
Aberrant Dopamine System Function in the Ferrous Amyloid Buthionine (FAB) Rat Model of Alzheimer’s Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
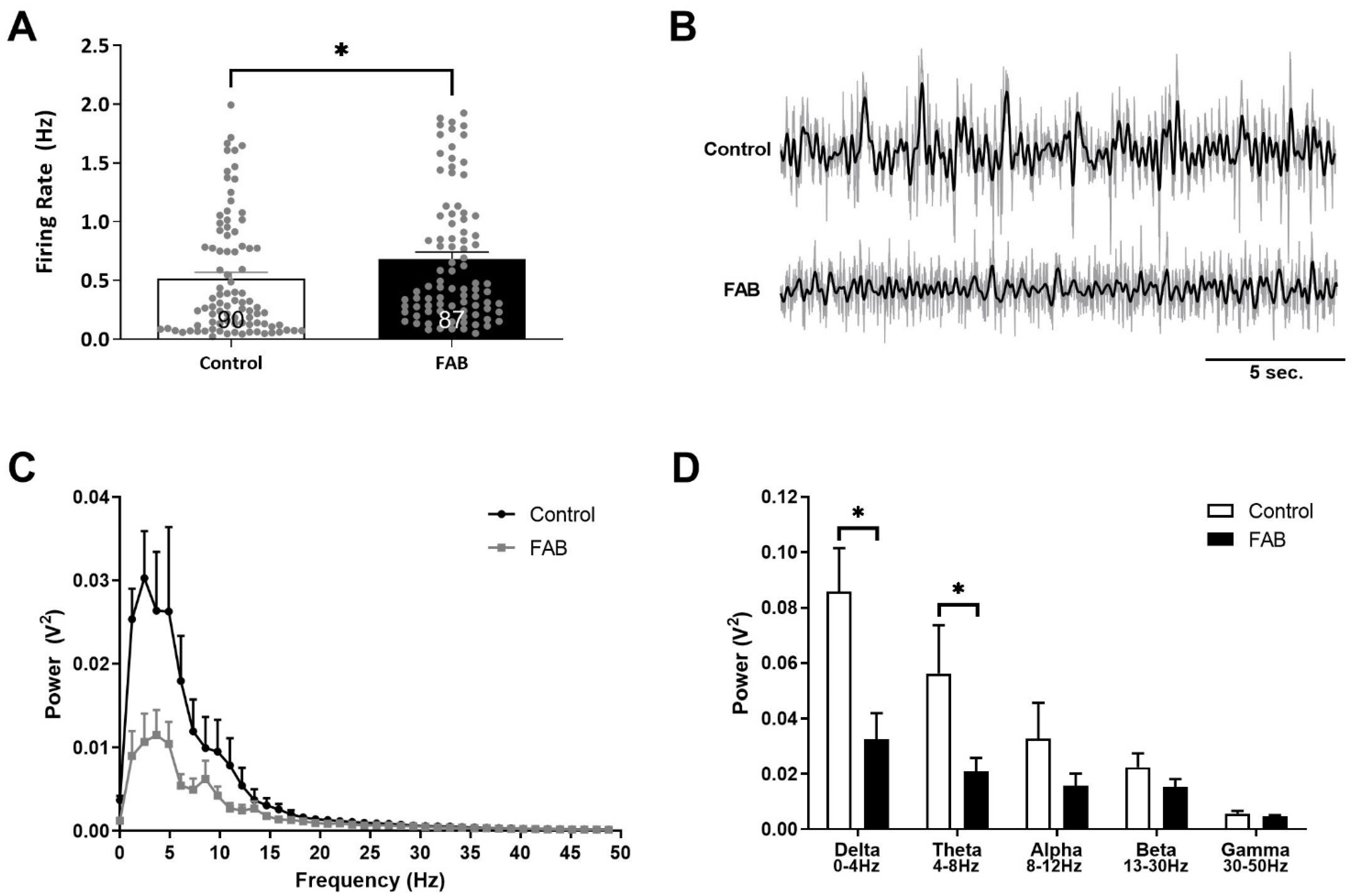
2.1. Firing Rates of Putative Pyramidal Neurons in the vHipp Were Increased, While Coordinated Neuronal Activity Was Significantly Decreased in FAB Rats
2.2. FAB Rats Displayed a Significant Increase in Dopamine Neuron Population Activity
2.3. FAB Rats Displayed Augmented Locomotor Activity and Deficits in Working Memory
2.4. FAB Rats Exhibited a Significant Decrease in Parvalbumin Protein in the Ventral Hippocampus
3. Discussion
4. Materials and Methods
4.1. Osmotic Minipump Survival Surgeries
4.2. In Vivo Electrophysiology and Local Field Potentials (LFPs)
4.3. MK-801-Induced Locomotor Response
4.4. Pre-Pulse Inhibition of Startle
4.5. Y-Maze Spontaneous Alternation Assay
4.6. Social Interaction (SI)
4.7. Western Blot
4.8. Histology
4.9. Analysis
4.10. Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ballard, C.; Walker, M. Neuropsychiatric aspects of Alzheimer’s disease. Curr. Psychiatry Rep. 1999, 1, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Haupt, M.; Kurz, A.; Janner, M. A 2-year follow-up of behavioural and psychological symptoms in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2000, 11, 147–152. [Google Scholar] [CrossRef]
- Ropacki, S.A.; Jeste, D.V. Epidemiology of and risk factors for psychosis of Alzheimer’s disease: A review of 55 studies published from 1990 to 2003. Am. J. Psychiatry 2005, 162, 2022–2030. [Google Scholar] [CrossRef]
- Emanuel, J.E.; Lopez, O.L.; Houck, P.R.; Becker, J.T.; Weamer, E.A.; Demichele-Sweet, M.A.; Kuller, L.; Sweet, R.A. Trajectory of cognitive decline as a predictor of psychosis in early Alzheimer disease in the cardiovascular health study. Am. J. Geriatr. Psychiatry 2011, 19, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Ballard, C.; Kales, H.C.; Lyketsos, C.; Aarsland, D.; Creese, B.; Mills, R.; Williams, H.; Sweet, R.A. Psychosis in Alzheimer’s Disease. Curr. Neurol. Neurosci. Rep. 2020, 20, 57. [Google Scholar] [CrossRef] [PubMed]
- Creese, B.; Ballard, C.; Jones, E. Cognitive impairment in studies of 5HTTLPR and psychosis in Alzheimer’s disease: A systematic review. Dement. Geriatr. Cogn. Disord. 2013, 35, 155–164. [Google Scholar] [CrossRef]
- Wilcock, G.K.; Ballard, C.G.; Cooper, J.A.; Loft, H. Memantine for agitation/aggression and psychosis in moderately severe to severe Alzheimer’s disease: A pooled analysis of 3 studies. J. Clin. Psychiatry 2008, 69, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Maust, D.T.; Kim, H.M.; Seyfried, L.S.; Chiang, C.; Kavanagh, J.; Schneider, L.S.; Kales, H.C. Antipsychotics, other psychotropics, and the risk of death in patients with dementia: Number needed to harm. JAMA Psychiatry 2015, 72, 438–445. [Google Scholar] [CrossRef] [Green Version]
- De Hert, M.; Detraux, J.; van Winkel, R.; Yu, W.; Correll, C.U. Metabolic and cardiovascular adverse effects associated with antipsychotic drugs. Nat. Rev. Endocrinol. 2011, 8, 114–126. [Google Scholar] [CrossRef]
- Coupland, C.A.C.; Hill, T.; Dening, T.; Morriss, R.; Moore, M.; Hippisley-Cox, J. Anticholinergic Drug Exposure and the Risk of Dementia: A Nested Case-Control Study. JAMA Intern. Med. 2019, 179, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Creese, B.; Da Silva, M.V.; Johar, I.; Ballard, C. The modern role of antipsychotics for the treatment of agitation and psychosis in Alzheimer’s disease. Expert Rev. Neurother. 2018, 18, 461–467. [Google Scholar] [CrossRef]
- Vik-Mo, A.O.; Bencze, J.; Ballard, C.; Hortobagyi, T.; Aarsland, D. Advanced cerebral amyloid angiopathy and small vessel disease are associated with psychosis in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2019, 90, 728–730. [Google Scholar] [CrossRef]
