
To Target or Not to Target Schistosoma mansoni Cyclic Nucleotide Phosphodiesterase 4A?
 , ,
, ,  , ,
, ,  , , add
Show full author list
, , add
Show full author list
Abstract
:1. Introduction
2. Results and Discussion
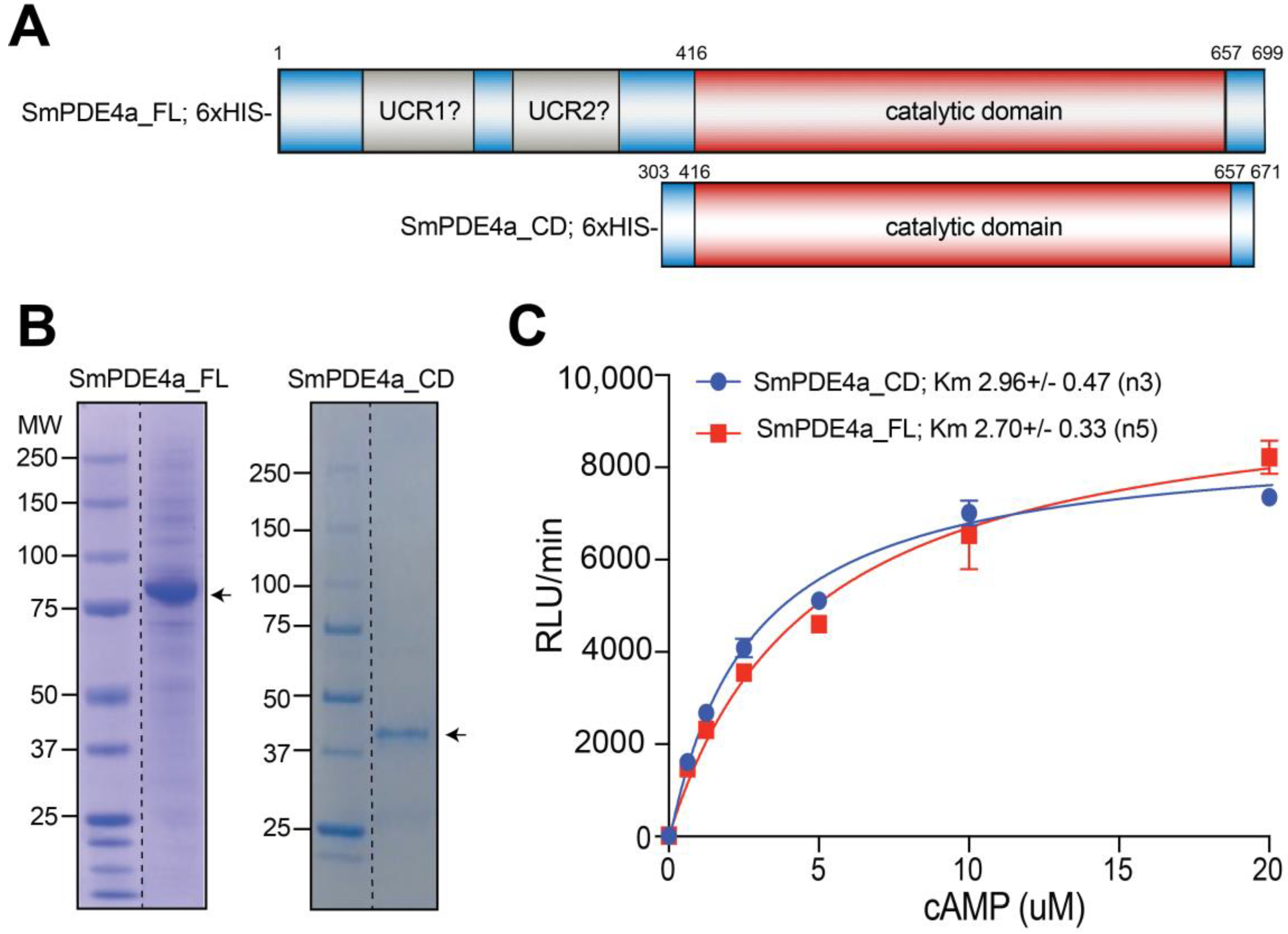
2.1. Protein Purification and Biochemical Characterization
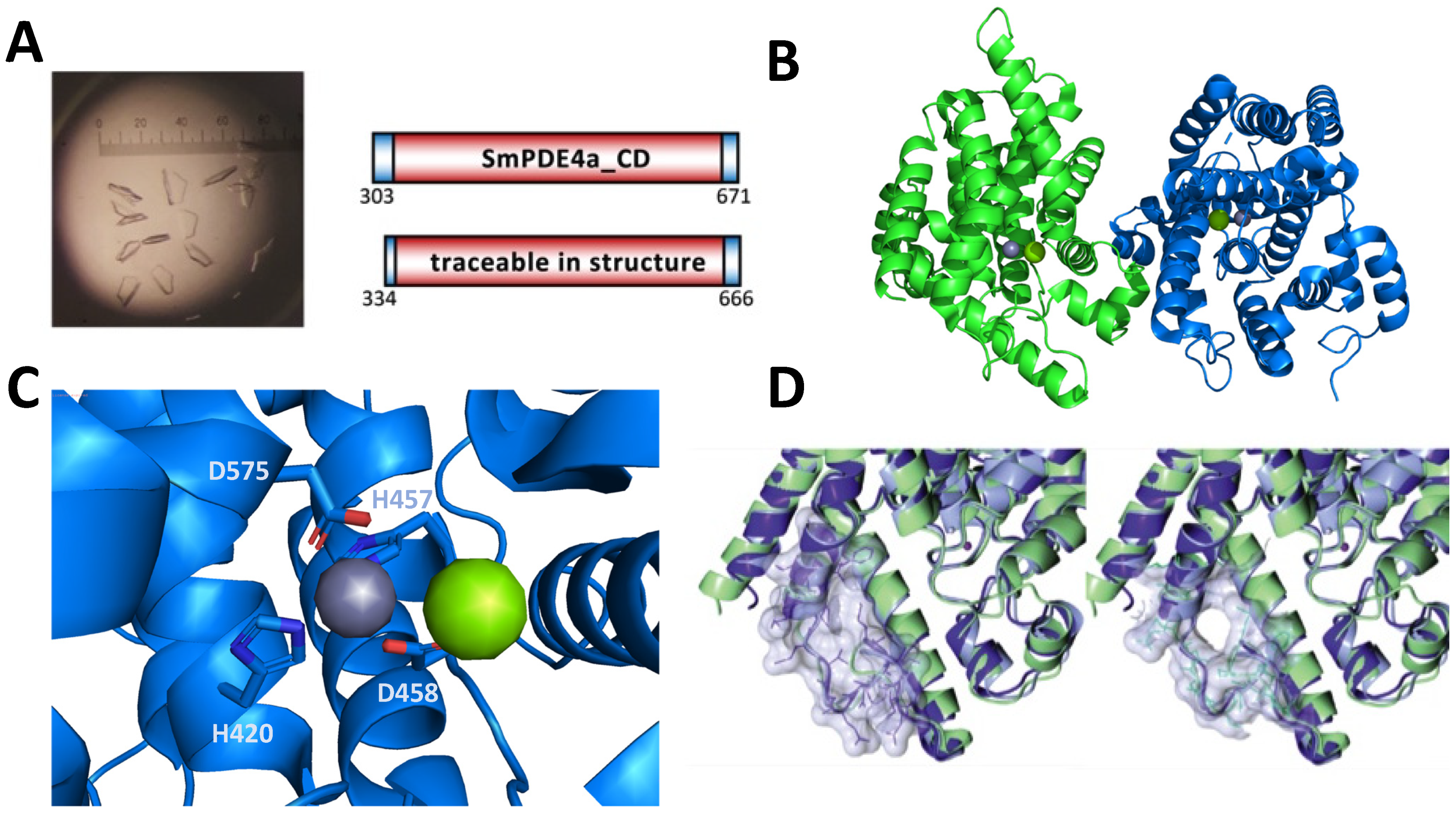
2.2. Crystallization and X-Ray Structure of SmPDE4A Catalytic Domain
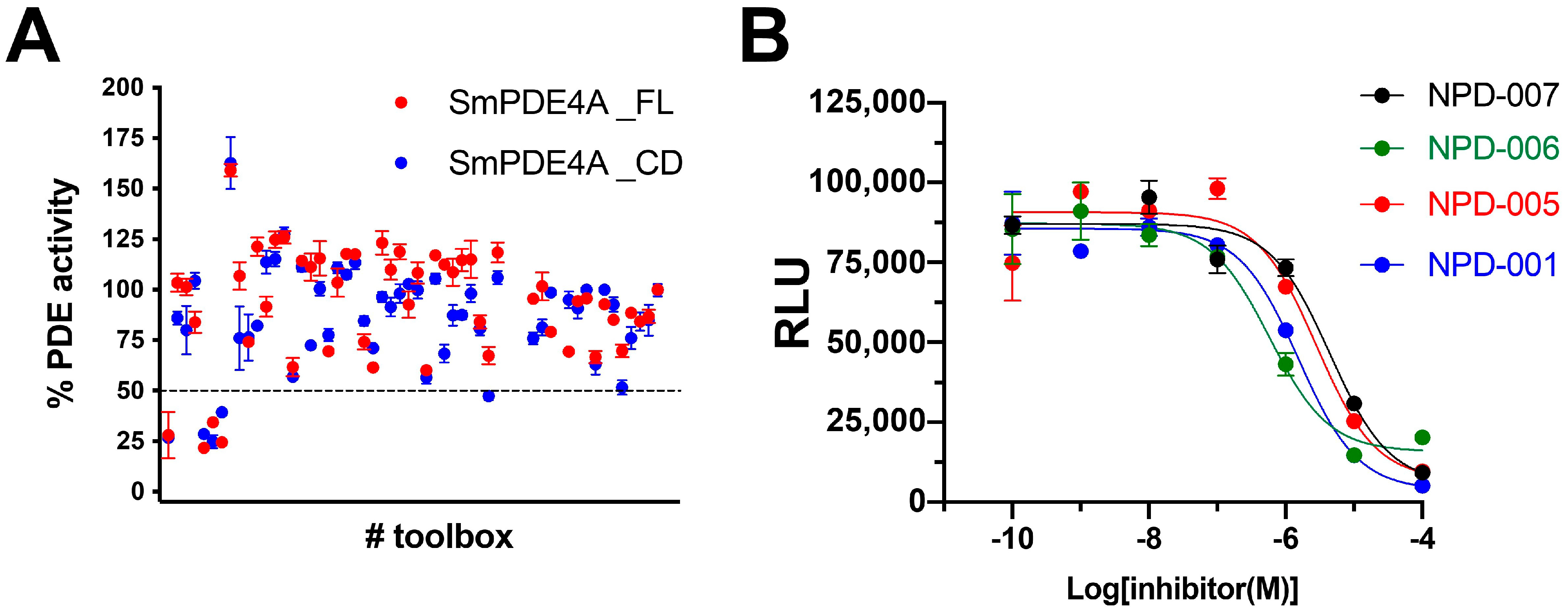
2.3. Screening of a PDE-Inhibitor Toolbox
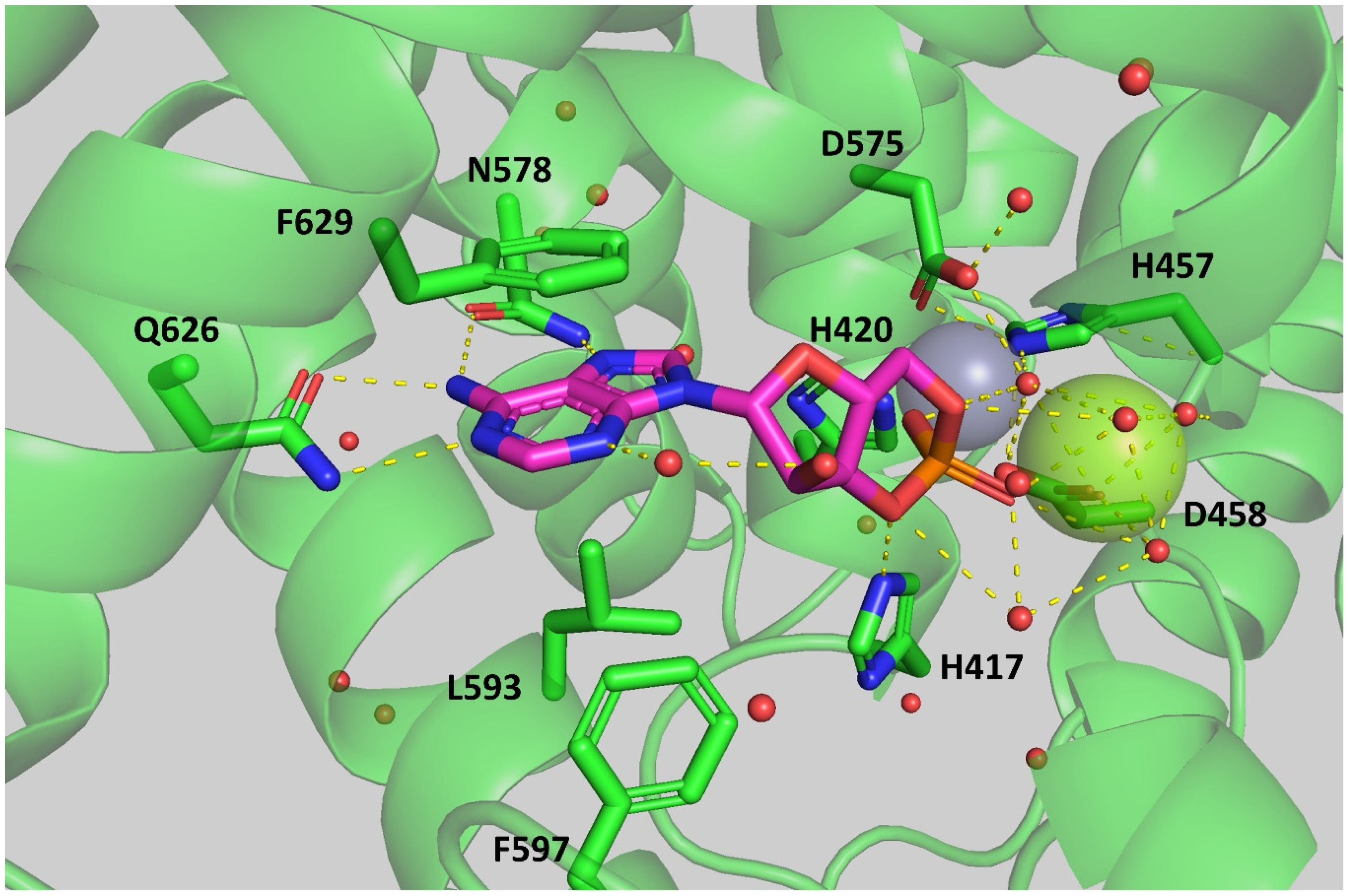
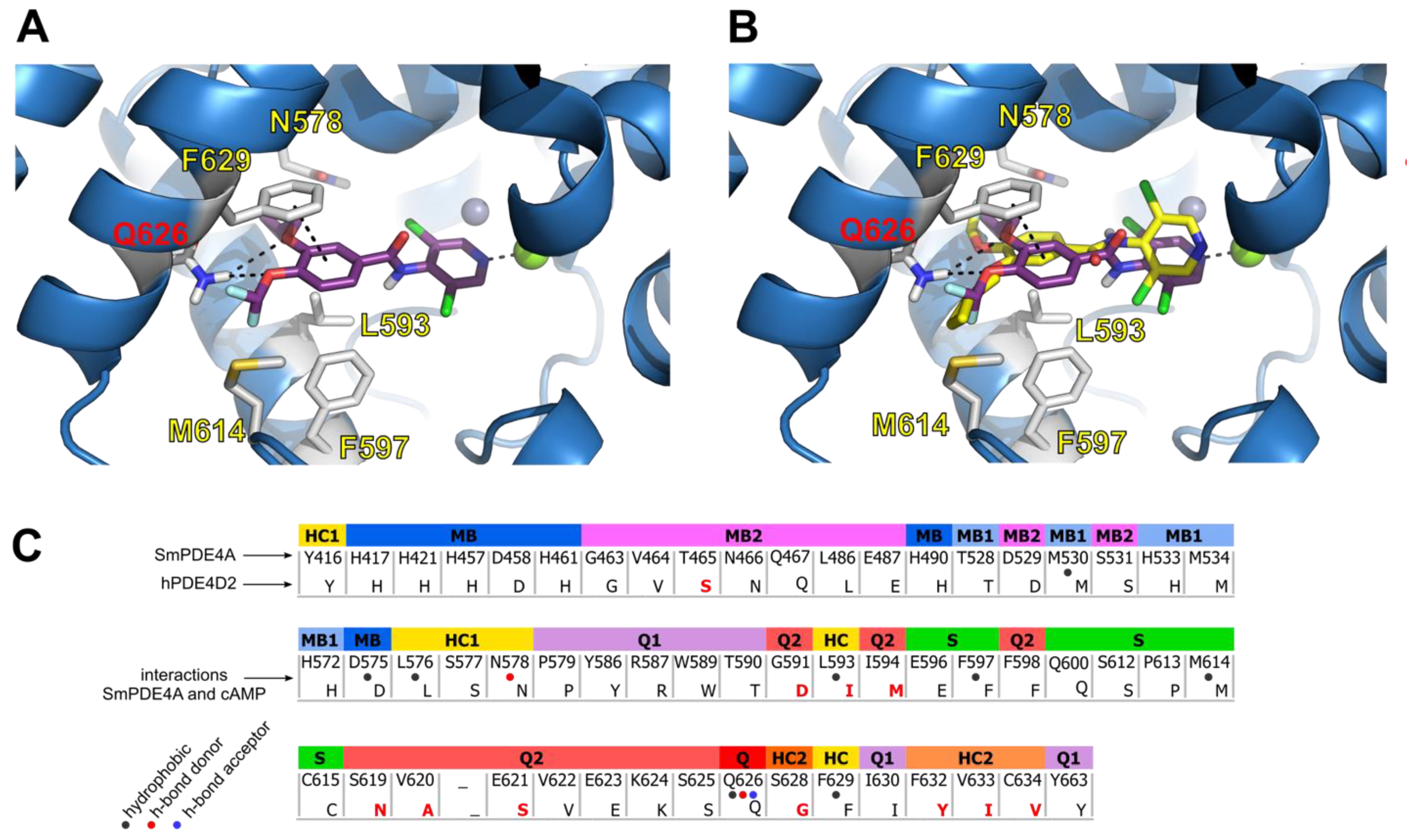
2.4. Docking of Roflumilast in the SmPDE4A Active Site
2.5. Evaluation of Roflumilast Analogues as SmPDE4A Inhibitors
2.6. Effect of Identified SmPDE4A Inhibitors on S. mansoni Survival and Ovipositioning
3. Materials and Methods
3.1. PDE4NPD Toolbox Generation
3.2. Expression and Purification of SmPDE4A Full-Length (FL) and SmPDE4A Catalytic Domain (CD)
3.3. Measurement of Phosphodiesterase Activity
3.4. PDE Inhibitor Screening
3.5. SmPDE4A Protein Crystallography
3.6. Docking of Roflumilast in the SmPDE4A Crystal Structure
3.7. Determination of Anti-Schistosomal Activity of SmPDE4A Inhibitors
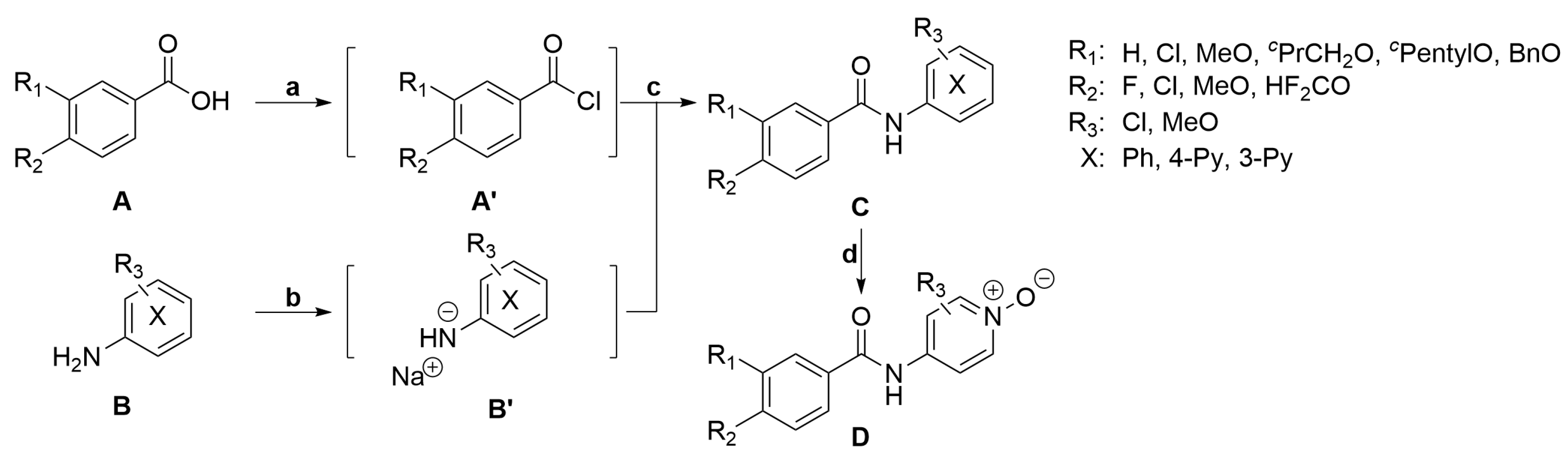
3.8. Synthesis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kyu, H.H.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, Regional, and National Disability-Adjusted Life-Years (DALYs) for 359 Diseases and Injuries and Healthy Life Expectancy (HALE) for 195 Countries and Territories, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1859–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, C.J.L.; Vos, T.; Lozano, R.; Naghavi, M.; Flaxman, A.D.