef]
- Gurwitz, D.; Haring, R.; Heldman, E.; Fraser, C.M.; Manor, D.; Fisher, A. Discrete activation of transduction pathways associated with acetylcholine ml receptor by several muscarinic ligands. Eur. J. Pharmacol. Mol. Pharmacol. 1994, 267, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.T.; Figueroa, K.W.; Liller, S.; Ehlert, F.J. Estimation of agonist activity at G protein-coupled receptors: Analysis of M2 muscarinic receptor signaling through Gi/o,Gs, and G15. J. Pharmacol. Exp. Ther. 2007, 321, 1193–1207. [Google Scholar] [CrossRef]
- Staus, D.P.; Hu, H.; Robertson, M.J.; Kleinhenz, A.L.W.; Wingler, L.M.; Capel, W.D.; Latorraca, N.R.; Lefkowitz, R.J.; Skiniotis, G. Structure of the M2 muscarinic receptor-beta-arrestin complex in a lipid nanodisc. Nature 2020, 579, 297–302. [Google Scholar] [CrossRef]
- Xu, J.; Hu, Y.; Kaindl, J.; Risel, P.; Hübner, H.; Maeda, S.; Niu, X.; Li, H.; Gmeiner, P.; Jin, C.; et al. Conformational Complexity and Dynamics in a Muscarinic Receptor Revealed by NMR Spectroscopy. Mol. Cell 2019, 75, 53–65.e7. [Google Scholar] [CrossRef]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the micro-opioid receptor-Gi protein complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef]
- El Daibani, A.; Paggi, J.M.; Kim, K.; Laloudakis, Y.D.; Popov, P.; Bernhard, S.M.; Krumm, B.E.; Olsen, R.H.J.; Diberto, J.; Carroll, F.I.; et al. Molecular mechanism of biased signaling at the kappa opioid receptor. Nat. Commun. 2023, 14, 1338. [Google Scholar] [CrossRef]
- Bock, A.; Merten, N.; Schrage, R.; Dallanoce, C.; Batz, J.; Klockner, J.; Schmitz, J.; Matera, C.; Simon, K.; Kebig, A.; et al. The allosteric vestibule of a seven transmembrane helical receptor controls G-protein coupling. Nat. Commun. 2012, 3, 1044. [Google Scholar] [CrossRef]
- Brannan, S.K.; Sawchak, S.; Miller, A.C.; Lieberman, J.A.; Paul, S.M.; Breier, A. Muscarinic Cholinergic Receptor Agonist and Peripheral Antagonist for Schizophrenia. N. Engl. J. Med. 2021, 384, 717–726. [Google Scholar] [CrossRef]
- Yang, B.; Yang, Y. Maqianzi (Strychnos), A poisonous medicinal native to the Western Regions. Chin. Med. Cult. 2019, 2, 44–47. [Google Scholar] [CrossRef]
- Pronin, A.N.; Wang, Q.; Slepak, V.Z. Teaching an Old Drug New Tricks: Agonism, Antagonism, and Biased Signaling of Pilocarpine through M3 Muscarinic Acetylcholine Receptor. Mol. Pharmacol. 2017, 92, 601–612. [Google Scholar] [CrossRef]
- Wingler, L.M.; Skiba, M.A.; McMahon, C.; Staus, D.P.; Kleinhenz, A.L.W.; Suomivuori, C.M.; Latorraca, N.R.; Dror, R.O.; Lefkowitz, R.J.; Kruse, A.C. Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science 2020, 367, 888–892. [Google Scholar] [CrossRef] [PubMed]
- Suomivuori, C.-M.; Latorraca, N.R.; Wingler, L.M.; Eismann, S.; King, M.C.; Kleinhenz, A.L.W.; Skiba, M.A.; Staus, D.P.; Kruse, A.C.; Lefkowitz, R.J.; et al. Molecular mechanism of biased signaling in a prototypical G protein–coupled receptor. Science 2020, 367, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Huang, W.; Aydin, D.; Paggi, J.M.; Seven, A.B.; Wang, H.; Chakraborty, S.; Che, T.; DiBerto, J.F.; Robertson, M.J.; et al. Insights into distinct signaling profiles of the microOR activated by diverse agonists. Nat. Chem. Biol. 2023, 19, 423–430. [Google Scholar] [CrossRef]