- Muhlbauer, V.; Mohler, R.; Dichter, M.N.; Zuidema, S.U.; Kopke, S.; Luijendijk, H.J. Antipsychotics for agitation and psychosis in people with Alzheimer’s disease and vascular dementia. Cochrane Database Syst. Rev. 2021, 12, CD013304. [Google Scholar] [CrossRef]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Van Hoesen, G.W.; Damasio, A.R.; Barnes, C.L. Alzheimer’s disease: Cell-specific pathology isolates the hippocampal formation. Science 1984, 225, 1168–1170. [Google Scholar] [CrossRef]
- Apostolova, L.G.; Dutton, R.A.; Dinov, I.D.; Hayashi, K.M.; Toga, A.W.; Cummings, J.L.; Thompson, P.M. Conversion of mild cognitive impairment to Alzheimer disease predicted by hippocampal atrophy maps. Arch. Neurol. 2006, 63, 693–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrella, J.R.; Krishnan, S.; Slavin, M.J.; Tran, T.T.; Murty, L.; Doraiswamy, P.M. Mild cognitive impairment: Evaluation with 4-T functional MR imaging. Radiology 2006, 240, 177–186. [Google Scholar] [CrossRef]
- Miller, S.L.; Fenstermacher, E.; Bates, J.; Blacker, D.; Sperling, R.A.; Dickerson, B.C. Hippocampal activation in adults with mild cognitive impairment predicts subsequent cognitive decline. J. Neurol. Neurosurg Psychiatry 2008, 79, 630–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodge, D.J.; Grace, A.A. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J. Neurosci. 2007, 27, 11424–11430. [Google Scholar] [CrossRef] [Green Version]
- Grace, A.A. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: A hypothesis for the etiology of schizophrenia. Neuroscience 1991, 41, 1–24. [Google Scholar] [CrossRef]
- Grace, A.A. Gating of information flow within the limbic system and the pathophysiology of schizophrenia. Brain Res. Brain Res. Rev. 2000, 31, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Maruszak, A.; Thuret, S. Why looking at the whole hippocampus is not enough-a critical role for anteroposterior axis, subfield and activation analyses to enhance predictive value of hippocampal changes for Alzheimer’s disease diagnosis. Front Cell Neurosci. 2014, 8, 95. [Google Scholar] [CrossRef] [Green Version]
- Fanselow, M.S.; Dong, H.W. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron 2010, 65, 7–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuno, F.; Minami, H.; Hattori, H.; Alzheimer’s Disease Neuroimaging, I. Relationship between neuropsychiatric symptoms and Alzheimer’s disease pathology: An in vivo positron emission tomography study. Int. J. Geriatr. Psychiatry 2021, 36, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Geda, Y.E.; Schneider, L.S.; Gitlin, L.N.; Miller, D.S.; Smith, G.S.; Bell, J.; Evans, J.; Lee, M.; Porsteinsson, A.; Lanctot, K.L.; et al. Neuropsychiatric symptoms in Alzheimer’s disease: Past progress and anticipation of the future. Alzheimers Dement 2013, 9, 602–608. [Google Scholar] [CrossRef] [Green Version]
- Krell-Roesch, J.; Vassilaki, M.; Mielke, M.M.; Kremers, W.K.; Lowe, V.J.; Vemuri, P.; Machulda, M.M.; Christianson, T.J.; Syrjanen, J.A.; Stokin, G.B.; et al. Cortical beta-amyloid burden, neuropsychiatric symptoms, and cognitive status: The Mayo Clinic Study of Aging. Transl. Psychiatry 2019, 9, 123. [Google Scholar] [CrossRef] [Green Version]
- Scaricamazza, E.; Colonna, I.; Sancesario, G.M.; Assogna, F.; Orfei, M.D.; Franchini, F.; Sancesario, G.; Mercuri, N.B.; Liguori, C. Neuropsychiatric symptoms differently affect mild cognitive impairment and Alzheimer’s disease patients: A retrospective observational study. Neurol. Sci. 2019, 40, 1377–1382. [Google Scholar] [CrossRef]
- Babulal, G.M.; Ghoshal, N.; Head, D.; Vernon, E.K.; Holtzman, D.M.; Benzinger, T.L.S.; Fagan, A.M.; Morris, J.C.; Roe, C.M. Mood Changes in Cognitively Normal Older Adults are Linked to Alzheimer Disease Biomarker Levels. Am. J. Geriatr. Psychiatry 2016, 24, 1095–1104. [Google Scholar] [CrossRef] [Green Version]
