
Delineation of G Protein-Coupled Receptor Kinase Phosphorylation Sites within the D1 Dopamine Receptor and Their Roles in Modulating β-Arrestin Binding and Activation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Results
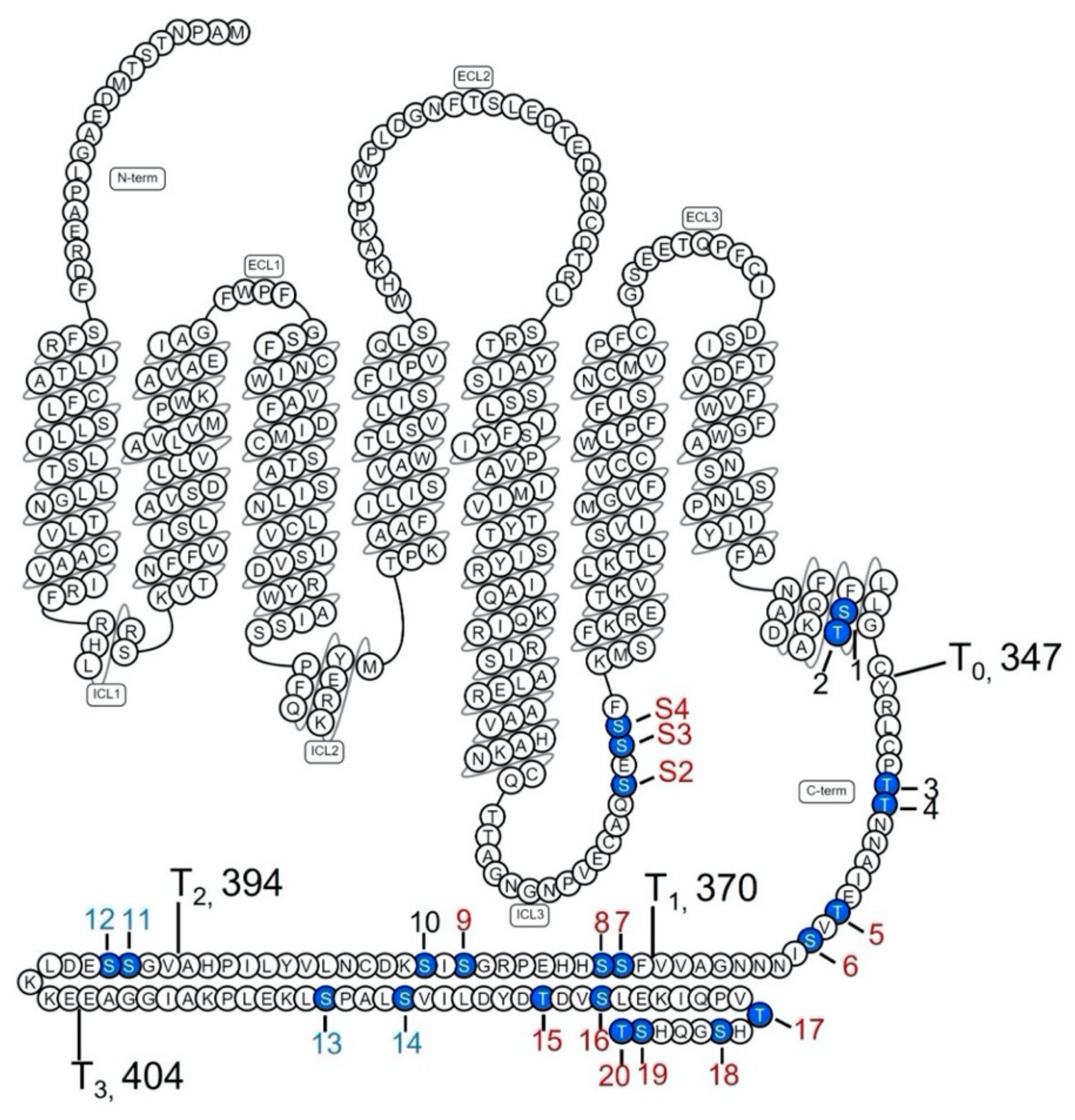
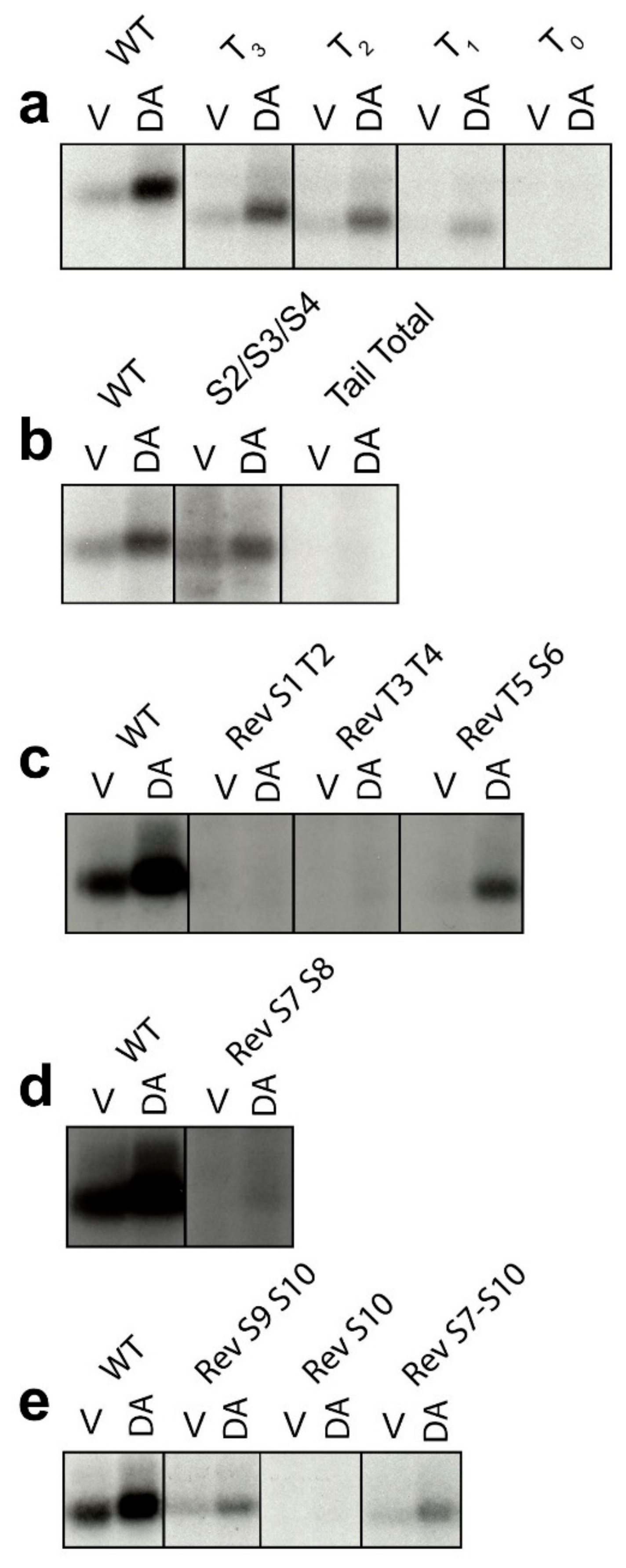
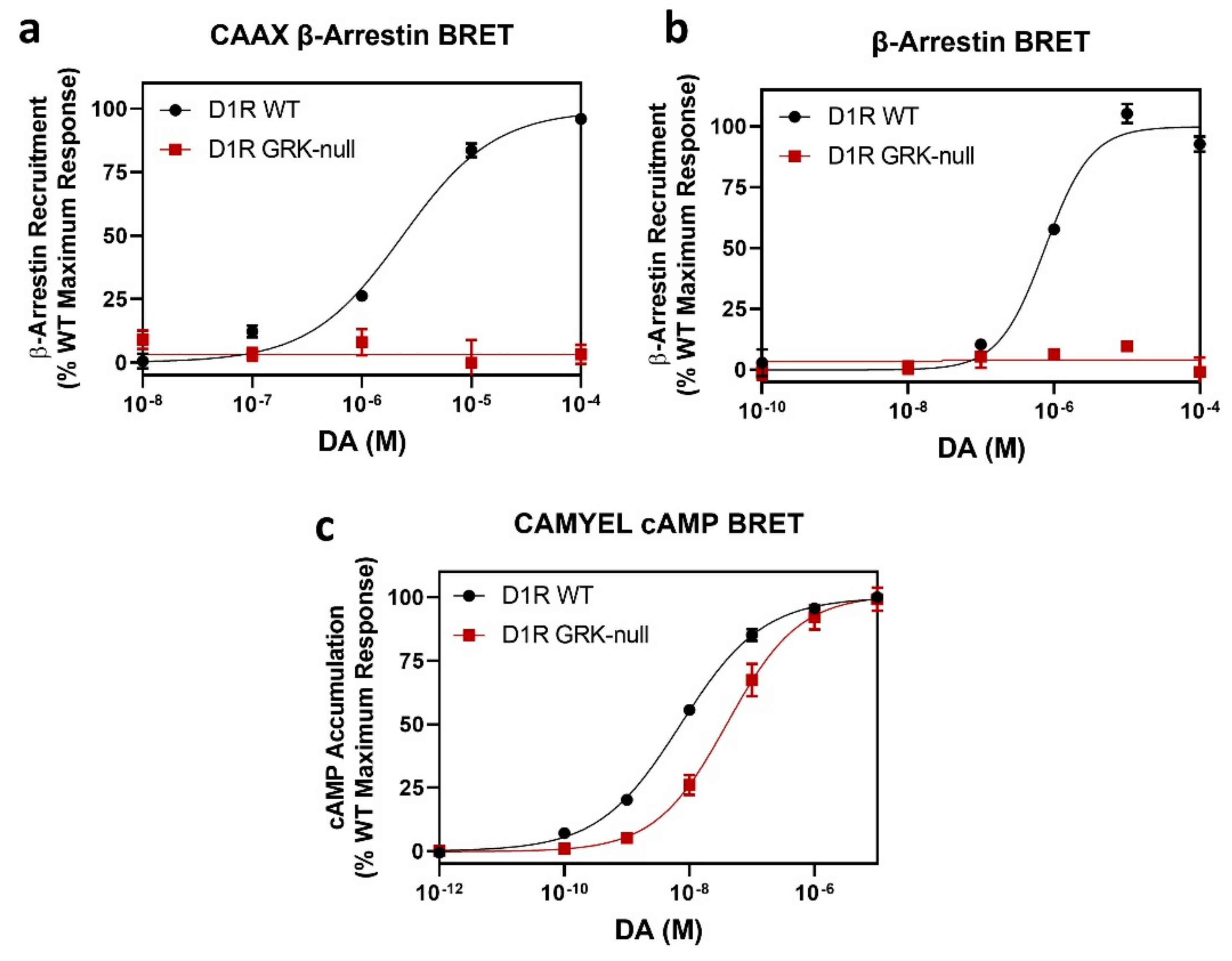
2.1. Determination of GRK-Mediated Phosphorylation Sites within the Rat D1R
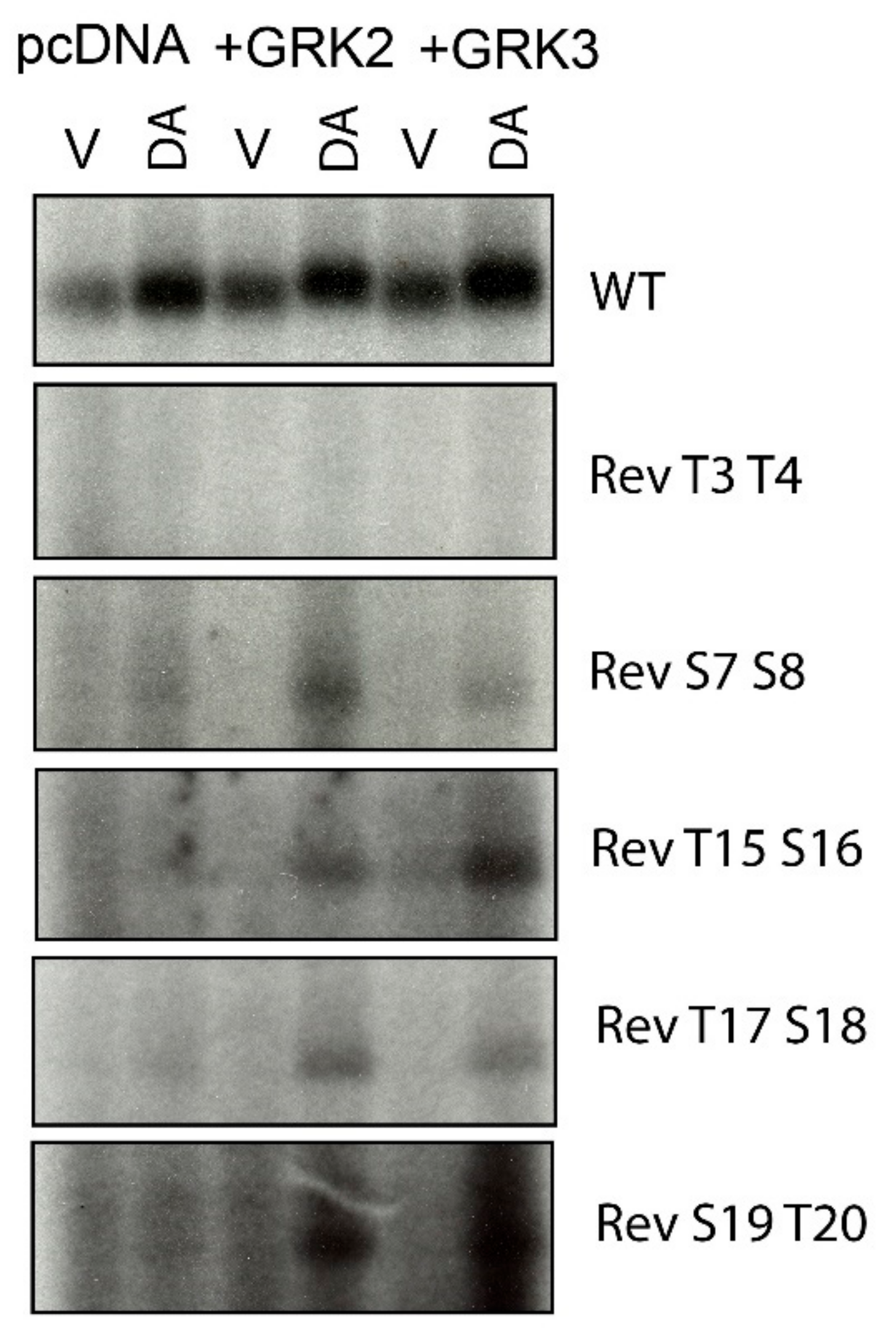
2.2. Overexpression of GRKs Can Enhance DA-Induced Phosphorylation of Specific D1R Residues
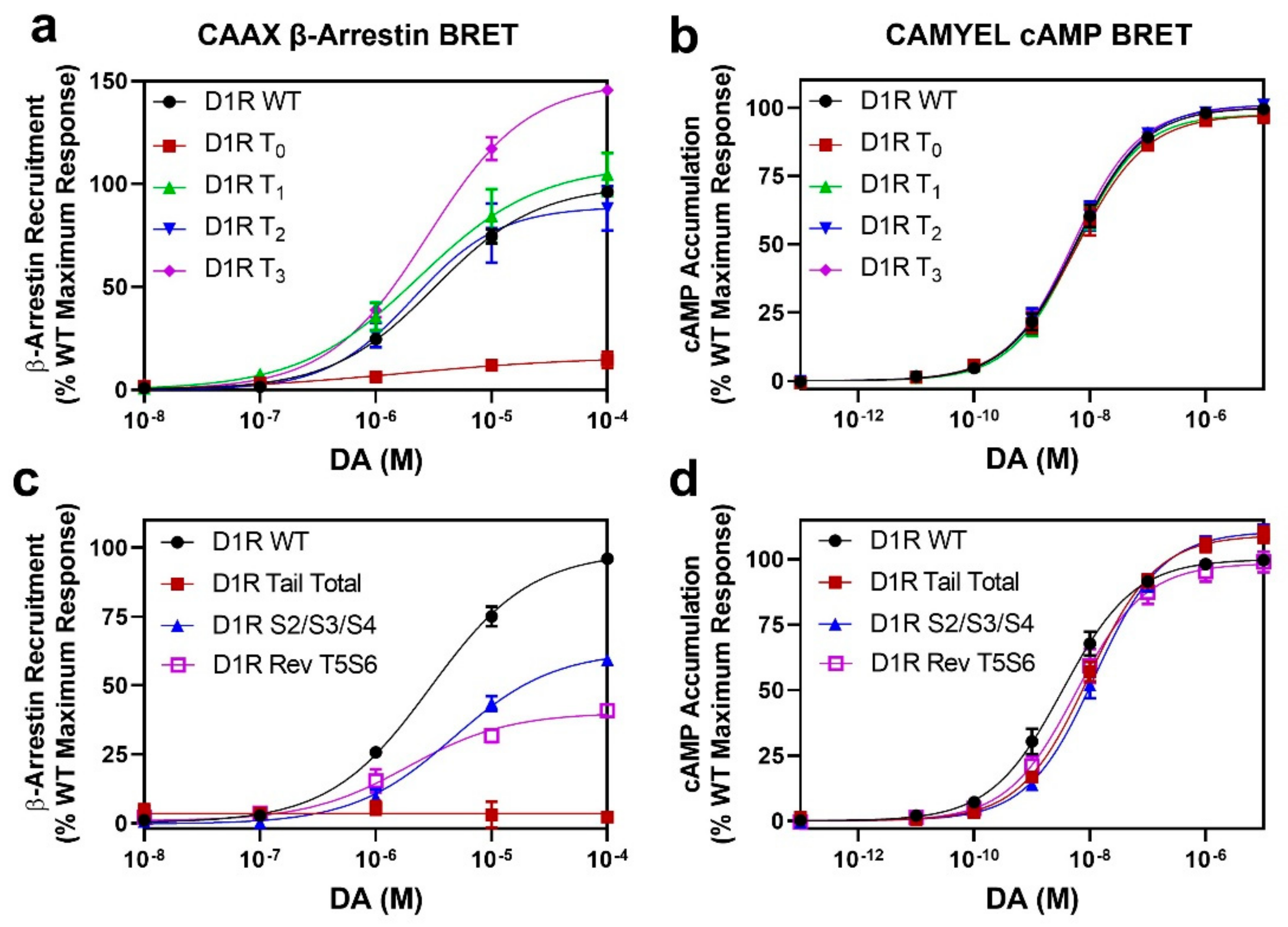
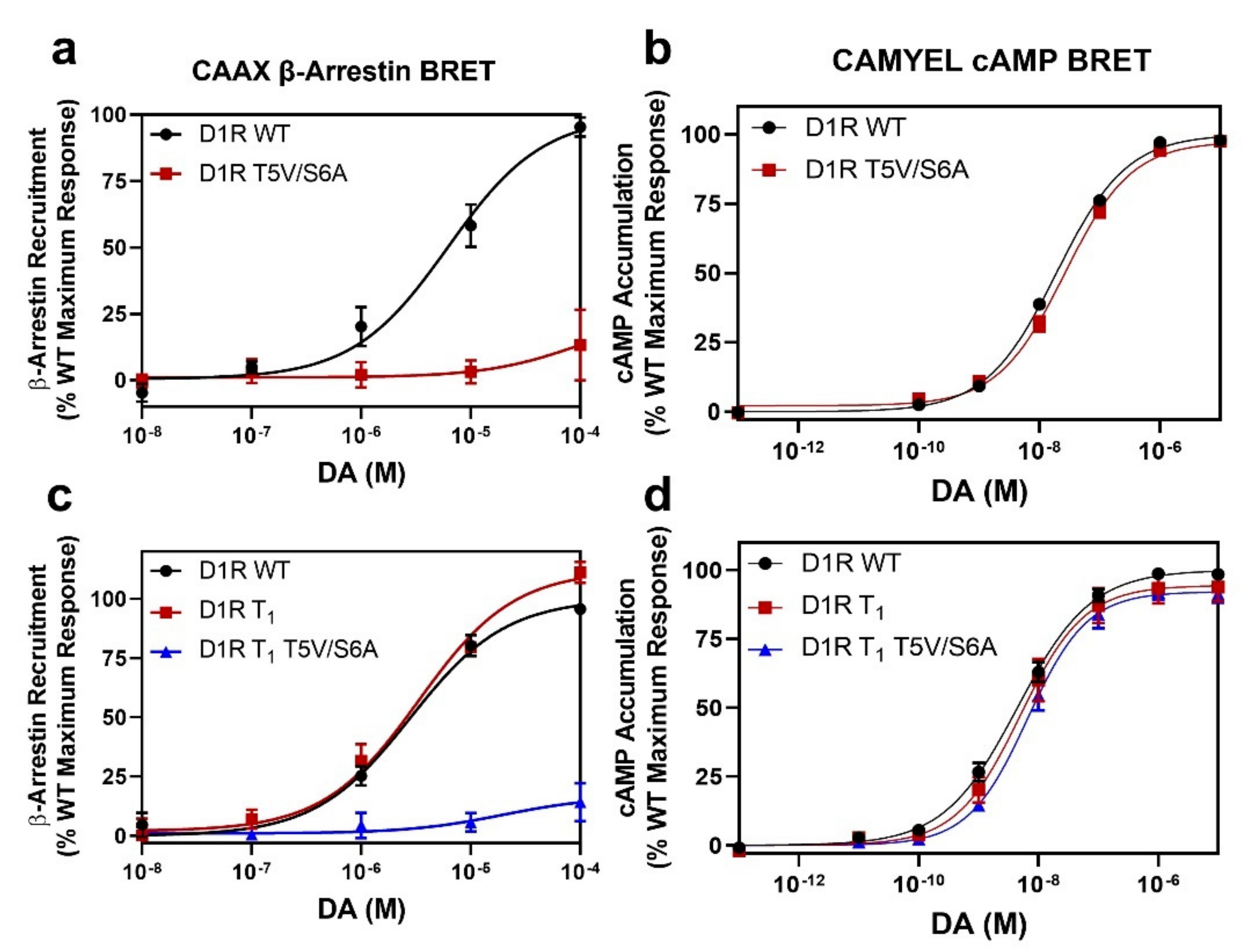
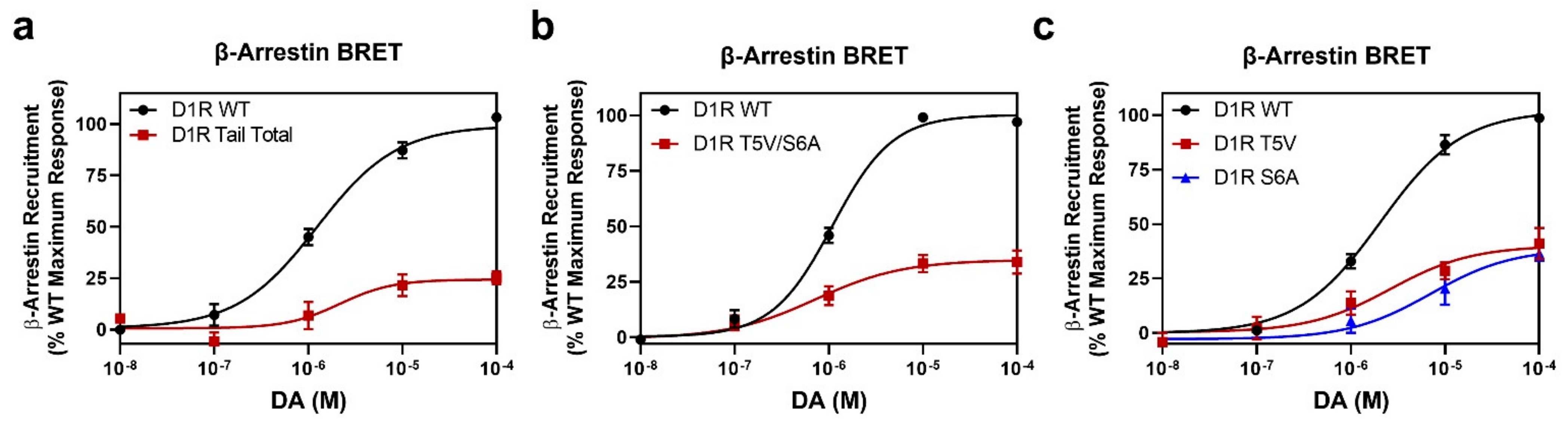
2.3. GRK Phosphorylation Sites in the D1R C-Terminus Are Critical for β-Arrestin Recruitment
2.4. Identification of Individual GRK Phosphorylation Sites That Are Responsible for β-Arrestin Recruitment
2.5. Mutation of the D1R GRK-Mediated Phosphorylation Sites Eliminate β-Arrestin Recruitment
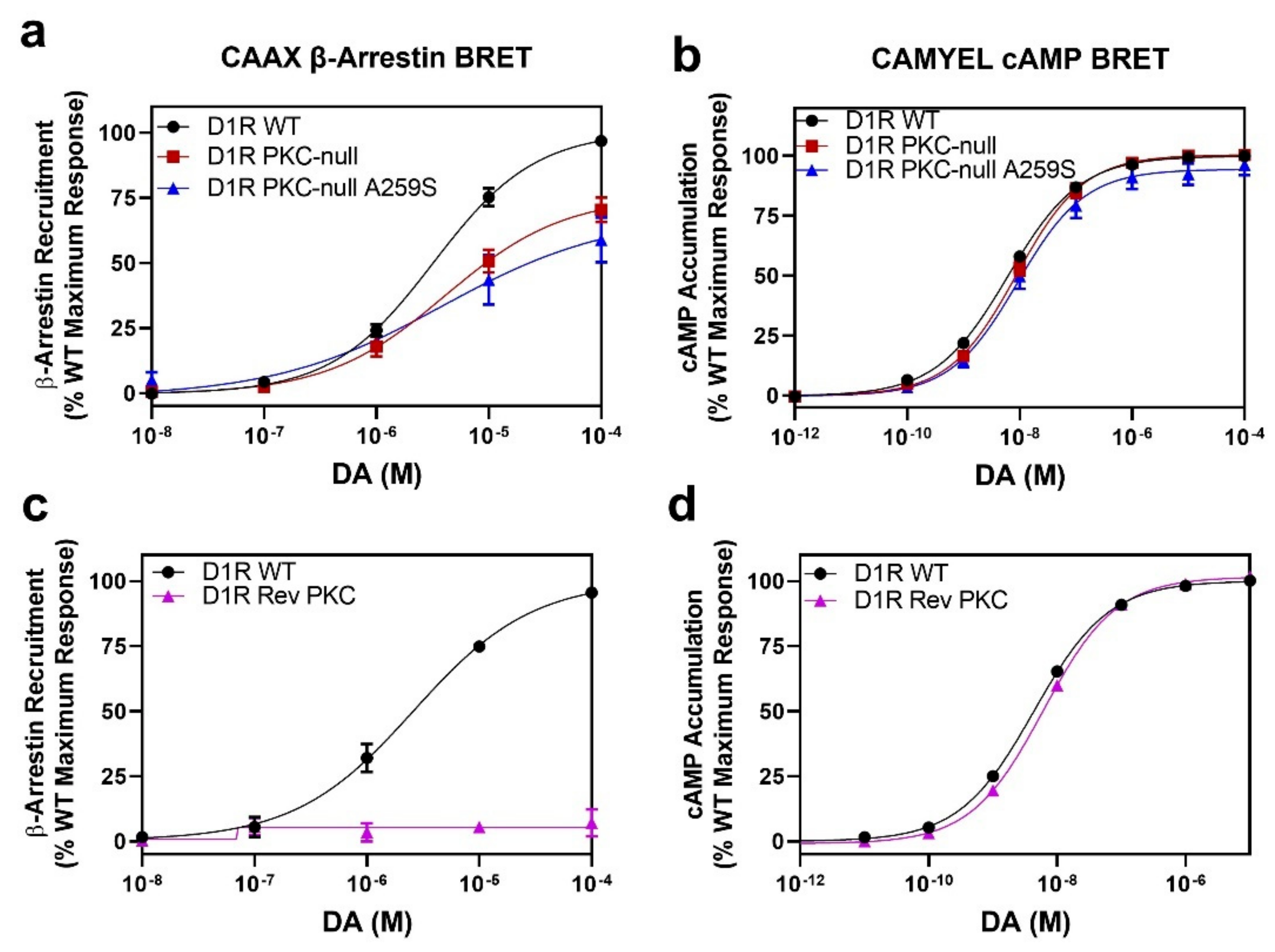
2.6. D1R Residues Phosphorylated by PKC Have Minimal Effects on Dopamine-Stimulated β-Arrestin Recruitment
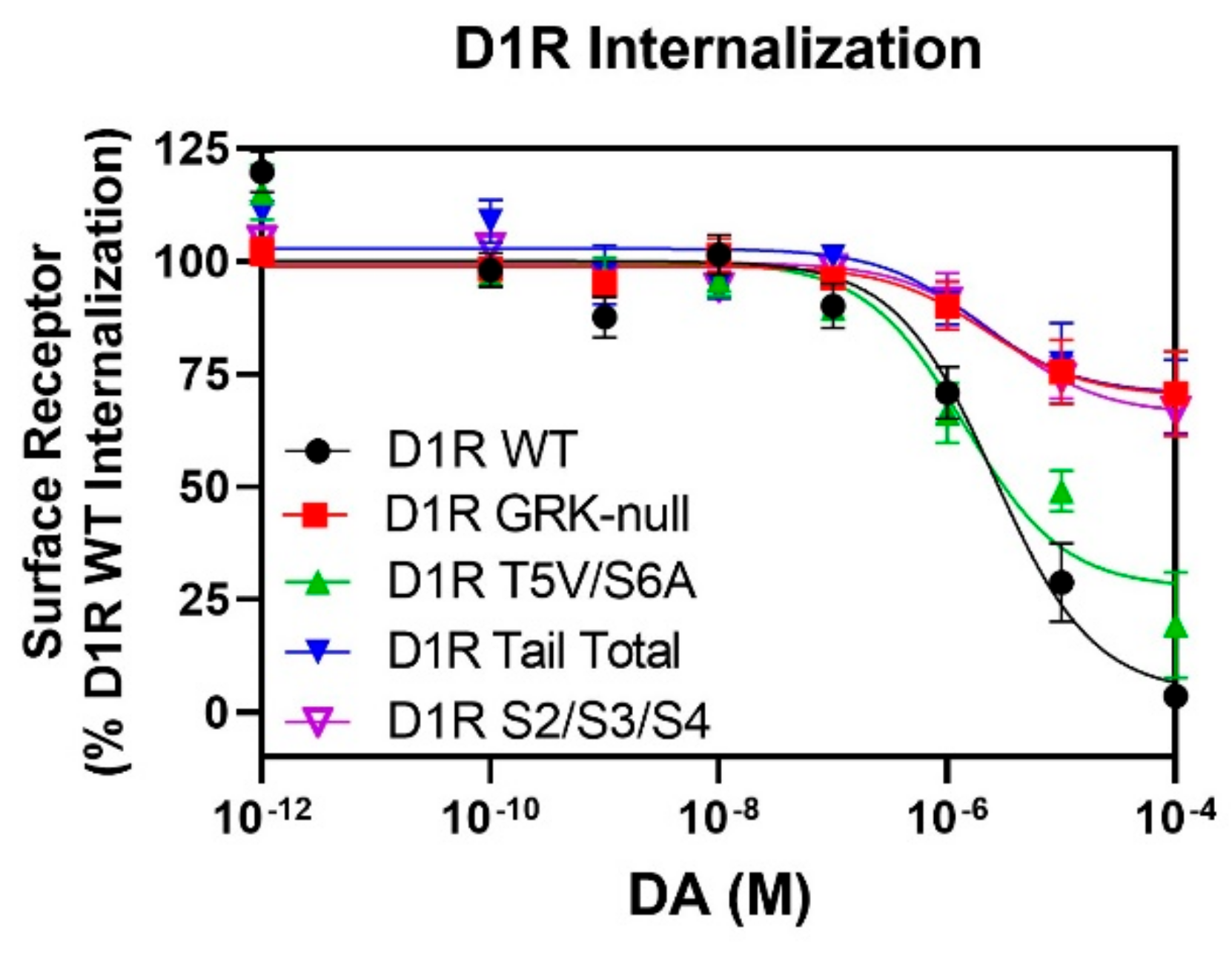
2.7. GRK-Mediated Phosphorylation of the S2/S3/S4 Cluster in ICL3 Is Necessary for DA-Stimulated D1R Internalization
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Transient Transfections
4.3. In Situ Phosphorylation Assays
4.4. Radioligand Binding for In Situ Phosphorylation Assays
4.5. Radioligand Saturation Binding Assays
4.6. CAAX β-Arrestin Recruitment BRET Assay
4.7. Direct β-Arrestin Recruitment BRET Assay
4.8. cAMP CAMYEL BRET Assay
4.9. Lyn Kinase BRET Internalization Assay
4.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sibley, D.R.; Monsma, F.J., Jr. Molecular biology of dopamine receptors. Trends Pharmacol. Sci. 1992, 13, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.M.; Gainetdinov, R.R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, J.M.; Espinoza, S.; Gainetdinov, R.R. Dopamine receptors - IUPHAR Review 13. Br. J. Pharmacol. 