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; et al. Disability-Adjusted Life Years (DALYs) for 291 Diseases and Injuries in 21 Regions, 1990–2010: A Systematic Analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2197–2223. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.S.; Biedermann, P.; Ekpo, U.F.; Garba, A.; Mathieu, E.; Midzi, N.; Mwinzi, P.; N’Goran, E.K.; Raso, G.; Assaré, R.K.; et al. Spatial Distribution of Schistosomiasis and Treatment Needs in Sub-Saharan Africa: A Systematic Review and Geostatistical Analysis. Lancet Infect. Dis. 2015, 15, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Cioli, D.; Pica-Mattoccia, L.; Basso, A.; Guidi, A. Schistosomiasis Control: Praziquantel Forever? Mol. Biochem. Parasitol. 2014, 195, 23–29. [Google Scholar] [CrossRef]
- Da Paixão Siqueira, L.; Fontes, D.A.F.; Aguilera, C.S.B.; Timóteo, T.R.R.; Ângelos, M.A.; Silva, L.C.P.B.B.; de Melo, C.G.; Rolim, L.A.; da Silva, R.M.F.; Neto, P.J.R. Schistosomiasis: Drugs Used and Treatment Strategies. Acta Trop. 2017, 176, 179–187. [Google Scholar] [CrossRef]
- Vale, N.; Gouveia, M.J.; Rinaldi, G.; Brindley, P.J.; Gärtner, F.; da Costa, J.M.C. Praziquantel for Schistosomiasis: Single-Drug Metabolism Revisited, Mode of Action, and Resistance. Antimicrob. Agents Chemother. 2017, 61, e02582-16. [Google Scholar] [CrossRef] [Green Version]
- Botros, S.; Sayed, H.; Amer, N.; El-Ghannam, M.; Bennett, J.L.; Day, T.A. Current Status of Sensitivity to Praziquantel in a Focus of Potential Drug Resistance in Egypt. Int. J. Parasitol. 2005, 35, 787–791. [Google Scholar] [CrossRef]
