
FZD7, Regulated by Non-CpG Methylation, Plays an Important Role in Immature Porcine Sertoli Cell Proliferation

Abstract
:1. Introduction
2. Results
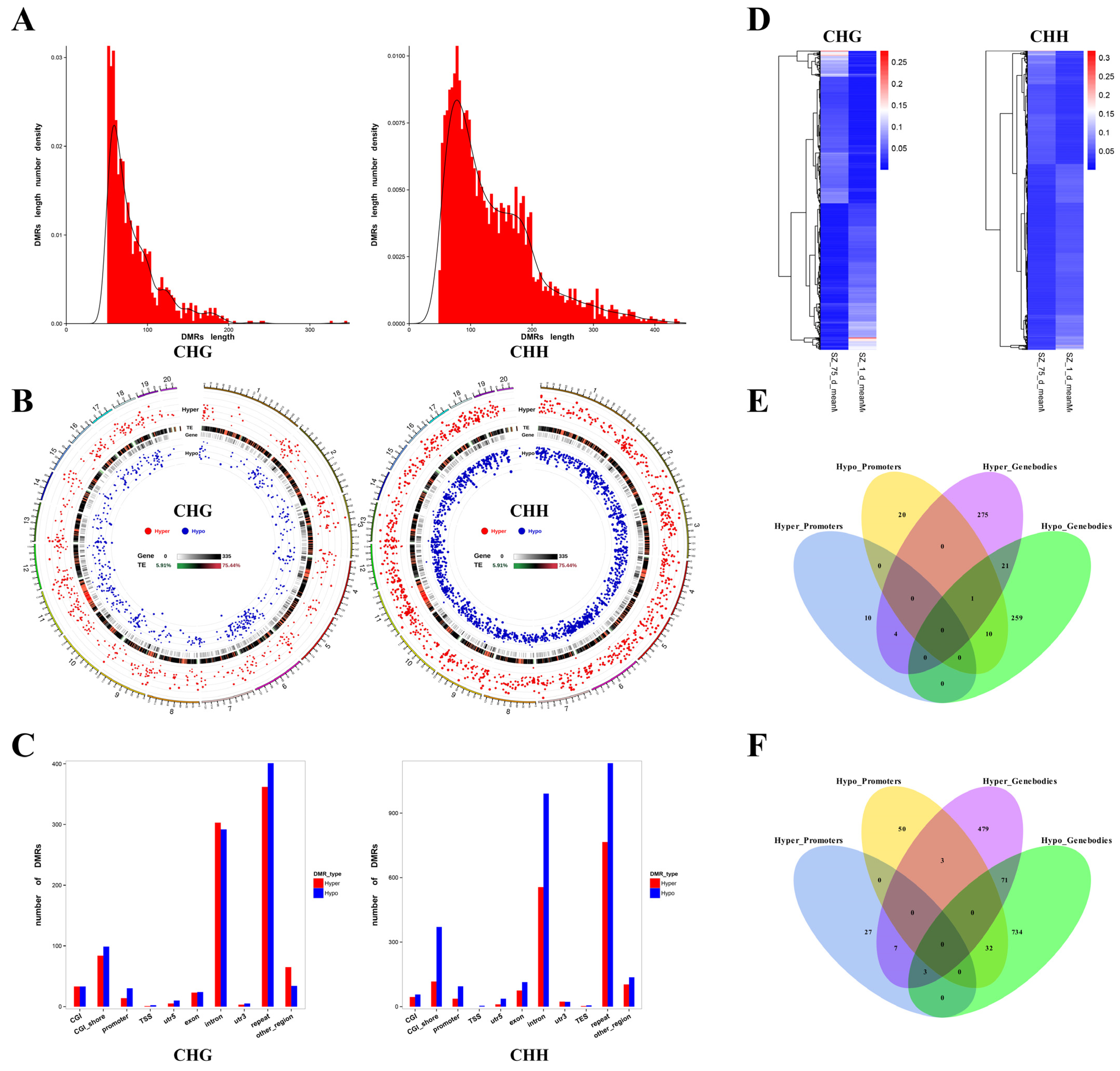