2015, 172, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Iverson, T.M.; Gurevich, V.V. Structural Basis of Arrestin-Dependent Signal Transduction. Trends Biochem. Sci. 2018, 43, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, E.V.; Gurevich, V.V. GRKs as Modulators of Neurotransmitter Receptors. Cells 2020, 10, 52. [Google Scholar] [CrossRef]
- Shukla, A.K.; Xiao, K.; Lefkowitz, R.J. Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem. Sci. 2011, 36, 457–469. [Google Scholar] [CrossRef] [Green Version]
- Sibley, D.R.; Peters, J.R.; Nambi, P.; Caron, M.G.; Lefkowitz, R.J. Desensitization of turkey erythrocyte adenylate cyclase. Beta-adrenergic receptor phosphorylation is correlated with attenuation of adenylate cyclase activity. J. Biol. Chem. 1984, 259, 9742–9749. [Google Scholar] [CrossRef]
- Sibley, D.R.; Strasser, R.H.; Caron, M.G.; Lefkowitz, R.J. Homologous desensitization of adenylate cyclase is associated with phosphorylation of the beta-adrenergic receptor. J. Biol. Chem. 1985, 260, 3883–3886. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019, 10, 125. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, E.V.; Tesmer, J.J.; Mushegian, A.; Gurevich, V.V. G protein-coupled receptor kinases: More than just kinases and not only for GPCRs. Pharmacol. Ther. 2012, 133, 40–69. [Google Scholar] [CrossRef] [Green Version]
- Benovic, J.L. Historical Perspective of the G Protein-Coupled Receptor Kinase Family. Cells 2021, 10, 555. [Google Scholar] [CrossRef]
- Sibley, D.R.; Benovic, J.L.; Caron, M.G.; Lefkowitz, R.J. Phosphorylation of cell surface receptors: A mechanism for regulating signal transduction pathways. Endocr. Rev. 1988, 9, 38–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rex, E.B.; Rankin, M.L.; Ariano, M.A.; Sibley, D.R. Ethanol regulation of D(1) dopamine receptor signaling is mediated by protein kinase C in an isozyme-specific manner. Neuropsychopharmacology 2008, 33, 2900–2911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankin, M.L.; Sibley, D.R. Constitutive phosphorylation by protein kinase C regulates D1 dopamine receptor signaling. J. Neurochem. 