- Daniel, P.B.; Walker, W.H.; Habener, J.F. Cyclic amp signaling and gene regulation. Annu. Rev. Nutr. 1998, 18, 353–383. [Google Scholar] [CrossRef]
- Pinner, N.A.; Hamilton, L.A.; Hughes, A. Roflumilast: A Phosphodiesterase-4 Inhibitor for the Treatment of Severe Chronic Obstructive Pulmonary Disease. Clin. Ther. 2012, 34, 56–66. [Google Scholar] [CrossRef]
- Calverley, P.M.; Rabe, K.F.; Goehring, U.M.; Kristiansen, S.; Fabbri, L.M.; Martinez, F.J. Roflumilast in Symptomatic Chronic Obstructive Pulmonary Disease: Two Randomised Clinical Trials. Lancet 2009, 374, 685–694. [Google Scholar] [CrossRef]
- Packer, M.; Carver, J.R.; Rodeheffer, R.J.; Ivanhoe, R.J.; DiBianco, R.; Zeldis, S.M.; Hendrix, G.H.; Bommer, W.J.; Elkayam, U.; Kukin, M.L.; et al. Effect of Oral Milrinone on Mortality in Severe Chronic Heart Failure. N. Engl. J. Med. 1991, 325, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Boolell, M.; Allen, M.J.; Ballard, S.A.; Gepi-Attee, S.; Muirhead, G.J.; Naylor, A.M.; Osterloh, I.H.; Gingell, C. Sildenafil: An Orally Active Type 5 Cyclic GMP-Specific Phosphodiesterase Inhibitor for the Treatment of Penile Erectile Dysfunction. Int. J. Impot. Res. 1996, 8, 47–52. [Google Scholar] [PubMed]
- Giembycz, M.A.; Field, S.K. Roflumilast: First Phosphodiesterase 4 Inhibitor Approved for Treatment of COPD. Drug Des. Dev. Ther. 2010, 4, 147–158. [Google Scholar]
- Seebeck, T.; Sterk, G.J.; Ke, H. Phosphodiesterase Inhibitors as a New Generation of Antiprotozoan Drugs: Exploiting the Benefit of Enzymes That Are Highly Conserved between Host and Parasite. Future Med. Chem. 2011, 3, 1289–1306. [Google Scholar] [CrossRef] [Green Version]