- Puzzo, D.; Privitera, L.; Fa, M.; Staniszewski, A.; Hashimoto, G.; Aziz, F.; Sakurai, M.; Ribe, E.M.; Troy, C.M.; Mercken, M.; et al. Endogenous amyloid-beta is necessary for hippocampal synaptic plasticity and memory. Ann. Neurol. 2011, 69, 819–830. [Google Scholar] [CrossRef] [Green Version]
- Puzzo, D.; Arancio, O. Amyloid-beta peptide: Dr. Jekyll or Mr. Hyde? J. Alzheimers Dis. 2013, 33 (Suppl. S1), S111-20. [Google Scholar]
- Palop, J.J.; Chin, J.; Roberson, E.D.; Wang, J.; Thwin, M.T.; Bien-Ly, N.; Yoo, J.; Ho, K.O.; Yu, G.Q.; Kreitzer, A.; et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 2007, 55, 697–711. [Google Scholar] [CrossRef] [Green Version]
- Andrews-Zwilling, Y.; Bien-Ly, N.; Xu, Q.; Li, G.; Bernardo, A.; Yoon, S.Y.; Zwilling, D.; Yan, T.X.; Chen, L.; Huang, Y. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci. 2010, 30, 13707–13717. [Google Scholar] [CrossRef] [Green Version]
- Verret, L.; Mann, E.O.; Hang, G.B.; Barth, A.M.; Cobos, I.; Ho, K.; Devidze, N.; Masliah, E.; Kreitzer, A.C.; Mody, I.; et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 2012, 149, 708–721. [Google Scholar] [CrossRef] [Green Version]
- Sosulina, L.; Mittag, M.; Geis, H.R.; Hoffmann, K.; Klyubin, I.; Qi, Y.; Steffen, J.; Friedrichs, D.; Henneberg, N.; Fuhrmann, F.; et al. Hippocampal hyperactivity in a rat model of Alzheimer’s disease. J. Neurochem. 2021, 157, 2128–2144. [Google Scholar] [CrossRef]
- Marin, O. Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 2012, 13, 107–120. [Google Scholar] [CrossRef]
- Arai, H.; Emson, P.C.; Mountjoy, C.Q.; Carassco, L.H.; Heizmann, C.W. Loss of parvalbumin-immunoreactive neurones from cortex in Alzheimer-type dementia. Brain Res. 1987, 418, 164–169. [Google Scholar] [CrossRef]
- Brady, D.R.; Mufson, E.J. Parvalbumin-immunoreactive neurons in the hippocampal formation of Alzheimer’s diseased brain. Neuroscience 1997, 80, 1113–1125. [Google Scholar] [CrossRef]
- Mikkonen, M.; Alafuzoff, I.; Tapiola, T.; Soininen, H.; Miettinen, R. Subfield- and layer-specific changes in parvalbumin, calretinin and calbindin-D28K immunoreactivity in the entorhinal cortex in Alzheimer’s disease. Neuroscience 1999, 92, 515–532. [Google Scholar] [CrossRef]
- Kawaguchi, Y. Physiological subgroups of nonpyramidal cells with specific morphological characteristics in layer II/III of rat frontal cortex. J. Neurosci. 1995, 15, 2638–2655. [Google Scholar] [CrossRef] [Green Version]
- Perez, S.M.; Lodge, D.J. Hippocampal interneuron transplants reverse aberrant dopamine system function and behavior in a rodent model of schizophrenia. Mol. Psychiatry 2013, 18, 1193–1198. [Google Scholar] [CrossRef] [Green Version]
- Boley, A.M.; Perez, S.M.; Lodge, D.J. A fundamental role for hippocampal parvalbumin in the dopamine hyperfunction associated with schizophrenia. Schizophr. Res. 2014, 157, 238–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donegan, J.J.; Tyson, J.A.; Branch, S.Y.; Beckstead, M.J.; Anderson, S.A.; Lodge, D.J. Stem cell-derived interneuron transplants as a treatment for schizophrenia: Preclinical validation in a rodent model. Mol. Psychiatry 2017, 22, 1492–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodge, D.J.; Behrens, M.M.; Grace, A.A. A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 2344–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, S.M.; Boley, A.; Lodge, D.J. Region specific knockdown of Parvalbumin or Somatostatin produces neuronal and behavioral deficits consistent with those observed in schizophrenia. Transl. Psychiatry 2019, 9, 264. [Google Scholar] [CrossRef] [Green Version]
- Lecanu, L.; Greeson, J.; Papadopoulos, V. Beta-amyloid and oxidative stress jointly induce neuronal death, amyloid deposits, gliosis, and memory impairment in the rat brain. Pharmacology 2006, 76, 19–33. [Google Scholar] [CrossRef]
- Shah, A.; Lodge, D.J. A loss of hippocampal perineuronal nets produces deficits in dopamine system function: Relevance to the positive symptoms of schizophrenia. Transl. Psychiatry 2013, 3, e215. [Google Scholar] [CrossRef] [Green Version]