2.1. Characterization of DMRs in Non-CpG Contexts
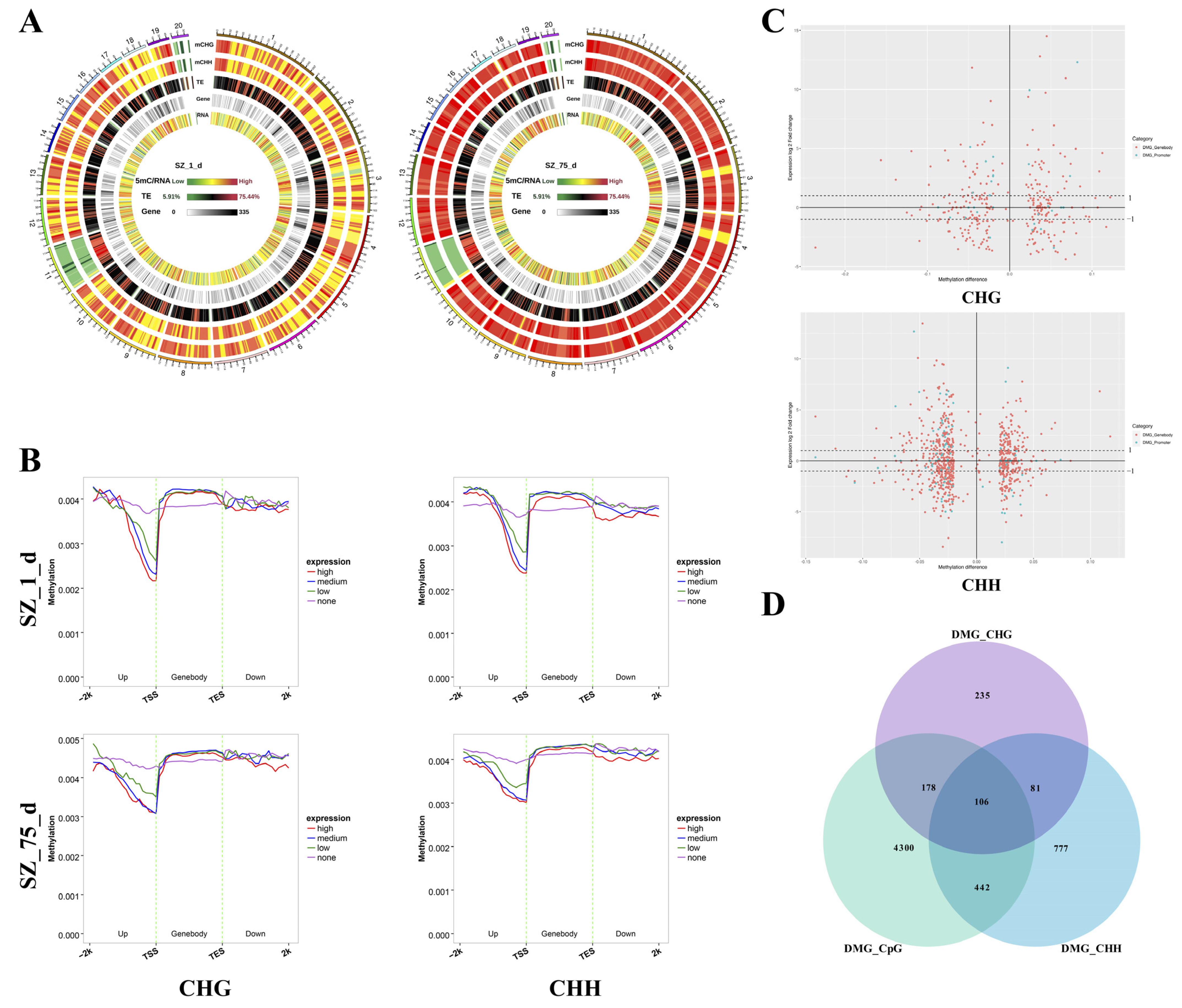
2.2. Identification of DMGs in Non-CpG Contexts