- Uprety, R.; Che, T.; Zaidi, S.A.; Grinnell, S.G.; Varga, B.R.; Faouzi, A.; Slocum, S.T.; Allaoa, A.; Varadi, A.; Nelson, M.; et al. Controlling opioid receptor functional selectivity by targeting distinct subpockets of the orthosteric site. eLife 2021, 10, e56519. [Google Scholar] [CrossRef]
- Hoffmann, C.; Nuber, S.; Zabel, U.; Ziegler, N.; Winkler, C.; Hein, P.; Berlot, C.H.; Bunemann, M.; Lohse, M.J. Comparison of the activation kinetics of the M3 acetylcholine receptor and a constitutively active mutant receptor in living cells. Mol. Pharmacol. 2012, 82, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, V.; Mosslein, N.; Bunemann, M. Structures in G proteins important for subtype selective receptor binding and subsequent activation. Commun. Biol. 2021, 4, 635. [Google Scholar] [CrossRef] [PubMed]
- Krasel, C.; Bunemann, M.; Lorenz, K.; Lohse, M.J. Beta-arrestin binding to the beta2-adrenergic receptor requires both receptor phosphorylation and receptor activation. J. Biol. Chem. 2005, 280, 9528–9535. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.C.; Lukasheva, V.; Le Gouill, C.; Kobayashi, H.; Breton, B.; Mailhot-Larouche, S.; Blondel-Tepaz, E.; Antunes Vieira, N.; Costa-Neto, C.; Heroux, M.; et al. BRET-based effector membrane translocation assay monitors GPCR-promoted and endocytosis-mediated Gq activation at early endosomes. Proc. Natl. Acad. Sci. USA 2021, 118, e2025846118. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T.; Christopoulos, A. Signalling bias in new drug discovery: Detection, quantification and therapeutic impact. Nat. Rev. Drug Discov. 2013, 12, 205–216. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Gαq (BRET) (Δlog (τ/KA)) | β-Arrestin (BRET) (Δlog (τ/KA)) | Gαq (Fluo4-AM) (Δlog (τ/KA)) | β-Arrestin (FRET) (Δlog (τ/KA)) |
|---|---|---|---|---|
| McN-A-343 | 1.85 ± 0.010 | 0.34 ± 0.040 | 1.20 ± 0.005 | −0.67 ± 0.020 |
| Iperoxo | 1.56 ± 0.090 | 1.27 ± 0.020 | 0.98 ± 0.003 | −0.03 ± 0.040 |
| Xanomeline | 1.58 ± 0.050 | 0.94 ± 0.030 | 0.32 ± 0.002 | −0.29 ± 0.020 |
| CCh | 0.26 ± 0.020 | 0.26 ± 0.040 | −1.09 ± 0.030 | −1.12 ± 0.040 |
| Pilocarpine | −3.20 ± 0.001 | −2.70 ± 0.002 | −2.61 ± 0.002 | −2.08 ± 0.030 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Yao, Y.; Wang, S.; Hou, Y.; Zhao, L.; Wang, H.; Chen, H.; Xu, J. Structural Insights into M1 Muscarinic Acetylcholine Receptor Signaling Bias between Gαq and β-Arrestin through BRET Assays and Molecular Docking. Int. J. Mol. Sci. 2023, 24, 7356. https://doi.org/10.3390/ijms24087356
Wang D, Yao Y, Wang S, Hou Y, Zhao L, Wang H, Chen H, Xu J. Structural Insights into M1 Muscarinic Acetylcholine Receptor Signaling Bias between Gαq and β-Arrestin through BRET Assays and Molecular Docking. International Journal of Molecular Sciences. 2023; 24(8):7356. https://doi.org/10.3390/ijms24087356
Chicago/Turabian StyleWang, Dongxue, Yunjin Yao, Shiqi Wang, Yifei Hou, Lanxue Zhao, Hao Wang, Hongzhuan Chen, and Jianrong Xu. 2023. "Structural Insights into M1 Muscarinic Acetylcholine Receptor Signaling Bias between Gαq and β-Arrestin through BRET Assays and Molecular Docking" International Journal of Molecular Sciences 24, no. 8: 7356. https://doi.org/10.3390/ijms24087356