- Perez, S.M.; Lodge, D.J. Convergent Inputs from the Hippocampus and Thalamus to the Nucleus Accumbens Regulate Dopamine Neuron Activity. J. NeuroSci. 2018, 38, 10607–10618. [Google Scholar] [CrossRef] [Green Version]
- Perez, S.M.; Aguilar, D.D.; Neary, J.L.; Carless, M.A.; Giuffrida, A.; Lodge, D.J. Schizophrenia-Like Phenotype Inherited by the F2 Generation of a Gestational Disruption Model of Schizophrenia. Neuropsychopharmacology 2016, 41, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Perez, S.M.; Chen, L.; Lodge, D.J. Alterations in dopamine system function across the estrous cycle of the MAM rodent model of schizophrenia. Psychoneuroendocrinology 2014, 47, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, D.D.; Chen, L.; Lodge, D.J. Increasing Endocannabinoid Levels in the Ventral Pallidum Restore Aberrant Dopamine Neuron Activity in the Subchronic PCP Rodent Model of Schizophrenia. Int. J. Neuropsychopharmacol. 2014. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; He, J.; Zhang, R.; Zhu, S.; Wang, J.; Kong, L.; Tan, Q.; Li, X.M. Sensorimotor gating and memory deficits in an APP/PS1 double transgenic mouse model of Alzheimer’s disease. Behav. Brain Res. 2012, 233, 237–243. [Google Scholar] [CrossRef]
- Jafari, Z.; Kolb, B.E.; Mohajerani, M.H. Prepulse inhibition of the acoustic startle reflex and P50 gating in aging and alzheimer’s disease. Ageing Res. Rev. 2020, 59, 101028. [Google Scholar] [CrossRef]
- Braff, D.L. Information processing and attention dysfunctions in schizophrenia. Schizophrenia Bulletin 1993, 19, 233–259. [Google Scholar] [CrossRef] [Green Version]
- Braff, D.L.; Geyer, M.A. Sensorimotor gating and schizophrenia. Human and animal model studies. Arch. Gen. Psychiatry 1990, 47, 181–188. [Google Scholar] [CrossRef]
- Braff, D.L.; Grillon, C.; Geyer, M.A. Gating and habituation of the startle reflex in schizophrenic patients. Arch. Gen. Psychiatry 1992, 49, 206–215. [Google Scholar] [CrossRef]
- Koppel, J.; Jimenez, H.; Azose, M.; D’Abramo, C.; Acker, C.; Buthorn, J.; Greenwald, B.S.; Lewis, J.; Lesser, M.; Liu, Z.; et al. Pathogenic tau species drive a psychosis-like phenotype in a mouse model of Alzheimer’s disease. Behav. Brain Res. 2014, 275, 27–33. [Google Scholar] [CrossRef]
- Perez, S.M.; Donegan, J.J.; Lodge, D.J. Effect of estrous cycle on schizophrenia-like behaviors in MAM exposed rats. Behav. Brain Res. 2019, 362, 258–265. [Google Scholar] [CrossRef]
- Ohno, M.; Sametsky, E.A.; Younkin, L.H.; Oakley, H.; Younkin, S.G.; Citron, M.; Vassar, R.; Disterhoft, J.F. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer’s disease. Neuron 2004, 41, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Kosel, F.; Munoz, P.T.; Yang, J.R.; Wong, A.A.; Franklin, T.B. Age-related changes in social behaviours in the 5xFAD mouse model of Alzheimer’s disease. Behav. Brain Res. 2019, 362, 160–172. [Google Scholar] [CrossRef]
- Chen, L.; Perez, S.M.; Lodge, D.J. An augmented dopamine system function is present prior to puberty in the methylazoxymethanol acetate rodent model of schizophrenia. Dev. Neurobiol. 2014, 74, 907–917. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.J.; Reynolds, G.P. A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophr. Res. 2002, 55, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Konradi, C.; Yang, C.K.; Zimmerman, E.I.; Lohmann, K.M.; Gresch, P.; Pantazopoulos, H.; Berretta, S.; Heckers, S. Hippocampal interneurons are abnormal in schizophrenia. Schizophr. Res. 2011, 131, 165–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, J.; Tabira, T.; Sano, M.; Nakayama, H.; Tateishi, J. Parvalbumin-immunoreactive neurons in the human central nervous system are decreased in Alzheimer’s disease. Acta Neuropathol. 1991, 81, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Tong, G.; Ballard, C. Treatment Combinations for Alzheimer’s Disease: Current and Future Pharmacotherapy Options. J. Alzheimers Dis. 2019, 67, 779–794. [Google Scholar] [CrossRef] [Green Version]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Gilley, D.W.; Whalen, M.E.; Wilson, R.S.; Bennett, D.A. Hallucinations and associated factors in Alzheimer’s disease. J. Neuropsychiatry Clin. Neurosci. 1991, 3, 371–376. [Google Scholar]