2010, 115, 1655–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rex, E.B.; Rankin, M.L.; Yang, Y.; Lu, Q.; Gerfen, C.R.; Jose, P.A.; Sibley, D.R. Identification of RanBP 9/10 as interacting partners for protein kinase C (PKC) gamma/delta and the D1 dopamine receptor: Regulation of PKC-mediated receptor phosphorylation. Mol. Pharmacol. 2010, 78, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Jackson, A.; Sedaghat, K.; Minerds, K.; James, C.; Tiberi, M. Opposing effects of phorbol-12-myristate-13-acetate, an activator of protein kinase C, on the signaling of structurally related human dopamine D1 and D5 receptors. J. Neurochem. 2005, 95, 1387–1400. [Google Scholar] [CrossRef]
- Wang, N.; Su, P.; Zhang, Y.; Lu, J.; Xing, B.; Kang, K.; Li, W.; Wang, Y. Protein kinase D1-dependent phosphorylation of dopamine D1 receptor regulates cocaine-induced behavioral responses. Neuropsychopharmacology 2014, 39, 1290–1301. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, N.; Su, P.; Lu, J.; Wang, Y. Disruption of dopamine D1 receptor phosphorylation at serine 421 attenuates cocaine-induced behaviors in mice. Neurosci. Bull. 2014, 30, 1025–1035. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.R.; Sun, P.H.; Ren, Z.X.; Meltzer, H.Y.; Zhen, X.C. GSK-3β Interacts with Dopamine D1 Receptor to Regulate Receptor Function: Implication for Prefrontal Cortical D1 Receptor Dysfunction in Schizophrenia. CNS Neurosci. Ther. 2017, 23, 174–187. [Google Scholar] [CrossRef]
- Jiang, D.; Sibley, D.R. Regulation of D(1) dopamine receptors with mutations of protein kinase phosphorylation sites: Attenuation of the rate of agonist-induced desensitization. Mol. Pharmacol. 1999, 56, 675–683. [Google Scholar]
- Ventura, A.L.; Sibley, D.R. Altered regulation of the D(1) dopamine receptor in mutant Chinese hamster ovary cells deficient in cyclic AMP-dependent protein kinase activity. J. Pharmacol. Exp. Ther. 2000, 293, 426–434. [Google Scholar]