- Oberholzer, M.; Marti, G.; Baresic, M.; Kunz, S.; Hemphill, A.; Seebeck, T. The Trypanosoma Brucei CAMP Phosphodiesterases TbrPDEBl and TbrPDEB2: Flagellar Enzymes That Are Essential for Parasite Virulence. FASEB J. 2007, 21, 720–731. [Google Scholar] [CrossRef]
- Orrling, K.M.; Jansen, C.; Vu, X.L.; Balmer, V.; Bregy, P.; Shanmugham, A.; England, P.; Bailey, D.; Cos, P.; Maes, L.; et al. Catechol Pyrazolinones as Trypanocidals: Fragment-Based Design, Synthesis, and Pharmacological Evaluation of Nanomolar Inhibitors of Trypanosomal Phosphodiesterase B1. J. Med. Chem. 2012, 55, 8745–8756. [Google Scholar] [CrossRef]
- Wang, C.; Ashton, T.D.; Gustafson, A.; Bland, N.D.; Ochiana, S.O.; Campbell, R.K.; Pollastri, M.P. Synthesis and Evaluation of Human Phosphodiesterases (PDE) 5 Inhibitor Analogs as Trypanosomal PDE Inhibitors. Part 1. Sildenafil Analogs. Bioorg. Med. Chem. Lett. 2012, 22, 2579–2581. [Google Scholar] [CrossRef] [Green Version]
- Blaazer, A.R.; Singh, A.K.; De Heuvel, E.; Edink, E.; Orrling, K.M.; Veerman, J.J.N.; Van Den Bergh, T.; Jansen, C.; Balasubramaniam, E.; Mooij, W.J.; et al. Targeting a Subpocket in Trypanosoma Brucei Phosphodiesterase B1 (TbrPDEB1) Enables the Structure-Based Discovery of Selective Inhibitors with Trypanocidal Activity. J. Med. Chem. 2018, 61, 3870–3888. [Google Scholar] [CrossRef] [Green Version]
- De Heuvel, E.; Singh, A.K.; Boronat, P.; Kooistra, A.J.; van der Meer, T.; Sadek, P.; Blaazer, A.R.; Shaner, N.C.; Bindels, D.S.; Caljon, G.; et al. Alkynamide Phthalazinones as a New Class of TbrPDEB1 Inhibitors (Part 2). Bioorg. Med. Chem. 2019, 27, 4013–4029. [Google Scholar] [CrossRef]