- Ballard, C.; Howard, R. Neuroleptic drugs in dementia: Benefits and harm. Nat. Rev. Neurosci. 2006, 7, 492–500. [Google Scholar] [CrossRef]
- Kales, H.C.; Valenstein, M.; Kim, H.M.; McCarthy, J.F.; Ganoczy, D.; Cunningham, F.; Blow, F.C. Mortality risk in patients with dementia treated with antipsychotics versus other psychiatric medications. Am. J. Psychiatry 2007, 164, 1568–1576; quiz 1623. [Google Scholar] [CrossRef]
- Kales, H.C.; Kim, H.M.; Zivin, K.; Valenstein, M.; Seyfried, L.S.; Chiang, C.; Cunningham, F.; Schneider, L.S.; Blow, F.C. Risk of mortality among individual antipsychotics in patients with dementia. Am. J. Psychiatry 2012, 169, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Abraham, R.; Moskvina, V.; Sims, R.; Hollingworth, P.; Morgan, A.; Georgieva, L.; Dowzell, K.; Cichon, S.; Hillmer, A.M.; O’Donovan, M.C.; et al. A genome-wide association study for late-onset Alzheimer’s disease using DNA pooling. BMC Med. Genom. 2008, 1, 44. [Google Scholar] [CrossRef]
- Piaceri, I.; Nacmias, B.; Sorbi, S. Genetics of familial and sporadic Alzheimer’s disease. Front. Biosci. 2013, 5, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Grupe, A.; Abraham, R.; Li, Y.; Rowland, C.; Hollingworth, P.; Morgan, A.; Jehu, L.; Segurado, R.; Stone, D.; Schadt, E.; et al. Evidence for novel susceptibility genes for late-onset Alzheimer’s disease from a genome-wide association study of putative functional variants. Hum. Mol. Genet. 2007, 16, 865–873. [Google Scholar] [CrossRef]
- Grant, W.B.; Campbell, A.; Itzhaki, R.F.; Savory, J. The significance of environmental factors in the etiology of Alzheimer’s disease. J. Alzheimers Dis. 2002, 4, 179–189. [Google Scholar] [CrossRef]
- Li, P.; Marshall, L.; Oh, G.; Jakubowski, J.L.; Groot, D.; He, Y.; Wang, T.; Petronis, A.; Labrie, V. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nat. Commun. 2019, 10, 2246. [Google Scholar] [CrossRef] [Green Version]
- Seeman, P.; Lee, T. Antipsychotic drugs: Direct correlation between clinical potency and presynaptic action on dopamine neurons. Science 1975, 188, 1217–1219. [Google Scholar] [CrossRef]
- Howes, O.D.; Kambeitz, J.; Kim, E.; Stahl, D.; Slifstein, M.; Abi-Dargham, A.; Kapur, S. The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch. Gen. Psychiatry 2012, 69, 776–786. [Google Scholar] [CrossRef] [Green Version]
- Abi-Dargham, A. Do we still believe in the dopamine hypothesis? New data bring new evidence. Int. J. Neuropsychopharmacol. 2004, 7 (Suppl. S1), S1–S5. [Google Scholar] [CrossRef]
- Laruelle, M.; Abi-Dargham, A. Dopamine as the wind of the psychotic fire: New evidence from brain imaging studies. J. Psychopharmacol. 1999, 13, 358–371. [Google Scholar] [CrossRef]
- Laruelle, M.; Abi-Dargham, A.; Gil, R.; Kegeles, L.; Innis, R. Increased dopamine transmission in schizophrenia: Relationship to illness phases. Biol. Psychiatry 1999, 46, 56–72. [Google Scholar] [CrossRef]
- Perez, S.M.; Carreno, F.R.; Frazer, A.; Lodge, D.J. Vagal nerve stimulation reverses aberrant dopamine system function in the methylazoxymethanol acetate rodent model of schizophrenia. J. Neurosci. 2014, 34, 9261–9267. [Google Scholar] [CrossRef] [Green Version]