- Mason, J.N.; Kozell, L.B.; Neve, K.A. Regulation of dopamine D(1) receptor trafficking by protein kinase A-dependent phosphorylation. Mol. Pharmacol. 2002, 61, 806–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiberi, M.; Nash, S.R.; Bertrand, L.; Lefkowitz, R.J.; Caron, M.G. Differential regulation of dopamine D1A receptor responsiveness by various G protein-coupled receptor kinases. J. Biol. Chem. 1996, 271, 3771–3778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, B.; Liu, Z.F.; Jiang, D.; Sibley, D.R. The role of phosphorylation/dephosphorylation in agonist-induced desensitization of D1 dopamine receptor function: Evidence for a novel pathway for receptor dephosphorylation. Mol. Pharmacol. 2001, 59, 310–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, A.; Iwasiow, R.M.; Chaar, Z.Y.; Nantel, M.F.; Tiberi, M. Homologous regulation of the heptahelical D1A receptor responsiveness: Specific cytoplasmic tail regions mediate dopamine-induced phosphorylation, desensitization and endocytosis. J. Neurochem. 2002, 82, 683–697. [Google Scholar] [CrossRef] [PubMed]
- Lamey, M.; Thompson, M.; Varghese, G.; Chi, H.; Sawzdargo, M.; George, S.R.; O’Dowd, B.F. Distinct residues in the carboxyl tail mediate agonist-induced desensitization and internalization of the human dopamine D1 receptor. J. Biol. Chem. 2002, 277, 9415–9421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, O.J.; Gardner, B.R.; Williams, D.B.; Marinec, P.S.; Cabrera, D.M.; Peters, J.D.; Mak, C.C.; Kim, K.M.; Sibley, D.R. The role of phosphorylation in D1 dopamine receptor desensitization: Evidence for a novel mechanism of arrestin association. J. Biol. Chem. 2004, 279, 7999–8010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankin, M.L.; Marinec, P.S.; Cabrera, D.M.; Wang, Z.; Jose, P.A.; Sibley, D.R. The D1 dopamine receptor is constitutively phosphorylated by G protein-coupled receptor kinase 4. Mol. Pharmacol. 2006, 69, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat, K.; Tiberi, M. Cytoplasmic tail of D1 dopaminergic receptor differentially regulates desensitization and phosphorylation by G protein-coupled receptor kinase 2 and 3. Cell Signal. 2011, 23, 180–192. [Google Scholar] [CrossRef]