- De Heuvel, E.; Singh, A.K.; Edink, E.; van der Meer, T.; van der Woude, M.; Sadek, P.; Krell-Jørgensen, M.P.; van den Bergh, T.; Veerman, J.; Caljon, G.; et al. Alkynamide Phthalazinones as a New Class of TbrPDEB1 Inhibitors. Bioorg. Med. Chem. 2019, 27, 3998–4012. [Google Scholar] [CrossRef]
- Wang, H.; Yan, Z.; Geng, J.; Kunz, S.; Seebeck, T.; Ke, H. Crystal Structure of the Leishmania Major Phosphodiesterase LmjPDEB1 and Insight into the Design of the Parasite-Selective Inhibitors. Mol. Microbiol. 2007, 66, 1029–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, B.L.; Harvey, K.L.; Stewart, R.J.; Azevedo, M.F.; Crabb, B.S.; Jennings, I.G.; Sanders, P.R.; Manallack, D.T.; Thompson, P.E.; Tonkin, C.J.; et al. Identification of Potent Phosphodiesterase Inhibitors That Demonstrate Cyclic Nucleotide-Dependent Functions in Apicomplexan Parasites. ACS Chem. Biol. 2015, 10, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Kunz, S.; Balmer, V.; Sterk, G.J.; Pollastri, M.P.; Leurs, R.; Müller, N.; Hemphill, A.; Spycher, C. The Single Cyclic Nucleotide-Specific Phosphodiesterase of the Intestinal Parasite Giardia Lamblia Represents a Potential Drug Target. PLoS Negl. Trop. Dis. 2017, 11, e0005891. [Google Scholar] [CrossRef] [Green Version]
- Taft, A.S.; Norante, F.A.; Yoshino, T.P. The Identification of Inhibitors of Schistosoma mansoni Miracidial Transformation by Incorporating a Medium-Throughput Small-Molecule Screen. Exp. Parasitol. 2010, 125, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Final Report Summary—PDE4NPD. Available online: https://cordis.europa.eu/project/id/602666/reporting (accessed on 6 November 2022).