- Perez, S.M.; Shah, A.; Asher, A.; Lodge, D.J. Hippocampal deep brain stimulation reverses physiological and behavioural deficits in a rodent model of schizophrenia. Int. J. Neuropsychopharmacol. 2013, 16, 1331–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, S.M.; McCoy, A.M.; Prevot, T.D.; Mian, M.Y.; Carreno, F.R.; Frazer, A.; Cook, J.M.; Sibille, E.; Lodge, D.J. Hippocampal α5-GABAA receptors modulate dopamine neuron activity in the rat ventral tegmental area. Biol. Psychiatry Glob. Open Sci. 2022, 3, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Gill, K.M.; Lodge, D.J.; Cook, J.M.; Aras, S.; Grace, A.A. A novel alpha5GABA(A)R-positive allosteric modulator reverses hyperactivation of the dopamine system in the MAM model of schizophrenia. Neuropsychopharmacology 2011, 36, 1903–1911. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.M.; Lodge, D.J. New approaches to the management of schizophrenia: Focus on aberrant hippocampal drive of dopamine pathways. Drug Des. Dev. Ther. 2014, 8, 887–896. [Google Scholar] [PubMed] [Green Version]
- Fries, P. Rhythms for Cognition: Communication through Coherence. Neuron 2015, 88, 220–235. [Google Scholar] [CrossRef] [Green Version]
- Buzsaki, G. Theta oscillations in the hippocampus. Neuron 2002, 33, 325–340. [Google Scholar] [CrossRef] [Green Version]
- Goutagny, R.; Krantic, S. Hippocampal oscillatory activity in Alzheimer’s disease: Toward the identification of early biomarkers? Aging Dis. 2013, 4, 134–140. [Google Scholar]
- Bondi, M.W.; Houston, W.S.; Eyler, L.T.; Brown, G.G. fMRI evidence of compensatory mechanisms in older adults at genetic risk for Alzheimer disease. Neurology 2005, 64, 501–508. [Google Scholar] [CrossRef] [Green Version]
- Quiroz, Y.T.; Budson, A.E.; Celone, K.; Ruiz, A.; Newmark, R.; Castrillon, G.; Lopera, F.; Stern, C.E. Hippocampal hyperactivation in presymptomatic familial Alzheimer’s disease. Ann. Neurol. 2010, 68, 865–875. [Google Scholar] [CrossRef] [Green Version]
- Bookheimer, S.Y.; Strojwas, M.H.; Cohen, M.S.; Saunders, A.M.; Pericak-Vance, M.A.; Mazziotta, J.C.; Small, G.W. Patterns of brain activation in people at risk for Alzheimer’s disease. N. Engl. J. Med. 2000, 343, 450–456. [Google Scholar] [CrossRef] [Green Version]
- Schobel, S.A.; Lewandowski, N.M.; Corcoran, C.M.; Moore, H.; Brown, T.; Malaspina, D.; Small, S.A. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch. Gen. Psychiatry 2009, 66, 938–946. [Google Scholar] [CrossRef] [Green Version]
- Heckers, S.; Rauch, S.L.; Goff, D.; Savage, C.R.; Schacter, D.L.; Fischman, A.J.; Alpert, N.M. Impaired recruitment of the hippocampus during conscious recollection in schizophrenia. Nat. Neurosci. 1998, 1, 318–323. [Google Scholar] [CrossRef]
- Malaspina, D.; Harkavy-Friedman, J.; Corcoran, C.; Mujica-Parodi, L.; Printz, D.; Gorman, J.M.; Van Heertum, R. Resting neural activity distinguishes subgroups of schizophrenia patients. Biol. Psychiatry 2004, 56, 931–937. [Google Scholar] [CrossRef] [Green Version]
- Lewis, D.A.; Hashimoto, T.; Volk, D.W. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 2005, 6, 312–324. [Google Scholar] [CrossRef]
- Inaguma, Y.; Shinohara, H.; Inagaki, T.; Kato, K. Immunoreactive parvalbumin concentrations in parahippocampal gyrus decrease in patients with Alzheimer’s disease. J. Neurol. Sci. 1992, 110, 57–61. [Google Scholar] [CrossRef]
- Behrens, M.M.; Ali, S.S.; Dao, D.N.; Lucero, J.; Shekhtman, G.; Quick, K.L.; Dugan, L.L. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science 2007, 318, 1645–1647. [Google Scholar] [CrossRef] [Green Version]
- Francois, J.; Ferrandon, A.; Koning, E.; Angst, M.J.; Sandner, G.; Nehlig, A. Selective reorganization of GABAergic transmission in neonatal ventral hippocampal-lesioned rats. Int. J. Neuropsychopharmacol. Off. Sci. J. Coll. Int. Neuropsychopharmacol. 2009, 12, 1097–1110. [Google Scholar] [CrossRef] [Green Version]
- Harte, M.K.; Powell, S.B.; Swerdlow, N.R.; Geyer, M.A.; Reynolds, G.P. Deficits in parvalbumin and calbindin immunoreactive cells in the hippocampus of isolation reared rats. J. Neural Transm. 2007, 114, 893–898. [Google Scholar] [CrossRef]