- Watanabe, H.; Xu, J.; Bengra, C.; Jose, P.A.; Felder, R.A. Desensitization of human renal D1 dopamine receptors by G protein-coupled receptor kinase 4. Kidney Int. 2002, 62, 790–798. [Google Scholar] [CrossRef] [Green Version]
- Jose, P.A.; Soares-da-Silva, P.; Eisner, G.M.; Felder, R.A. Dopamine and G protein-coupled receptor kinase 4 in the kidney: Role in blood pressure regulation. Biochim. Biophys. Acta 2010, 1802, 1259–1267. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Hall, J.E.; Jose, P.A.; Chen, K.; Zeng, C. Comprehensive insights in GRK4 and hypertension: From mechanisms to potential therapeutics. Pharmacol. Ther. 2022, 239, 108194. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, A.J.; Mordalski, S.; Pándy-Szekeres, G.; Esguerra, M.; Mamyrbekov, A.; Munk, C.; Keserű, G.M.; Gloriam, D.E. GPCRdb in 2021: Integrating GPCR sequence, structure and function. Nucleic Acids Res. 2021, 49, D335–D343. [Google Scholar] [CrossRef] [PubMed]
- Drube, J.; Haider, R.S.; Matthees, E.S.F.; Reichel, M.; Zeiner, J.; Fritzwanker, S.; Ziegler, C.; Barz, S.; Klement, L.; Filor, J.; et al. GPCR kinase knockout cells reveal the impact of individual GRKs on arrestin binding and GPCR regulation. Nat. Commun. 2022, 13, 540. [Google Scholar] [CrossRef] [PubMed]
- Namkung, Y.; Le Gouill, C.; Lukashova, V.; Kobayashi, H.; Hogue, M.; Khoury, E.; Song, M.; Bouvier, M.; Laporte, S.A. Monitoring G protein-coupled receptor and β-arrestin trafficking in live cells using enhanced bystander BRET. Nat. Commun. 2016, 7, 12178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumova, K.; Iwasiow, R.M.; Tiberi, M. Insight into the mechanism of dopamine D1-like receptor activation. Evidence for a molecular interplay between the third extracellular loop and the cytoplasmic tail. J. Biol. Chem. 2003, 278, 8146–8153. [Google Scholar] [CrossRef] [Green Version]