- Munday, J.C.; Kunz, S.; Kalejaiye, T.D.; Siderius, M.; Schroeder, S.; Paape, D.; Alghamdi, A.H.; Abbasi, Z.; Huang, S.X.; Donachie, A.-M.; et al. Cloning and Functional Complementation of Ten Schistosoma mansoni Phosphodiesterases Expressed in the Mammalian Host Stages. PLoS Negl. Trop. Dis. 2020, 14, e0008447. [Google Scholar] [CrossRef] [PubMed]
- Long, T.; Rojo-Arreola, L.; Shi, D.; El-Sakkary, N.; Jarnagin, K.; Rock, F.; Meewan, M.; Rascón, A.A.; Lin, L.; Cunningham, K.A.; et al. Phenotypic, Chemical and Functional Characterization of Cyclic Nucleotide Phosphodiesterase 4 (PDE4) as a Potential Anthelmintic Drug Target. PLoS Negl. Trop. Dis. 2017, 11, e0005680. [Google Scholar] [CrossRef] [Green Version]
- Pagès, L.; Gavaldà, A.; Lehner, M.D. PDE4 Inhibitors: A Review of Current Developments (2005–2009). Expert Opin. Ther. Pat. 2009, 19, 1501–1519. [Google Scholar] [CrossRef] [PubMed]
- Zebda, R.; Paller, A.S. Phosphodiesterase 4 Inhibitors. J. Am. Acad. Dermatol. 2018, 78, S43–S52. [Google Scholar] [CrossRef] [PubMed]
- Sebastián-Pérez, V.; Schroeder, S.; Munday, J.C.; van der Meer, T.; Zaldívar-Díez, J.; Siderius, M.; de Koning, H.P.; Brown, D.; Martínez, A.; Campillo, N.E.; et al. Discovery of Novel Schistosoma mansoni PDE4A Inhibitors as Potential Agents against Schistosomiasis. Future Med. Chem. 2019, 11, 1703–1720. [Google Scholar] [CrossRef]
- Rose, A.S.; Bradley, A.R.; Valasatava, Y.; Duarte, J.M.; Prlić, A.; Rose, P.W. NGL Viewer: Web-Based Molecular Graphics for Large Complexes. Bioinformatics 2018, 34, 3755–3758. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.X.; Hassell, A.M.; Vanderwall, D.; Lambert, M.H.; Holmes, W.D.; Luther, M.A.; Rocque, W.J.; Milburn, M.V.; Zhao, Y.; Ke, H.; et al. Atomic Structure of PDE4: Insights into Phosphodiesterase Mechanism and Specificity. Science 2000, 288, 1822–1825. [Google Scholar] [CrossRef]
- Wang, H.; Kunz, S.; Chen, G.; Seebeck, T.; Wan, Y.; Robinson, H.; Martinelli, S.; Ke, H. Biological and Structural Characterization of Trypanosoma Cruzi Phosphodiesterase C and Implications for Design of Parasite Selective Inhibitors. J. Biol. Chem. 2012, 287, 11788–11797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, C.; Wang, H.; Kooistra, A.J.; De Graaf, C.; Orrling, K.M.; Tenor, H.; Seebeck, T.; Bailey, D.; De Esch, I.J.P.; Ke, H.; et al. Discovery of Novel Trypanosoma Brucei Phosphodiesterase B1 Inhibitors by Virtual Screening against the Unliganded TbrPDEB1 Crystal Structure. J. Med. Chem. 2013, 56, 2087–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, C.; Kooistra, A.J.; Kanev, G.K.; Leurs, R.; De Esch, I.J.P.; De Graaf, C. PDEStrIAn: A Phosphodiesterase Structure and Ligand Interaction Annotated Database As a Tool for Structure-Based Drug Design. J. Med. Chem. 2016, 59, 7029–7065. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Card, G.L.; England, B.P.; Suzuki, Y.; Fong, D.; Powell, B.; Lee, B.; Luu, C.; Tabrizizad, M.; Gillette, S.; Ibrahim, P.N.; et al. Structural Basis for the Activity of Drugs That Inhibit Phosphodiesterases. Structure 2004, 12, 2233–2247. [Google Scholar] [CrossRef] [Green Version]
- Botros, S.S.; El-Lakkany, N.M.; Seif el-Din, S.H.; William, S.; Sabra, A.N.; Hammam, O.A.; de Koning, H.P. The Phosphodiesterase-4 Inhibitor Roflumilast Impacts Schistosoma mansoni Ovipositing in Vitro but Displays Only Modest Antischistosomal Activity In Vivo. Exp. Parasitol. 2020, 208, 107793. [Google Scholar] [CrossRef] [PubMed]
- Freitag, A.; Wessier, I.; Racké, K. Phosphodiesterase Inhibitors Suppress A2-Adrenoceptor-Mediated 5-Hydroxytryptamine Release from Tracheae of Newborn Rabbits. Eur. J. Pharmacol. 1998, 354, 67–71. [Google Scholar] [CrossRef]
- Francis, S.H.; Sekhar, K.R.; Ke, H.; Corbin, J.D. Inhibition of Cyclic Nucleotide Phosphodiesterases by Methylxanthines and Related Compounds. In Handbook of Experimental Pharmacology; Fredholm, B.B., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 93–133. ISBN 978-3-642-13443-2. [Google Scholar]
- Thompson, W.J. Cyclic Nucleotide Phosphodiesterases: Pharmacology, Biochemistry and Function. Pharmacol. Ther. 1991, 51, 13–33. [Google Scholar] [CrossRef]
- Menniti, F.S.; Faraci, W.S.; Schmidt, C.J. Phosphodiesterases in the CNS: Targets for Drug Development. Nat. Rev. Drug Discov. 2006, 5, 660–670. [Google Scholar] [CrossRef]
- Bell, A.S.; Terrett, N.K. Pyrazolo[4,3-d]Pyrimidine Derivatives and Pharmaceutical Compositions Containing Them. Patent EP0911333A1, 28 April 1999. [Google Scholar]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in Targeting Cyclic Nucleotide Phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [Green Version]
- Ruppert, D.; Weithmann, K.U. HL 725, an Extremely Potent Inhibitor of Platelet Phosphodiesterase and Induced Platelet Aggregation in Vitro. Life Sci. 1982, 31, 2037–2043. [Google Scholar] [CrossRef]