- Ali, F.; Baringer, S.L.; Neal, A.; Choi, E.Y.; Kwan, A.C. Parvalbumin-Positive Neuron Loss and Amyloid-beta Deposits in the Frontal Cortex of Alzheimer’s Disease-Related Mice. J. Alzheimers Dis. 2019, 72, 1323–1339. [Google Scholar] [CrossRef]
- Zallo, F.; Gardenal, E.; Verkhratsky, A.; Rodriguez, J.J. Loss of calretinin and parvalbumin positive interneurones in the hippocampal CA1 of aged Alzheimer’s disease mice. Neurosci. Lett. 2018, 681, 19–25. [Google Scholar] [CrossRef]
- Floresco, S.B.; Todd, C.L.; Grace, A.A. Glutamatergic afferents from the hippocampus to the nucleus accumbens regulate activity of ventral tegmental area dopamine neurons. J. Neurosci. 2001, 21, 4915–4922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floresco, S.B.; West, A.R.; Ash, B.; Moore, H.; Grace, A.A. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat. Neurosci. 2003, 6, 968–973. [Google Scholar] [CrossRef] [PubMed]
- Grace, A.A.; Floresco, S.B.; Goto, Y.; Lodge, D.J. Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci. 2007, 30, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Grace, A.A.; Bunney, B.S. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons--1. Identification and characterization. Neuroscience 1983, 10, 301–315. [Google Scholar] [CrossRef]
- Grace, A.A.; Bunney, B.S. The control of firing pattern in nigral dopamine neurons: Burst firing. J. Neurosci. 1984, 4, 2877–2890. [Google Scholar] [CrossRef]
- Grace, A.A.; Bunney, B.S. The control of firing pattern in nigral dopamine neurons: Single spike firing. J. Neurosci. 1984, 4, 2866–2876. [Google Scholar] [CrossRef] [Green Version]
- Grace, A.A.; Bunney, B.S. Opposing effects of striatonigral feedback pathways on midbrain dopamine cell activity. Brain Res. 1985, 333, 271–284. [Google Scholar] [CrossRef]
- Lodge, D.J.; Grace, A.A. The hippocampus modulates dopamine neuron responsivity by regulating the intensity of phasic neuron activation. Neuropsychopharmacology 2006, 31, 1356–1361. [Google Scholar] [CrossRef] [Green Version]
- Lodge, D.J.; Grace, A.A. Hippocampal dysfunction and disruption of dopamine system regulation in an animal model of schizophrenia. Neurotox Res. 2008, 14, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Le Pen, G.; Gourevitch, R.; Hazane, F.; Hoareau, C.; Jay, T.M.; Krebs, M.O. Peri-pubertal maturation after developmental disturbance: A model for psychosis onset in the rat. Neuroscience 2006, 143, 395–405. [Google Scholar] [CrossRef]
- Moore, H.; Jentsch, J.D.; Ghajarnia, M.; Geyer, M.A.; Grace, A.A. A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: Implications for the neuropathology of schizophrenia. Biol. Psychiatry 2006, 60, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Flagstad, P.; Mork, A.; Glenthoj, B.Y.; van Beek, J.; Michael-Titus, A.T.; Didriksen, M. Disruption of neurogenesis on gestational day 17 in the rat causes behavioral changes relevant to positive and negative schizophrenia symptoms and alters amphetamine-induced dopamine release in nucleus accumbens. Neuropsychopharmacology 2004, 29, 2052–2064. [Google Scholar] [CrossRef]
- White, I.M.; Whitaker, C.; White, W. Amphetamine-induced hyperlocomotion in rats: Hippocampal modulation of the nucleus accumbens. Hippocampus 2006, 16, 596–603. [Google Scholar] [CrossRef]
- Bjorklund, L.M.; Sanchez-Pernaute, R.; Chung, S.; Andersson, T.; Chen, I.Y.; McNaught, K.S.; Brownell, A.L.; Jenkins, B.G.; Wahlestedt, C.; Kim, K.S.; et al. Embryonic stem cells develop into functional dopaminergic neurons after transplantation in a Parkinson rat model. Proc. Natl. Acad. Sci. USA 2002, 99, 2344–2349. [Google Scholar] [CrossRef] [Green Version]
- Hyland, B.I.; Reynolds, J.N.; Hay, J.; Perk, C.G.; Miller, R. Firing modes of midbrain dopamine cells in the freely moving rat. Neuroscience 2002, 114, 475–492. [Google Scholar] [CrossRef]