- Plouffe, B.; Yang, X.; Tiberi, M. The third intracellular loop of D1 and D5 dopaminergic receptors dictates their subtype-specific PKC-induced sensitization and desensitization in a receptor conformation-dependent manner. Cell Signal. 2012, 24, 106–118. [Google Scholar] [CrossRef]
- Kouhen, O.M.; Wang, G.; Solberg, J.; Erickson, L.J.; Law, P.Y.; Loh, H.H. Hierarchical phosphorylation of delta-opioid receptor regulates agonist-induced receptor desensitization and internalization. J. Biol. Chem. 2000, 275, 36659–36664. [Google Scholar] [CrossRef] [Green Version]
- Palmer, T.M.; Stiles, G.L. Identification of threonine residues controlling the agonist-dependent phosphorylation and desensitization of the rat A(3) adenosine receptor. Mol. Pharmacol. 2000, 57, 539–545. [Google Scholar] [CrossRef] [Green Version]
- Just, S.; Illing, S.; Trester-Zedlitz, M.; Lau, E.K.; Kotowski, S.J.; Miess, E.; Mann, A.; Doll, C.; Trinidad, J.C.; Burlingame, A.L.; et al. Differentiation of opioid drug effects by hierarchical multi-site phosphorylation. Mol. Pharmacol. 2013, 83, 633–639. [Google Scholar] [CrossRef] [Green Version]
- Mueller, W.; Schütz, D.; Nagel, F.; Schulz, S.; Stumm, R. Hierarchical organization of multi-site phosphorylation at the CXCR4 C terminus. PLoS ONE 2013, 8, e64975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urs, N.M.; Daigle, T.L.; Caron, M.G. A dopamine D1 receptor-dependent β-arrestin signaling complex potentially regulates morphine-induced psychomotor activation but not reward in mice. Neuropsychopharmacology 2011, 36, 551–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urs, N.M.; Bido, S.; Peterson, S.M.; Daigle, T.L.; Bass, C.E.; Gainetdinov, R.R.; Bezard, E.; Caron, M.G. Targeting β-arrestin2 in the treatment of L-DOPA-induced dyskinesia in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2015, 112, E2517–E2526. [Google Scholar] [CrossRef] [Green Version]