- Sircar, I.; Steffen, R.P.; Bobowski, G.; Burke, S.E.; Newton, R.S.; Weishaar, R.E.; Bristol, J.A.; Evans, D.B. Cardiotonic Agents. 9. Synthesis and Biological Evaluation of a Series of (E)-4,5-Dihydro-6-[2-[4-(1H-Imidazol-1-Yl)Phenyl]Ethenyl]-3(2H)-Pyridazinones: A Novel Class of Compounds with Positive Inotropic, Antithrombotic, and Vasodilatory Activities for Th. J. Med. Chem. 1989, 32, 342–350. [Google Scholar] [CrossRef]
- Sudo, T.; Tachibana, K.; Toga, K.; Tochizawa, S.; Inoue, Y.; Kimura, Y.; Hidaka, H. Potent Effects of Novel Anti-Platelet Aggregatory Cilostamide Analogues on Recombinant Cyclic Nucleotide Phosphodiesterase Isozyme Activity. Biochem. Pharmacol. 2000, 59, 347–356. [Google Scholar] [CrossRef]
- Mata, M.; Pallardo, F.; Morcillo, E.J.; Cortijo, J. Piclamilast Inhibits the Pro-Apoptotic and Anti-Proliferative Responses of A549 Cells Exposed to H2O2 via Mechanisms Involving AP-1 Activation. Free Radic. Res. 2012, 46, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Sterk, G.J.; Hatzelmann, A.; Barsig, J.; Marx, D.; Kley, H.-P.; Christiaans, J.A.M.; Menge, W.M.P.B. Pyrrolidinedione Substituted Piperidine-Phthalazones as Pde4. Inhibitors. Patent PL373645A1, 5 September 2005. [Google Scholar]
- Vellas, B.; Sol, O.; Snyder, P.J.; Ousset, P.-J.; Haddad, R.; Maurin, M.; Lemarie, J.-C.; Desire, L.; Pando, M.P. EHT0202 in Alzheimers Disease: A 3-Month, Randomized, Placebo- Controlled, Double-Blind Study. Curr. Alzheimer Res. 2011, 8, 203–212. [Google Scholar] [CrossRef]
- Sterk, G.J.; Hatzelmann, A.; Marx, D.; Steinhilber, W. Phthalazinones Derivatives Useful as Pde4/7. Inhibitors. Patent SK14342003A3, 4 May 2004. [Google Scholar]
- De Koning, H.P.; Gould, M.K.; Sterk, G.J.; Tenor, H.; Kunz, S.; Luginbuehl, E.; Seebeck, T. Pharmacological Validation of Trypanosoma Brucei Phosphodiesterases as Novel Drug Targets. J. Infect. Dis. 2012, 206, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Weinbrenner, S.; Dunkern, T.; Marx, D.; Schmidt, B.; Stenget, T.; Flockerzi, D.; Kautz, U.; Hauser, D.; Diefenbach, J.; Christiaans, J.A.M.; et al. 6-Benzyl-2,3,4,7-Tetrahydro-Indolo[2,3-c]Quinoline Compounds Useful as Pde5. Inhibitors. Patent WO2008095835A1, 14 August 2008. [Google Scholar]
- Haning, H.; Niewöhner, U.; Schenke, T.; Lampe, T.; Hillisch, A.; Bischoff, E. Comparison of Different Heterocyclic Scaffolds as Substrate Analog PDE5 Inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 3900–3907. [Google Scholar] [CrossRef]
- Choi, S.-H.; Choi, D.-H.; Song, K.-S.; Shin, K.-H.; Chun, B.-G. Zaprinast, an Inhibitor of CGMP-Selective Phosphodiesterases, Enhances the Secretion of TNF-α and IL-1β and the Expression of INOS and MHC Class II Molecules in Rat Microglial Cells. J. Neurosci. Res. 2002, 67, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.J.; Cieslinski, L.B.; Newton, R.; Donnelly, L.E.; Fenwick, P.S.; Nicholson, A.G.; Barnes, P.J.; Barnette, M.S.; Giembycz, M.A. Discovery of BRL 50481 [3-(<Em>N,N</Em>-Dimethylsulfonamido)-4-Methyl-Nitrobenzene], a Selective Inhibitor of Phosphodiesterase 7: In Vitro Studies in Human Monocytes, Lung Macrophages, and CD8<Sup>+</Sup> T-Lymphocytes. Mol. Pharmacol. 2004, 66, 1679LP–1689. [Google Scholar] [CrossRef] [Green Version]
- Redondo, M.; Brea, J.; Perez, D.I.; Soteras, I.; Val, C.; Perez, C.; Morales-García, J.A.; Alonso-Gil, S.; Paul-Fernandez, N.; Martin-Alvarez, R.; et al. Effect of Phosphodiesterase 7 (PDE7) Inhibitors in Experimental Autoimmune Encephalomyelitis Mice. Discovery of a New Chemically Diverse Family of Compounds. J. Med. Chem. 2012, 55, 3274–3284. [Google Scholar] [CrossRef] [Green Version]
- García, A.M.; Brea, J.; Morales-García, J.A.; Perez, D.I.; González, A.; Alonso-Gil, S.; Gracia-Rubio, I.; Ros-Simó, C.; Conde, S.; Cadavid, M.I.; et al. Modulation of CAMP-Specific PDE without Emetogenic Activity: New Sulfide-Like PDE7 Inhibitors. J. Med. Chem. 2014, 57, 8590–8607. [Google Scholar] [CrossRef] [PubMed]
- DeNinno, M.P.; Wright, S.W.; Visser, M.S.; Etienne, J.B.; Moore, D.E.; Olson, T.V.; Rocke, B.N.; Andrews, M.P.; Zarbo, C.; Millham, M.L.; et al. 1,5-Substituted Nipecotic Amides: Selective PDE8 Inhibitors Displaying Diastereomer-Dependent Microsomal Stability. Bioorg. Med. Chem. Lett. 2011, 21, 3095–3098. [Google Scholar] [CrossRef] [PubMed]
- Malamas, M.S.; Ni, Y.; Erdei, J.; Stange, H.; Schindler, R.; Lankau, H.-J.; Grunwald, C.; Fan, K.Y.; Parris, K.; Langen, B.; et al. Highly Potent, Selective, and Orally Active Phosphodiesterase 10A Inhibitors. J. Med. Chem. 2011, 54, 7621–7638. [Google Scholar] [CrossRef]
- Ceyhan, O.; Birsoy, K.; Hoffman, C.S. Identification of Biologically Active PDE11-Selective Inhibitors Using a Yeast-Based High-Throughput Screen. Chem. Biol. 2012, 19, 155–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Integrated DNA Technologies Tool. Available online: https://www.idtdna.com/CodonOpt (accessed on 31 March 2023).