- Carreno, F.R.; Collins, G.T.; Frazer, A.; Lodge, D.J. Selective Pharmacological Augmentation of Hippocampal Activity Produces a Sustained Antidepressant-Like Response without Abuse-Related or Psychotomimetic Effects. Int. J. Neuropsychopharmacol. 2017, 20, 504–509. [Google Scholar] [CrossRef] [Green Version]
- Rohleder, C.; Wiedermann, D.; Neumaier, B.; Drzezga, A.; Timmermann, L.; Graf, R.; Leweke, F.M.; Endepols, H. The Functional Networks of Prepulse Inhibition: Neuronal Connectivity Analysis Based on FDG-PET in Awake and Unrestrained Rats. Front. Behav. Neurosci. 2016, 10, 148. [Google Scholar] [CrossRef] [Green Version]
- Cano, J.C.; Huang, W.; Fenelon, K. The amygdala modulates prepulse inhibition of the auditory startle reflex through excitatory inputs to the caudal pontine reticular nucleus. BMC Biol. 2021, 19, 116. [Google Scholar] [CrossRef]
- Hammer, T.B.; Oranje, B.; Skimminge, A.; Aggernaes, B.; Ebdrup, B.H.; Glenthoj, B.; Baare, W. Structural brain correlates of sensorimotor gating in antipsychotic-naive men with first-episode schizophrenia. J. Psychiatry Neurosci. 2013, 38, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Tanimizu, T.; Kenney, J.W.; Okano, E.; Kadoma, K.; Frankland, P.W.; Kida, S. Functional Connectivity of Multiple Brain Regions Required for the Consolidation of Social Recognition Memory. J. Neurosci. 2017, 37, 4103–4116. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Jiang, M.; Liu, X.; Sun, Y.; Yang, L.; Yang, Q.; Bai, Z. Neural Circuits for Social Interactions: From Microcircuits to Input-Output Circuits. Front. Neural Circuits 2021, 15, 768294. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.M.; Prevot, T.D.; Mian, M.Y.; Cook, J.M.; Frazer, A.; Sibille, E.L.; Carreno, F.R.; Lodge, D.J. Positive allosteric modulation of alpha5-GABAA receptors reverses stress-induced alterations in dopamine system function and prepulse inhibition of startle. Int. J. Neuropsychopharmacol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Prevot, T.D.; Li, G.; Vidojevic, A.; Misquitta, K.A.; Fee, C.; Santrac, A.; Knutson, D.E.; Stephen, M.R.; Kodali, R.; Zahn, N.M.; et al. Novel Benzodiazepine-Like Ligands with Various Anxiolytic, Antidepressant, or Pro-Cognitive Profiles. Mol. Neuropsychiatry 2019, 5, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Prevot, T.D.; Sumitomo, A.; Tomoda, T.; Knutson, D.E.; Li, G.; Mondal, P.; Banasr, M.; Cook, J.M.; Sibille, E. Reversal of Age-Related Neuronal Atrophy by alpha5-GABAA Receptor Positive Allosteric Modulation. Cereb. Cortex 2021, 31, 1395–1408. [Google Scholar] [CrossRef]
- Trulson, M.E.; Preussler, D.W. Dopamine-containing ventral tegmental area neurons in freely moving cats: Activity during the sleep-waking cycle and effects of stress. Exp. Neurol. 1984, 83, 367–377. [Google Scholar] [CrossRef]
- Ungless, M.A.; Grace, A.A. Are you or aren’t you? Challenges associated with physiologically identifying dopamine neurons. Trends Neurosci. 2012, 35, 422–430. [Google Scholar] [CrossRef] [Green Version]
- Cecchi, C. Emx2: A gene responsible for cortical development, regionalization and area specification. Gene 2002, 291, 1–9. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 2nd ed.; Academic Press Australia: Sydney, Austrilia, 1986. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez, S.M.; Boley, A.M.; McCoy, A.M.; Lodge, D.J. Aberrant Dopamine System Function in the Ferrous Amyloid Buthionine (FAB) Rat Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 7196. https://doi.org/10.3390/ijms24087196
Perez SM, Boley AM, McCoy AM, Lodge DJ. Aberrant Dopamine System Function in the Ferrous Amyloid Buthionine (FAB) Rat Model of Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(8):7196. https://doi.org/10.3390/ijms24087196
Chicago/Turabian StylePerez, Stephanie M., Angela M. Boley, Alexandra M. McCoy, and Daniel J. Lodge. 2023. "Aberrant Dopamine System Function in the Ferrous Amyloid Buthionine (FAB) Rat Model of Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 8: 7196. https://doi.org/10.3390/ijms24087196