- Kaya, A.I.; Perry, N.A.; Gurevich, V.V.; Iverson, T.M. Phosphorylation barcode-dependent signal bias of the dopamine D1 receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 14139–14149. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lee, S.M.; Imamura, F.; Gowda, K.; Amin, S.; Mailman, R.B. D1 dopamine receptors intrinsic activity and functional selectivity affect working memory in prefrontal cortex. Mol. Psychiatry 2021, 26, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y. Functional Selectivity of Dopamine D(1) Receptor Signaling: Retrospect and Prospect. Int. J. Mol. Sci. 2021, 22, 1914. [Google Scholar] [CrossRef]
- Yang, Y.; Lewis, M.M.; Huang, X.; Dokholyan, N.V.; Mailman, R.B. Dopamine D(1) receptor-mediated β-arrestin signaling: Insight from pharmacology, biology, behavior, and neurophysiology. Int. J. Biochem. Cell Biol. 2022, 148, 106235. [Google Scholar] [CrossRef]
- Seyedabadi, M.; Gharghabi, M.; Gurevich, E.V.; Gurevich, V.V. Receptor-Arrestin Interactions: The GPCR Perspective. Biomolecules 2021, 11, 218. [Google Scholar] [CrossRef]
- Latorraca, N.R.; Wang, J.K.; Bauer, B.; Townshend, R.J.L.; Hollingsworth, S.A.; Olivieri, J.E.; Xu, H.E.; Sommer, M.E.; Dror, R.O. Molecular mechanism of GPCR-mediated arrestin activation. Nature 2018, 557, 452–456. [Google Scholar] [CrossRef]
- Nobles, K.N.; Xiao, K.; Ahn, S.; Shukla, A.K.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.Y.; Bressler, E.A.; Hara, M.R.; et al. Distinct phosphorylation sites on the β(2)-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal. 2011, 4, ra51. [Google Scholar] [CrossRef] [Green Version]
- Latorraca, N.R.; Masureel, M.; Hollingsworth, S.A.; Heydenreich, F.M.; Suomivuori, C.M.; Brinton, C.; Townshend, R.J.L.; Bouvier, M.; Kobilka, B.K.; Dror, R.O. How GPCR Phosphorylation Patterns Orchestrate Arrestin-Mediated Signaling. Cell 2020, 183, 1813–1825.e1818. [Google Scholar] [CrossRef] [PubMed]