- Berrow, N.S.; Alderton, D.; Sainsbury, S.; Nettleship, J.; Assenberg, R.; Rahman, N.; Stuart, D.I.; Owens, R.J. A Versatile Ligation-Independent Cloning Method Suitable for High-Throughput Expression Screening Applications. Nucleic Acids. Res. 2007, 35, e45. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr. Sect. D 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. Sect. D 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Duvall, R.H.; DsWitt, W.B. An Improved Perfusion Technique for Recovering Adult Schistosomes from Laboratory Animals. Am. J. Trop. Med. Hyg. 1967, 16, 483–486. [Google Scholar] [CrossRef]
- Pica-Mattoccia, L.; Cioli, D. Sex- and Stage-Related Sensitivity of Schistosoma mansoni to In Vivo and In Vitro Praziquantel Treatment. Int. J. Parasitol. 2004, 34, 527–533. [Google Scholar] [CrossRef]
- Botros, S.; Pica-Mattoccia, L.; William, S.; El-Lakkani, N.; Cioli, D. Effect of Praziquantel on the Immature Stages of Schistosoma haematobium . Int. J. Parasitol. 2005, 35, 1453–1457. [Google Scholar] [CrossRef]
- Ashton, M.J.; Cook, D.C.; Fenton, G.; Karlsson, J.A.; Palfreyman, M.N.; Raeburn, D.; Ratcliffe, A.J.; Souness, J.E.; Thurairatnam, S.; Vicker, N. Selective Type IV Phosphodiesterase Inhibitors as Antiasthmatic Agents. The Syntheses and Biological Activities of 3-(Cyclopentyloxy)-4-Methoxybenzamides and Analogues. J. Med. Chem. 1994, 37, 1696–1703. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Huang, P.; Liu, S.; Sima, L.; Chen, L.; Wang, D. A Convenient Method for the Synthesis of Roflumilast. Res Chem. Intermed. 2013, 39, 2107–2113. [Google Scholar] [CrossRef]
- Felding, J.; Sørensen, M.D.; Poulsen, T.D.; Larsen, J.; Andersson, C.; Refer, P.; Engell, K.; Ladefoged, L.G.; Thormann, T.; Vinggaard, A.M.; et al. Discovery and Early Clinical Development of 2-{6-[2-(3,5-Dichloro-4- Pyridyl)Acetyl]-2,3-Dimethoxyphenoxy}- N -Propylacetamide (LEO 29102), a Soft-Drug Inhibitor of Phosphodiesterase 4 for Topical Treatment of Atopic Dermatitis. J. Med. Chem. 2014, 57, 5893–5903. [Google Scholar] [CrossRef] [PubMed]
- Bland, N.D.; Wang, C.; Tallman, C.; Gustafson, A.E.; Wang, Z.; Ashton, T.D.; Ochiana, S.O.; McAllister, G.; Cotter, K.; Fang, A.P.; et al. Pharmacological Validation of Trypanosoma Brucei Phosphodiesterases B1 and B2 as Druggable Targets for African Sleeping Sickness. J. Med. Chem. 2011, 54, 8188–8194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.; Xiao, Z. Preparation Method of Roflumilast N-Oxide. C.N. Patent 106256820A, 28 December 2016. [Google Scholar]
- Zhou, Z.Z.; Ge, B.C.; Zhong, Q.P.; Huang, C.; Cheng, Y.F.; Yang, X.M.; Wang, H.T.; Xu, J.P. Development of Highly Potent Phosphodiesterase 4 Inhibitors with Anti-Neuroinflammation Potential: Design, Synthesis, and Structure-Activity Relationship Study of Catecholamides Bearing Aromatic Rings. Eur. J. Med. Chem. 2016, 124, 372–379. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp No. | Structure | M.W. | cLogP a | PSA a | SmPDE4A_CD pKi b |
|---|---|---|---|---|---|
| NPD-0001 |  | 584.7 | 5.8 | 114.8 | 6.9 ± 0.2 |
| NPD-0007 |  | 508.6 | 0.3 | 108.8 | 6.3 ± 0.1 |
| NPD-0006 (roflumilast) |  | 403.2 | 4.5 | 60.5 | 7.1 ± 0.1 |
| NPD-0005 (piclamilast) |  | 381.3 | 4.1 | 60.5 | 6.5 ± 0.2 |

| Comp No. | R1 | R2 | M.W. | cLogP a | PSA a | SmPDE4A_CD pKi b or Inhibition at 10 µM (%) |
|---|---|---|---|---|---|---|
| roflumilast |  | HF2CO | 403.2 | 4.5 | 60.5 | 7.1 ± 0.1 |
| piclamilast |  | MeO | 419.2 | 2.4 | 74.5 | 7.2 ± 0.0 |
| NPD-3094 |  | MeO | 368.8 | 3.8 | 60.5 | 6.4 ± 0.1 |
| NPD-3093 |  | F | 368.8 | 3.8 | 60.5 | 5.6 ± 0.0 |
| NPD-3096 |  | Cl | 334.3 | 3.2 | 60.5 | 6.1 ± 0.1 |
| NPD-0546 |  | MeO | 367.8 | 5.1 | 47.6 | 8% |
| NPD-0446 | MeO | MeO | 367.8 | 5.1 | 47.6 | 5.4 ± 0.0 |
| NPD-1180 | Cl | MeO | 397.8 | 4.9 | 56.8 | 9% |
| NPD-0548 | H | MeO | 297.1 | 2.9 | 51.2 | 16% |

| Comp No. | R | M.W. | cLogP a | PSA a | SmPDE4A_CD pKi b or Inhibition at 10 µM (%) |
|---|---|---|---|---|---|
| roflumilast |  | 403.2 | 4.5 | 60.5 | 7.1 ± 0.1 |
| NPD-2980 |  | 419.2 | 2.4 | 74.5 | 7.2 ± 0.0 |
| NPD-1204 |  | 368.8 | 3.8 | 60.5 | 6.6 ± 0.1 |
| NPD-1205 |  | 368.8 | 3.8 | 60.5 | 21% |
| NPD-3076 |  | 334.3 | 3.2 | 60.5 | 46% |
| NPD-3080 |  | 367.8 | 5.1 | 47.6 | 5.7 ± 0.1 |
| NPD-3075 |  | 367.8 | 5.1 | 47.6 | 4% |
| NPD-3000 |  | 397.8 | 4.9 | 56.8 | 15% |
| Compound | SmPDE4A_CD pKi | Dose (µM) | %Worm Killing (Male) | %Worm Killing (Female) | % Oviposition Reduction | MRC-5 pIC50 |
|---|---|---|---|---|---|---|
| roflumilast a | 7.1 ± 0.1 | 100 | 0 | 0 | 52 * | <4.2 |
| 50 | 0 | 0 | 49 * | |||
| 25 | 0 | 0 | 42 * | |||
| 10 | 0 | 0 | 39 * | |||
| 5 | 0 | 0 | 35 * | |||
| NPD-0001 | 6.9 ± 0.2 | 100 | 63 | 0 | 100 * | 5.1 ± 0.1 |
| 50 | 0 | 0 | 23 | |||
| 25 | 0 | 0 | 13 | |||
| 10 | 0 | 0 | 0 | |||
| 5 | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Y.; Schroeder, S.; Kanev, G.K.; Botros, S.S.; William, S.; Sabra, A.-N.A.; Maes, L.; Caljon, G.; Gil, C.; Martinez, A.; et al. To Target or Not to Target Schistosoma mansoni Cyclic Nucleotide Phosphodiesterase 4A? Int. J. Mol. Sci. 2023, 24, 6817. https://doi.org/10.3390/ijms24076817
Zheng Y, Schroeder S, Kanev GK, Botros SS, William S, Sabra A-NA, Maes L, Caljon G, Gil C, Martinez A, et al. To Target or Not to Target Schistosoma mansoni Cyclic Nucleotide Phosphodiesterase 4A? International Journal of Molecular Sciences. 2023; 24(7):6817. https://doi.org/10.3390/ijms24076817
Chicago/Turabian StyleZheng, Yang, Susanne Schroeder, Georgi K. Kanev, Sanaa S. Botros, Samia William, Abdel-Nasser A. Sabra, Louis Maes, Guy Caljon, Carmen Gil, Ana Martinez, and et al. 2023. "To Target or Not to Target Schistosoma mansoni Cyclic Nucleotide Phosphodiesterase 4A?" International Journal of Molecular Sciences 24, no. 7: 6817. https://doi.org/10.3390/ijms24076817