- Haider, R.S.; Matthees, E.S.F.; Drube, J.; Reichel, M.; Zabel, U.; Inoue, A.; Chevigné, A.; Krasel, C.; Deupi, X.; Hoffmann, C. β-arrestin1 and 2 exhibit distinct phosphorylation-dependent conformations when coupling to the same GPCR in living cells. Nat. Commun. 2022, 13, 5638. [Google Scholar] [CrossRef] [PubMed]
- Macey, T.A.; Liu, Y.; Gurevich, V.V.; Neve, K.A. Dopamine D1 receptor interaction with arrestin3 in neostriatal neurons. J. Neurochem. 2005, 93, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Masureel, M.; Qu, Q.; Janetzko, J.; Inoue, A.; Kato, H.E.; Robertson, M.J.; Nguyen, K.C.; Glenn, J.S.; Skiniotis, G.; et al. Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 2020, 579, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.I.; Collins, J.; Davis, R.; Lin, K.M.; DeCamp, D.; Roach, T.; Hsueh, R.; Rebres, R.A.; Ross, E.M.; Taussig, R.; et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Biol. Chem. 2007, 282, 10576–10584. [Google Scholar] [CrossRef] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moritz, A.E.; Madaras, N.S.; Rankin, M.L.; Inbody, L.R.; Sibley, D.R. Delineation of G Protein-Coupled Receptor Kinase Phosphorylation Sites within the D1 Dopamine Receptor and Their Roles in Modulating β-Arrestin Binding and Activation. Int. J. Mol. Sci. 2023, 24, 6599. https://doi.org/10.3390/ijms24076599
Moritz AE, Madaras NS, Rankin ML, Inbody LR, Sibley DR. Delineation of G Protein-Coupled Receptor Kinase Phosphorylation Sites within the D1 Dopamine Receptor and Their Roles in Modulating β-Arrestin Binding and Activation. International Journal of Molecular Sciences. 2023; 24(7):6599. https://doi.org/10.3390/ijms24076599
Chicago/Turabian StyleMoritz, Amy E., Nora S. Madaras, Michele L. Rankin, Laura R. Inbody, and David R. Sibley. 2023. "Delineation of G Protein-Coupled Receptor Kinase Phosphorylation Sites within the D1 Dopamine Receptor and Their Roles in Modulating β-Arrestin Binding and Activation" International Journal of Molecular Sciences 24, no. 7: 6599. https://doi.org/10.3390/ijms24076599