2.3. The Correlation between DNA Methylation and Gene Expression
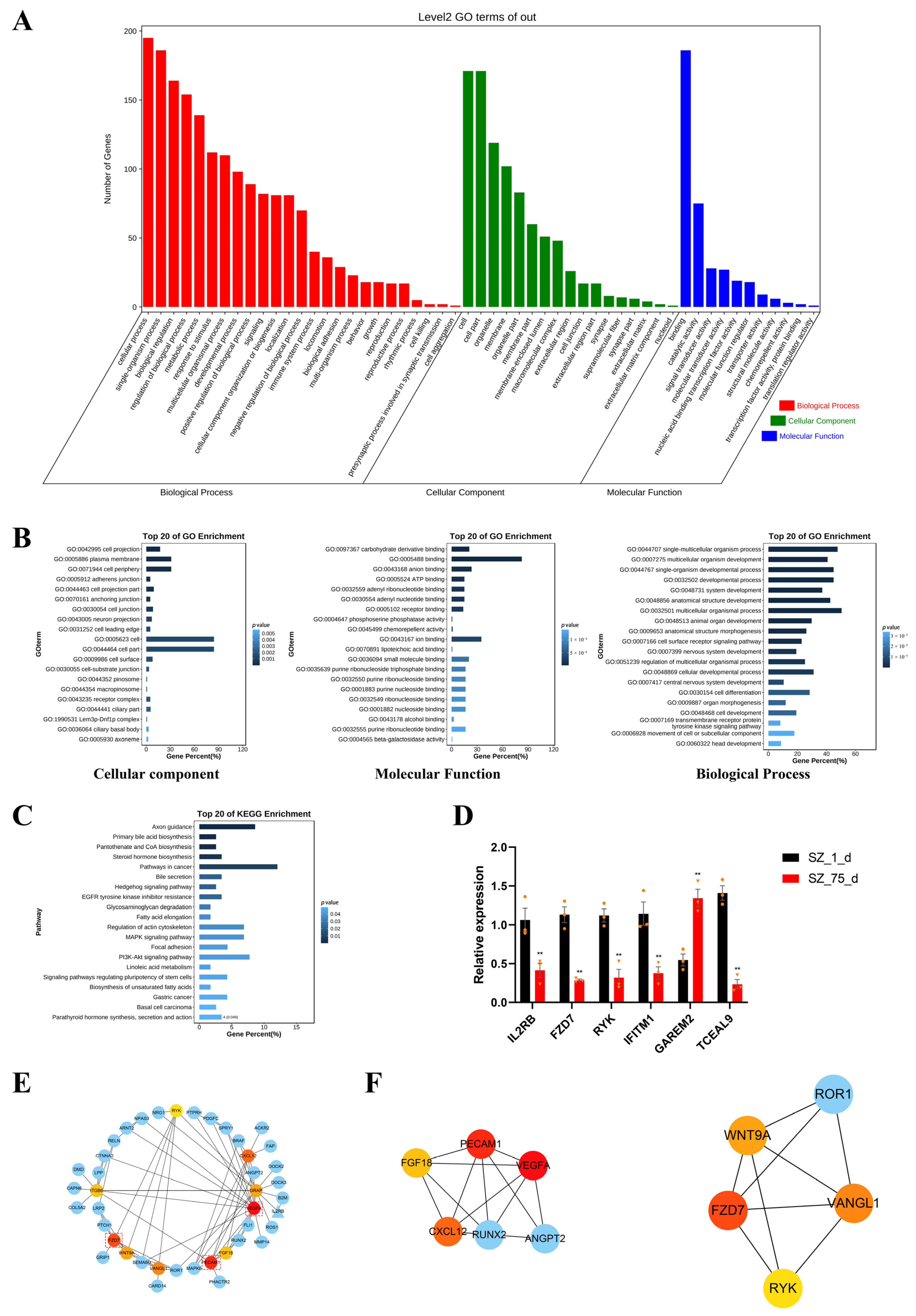
2.4. Enrichment Analyses
2.5. Screening of Hub Genes Regulated by Non-CpG Methylation
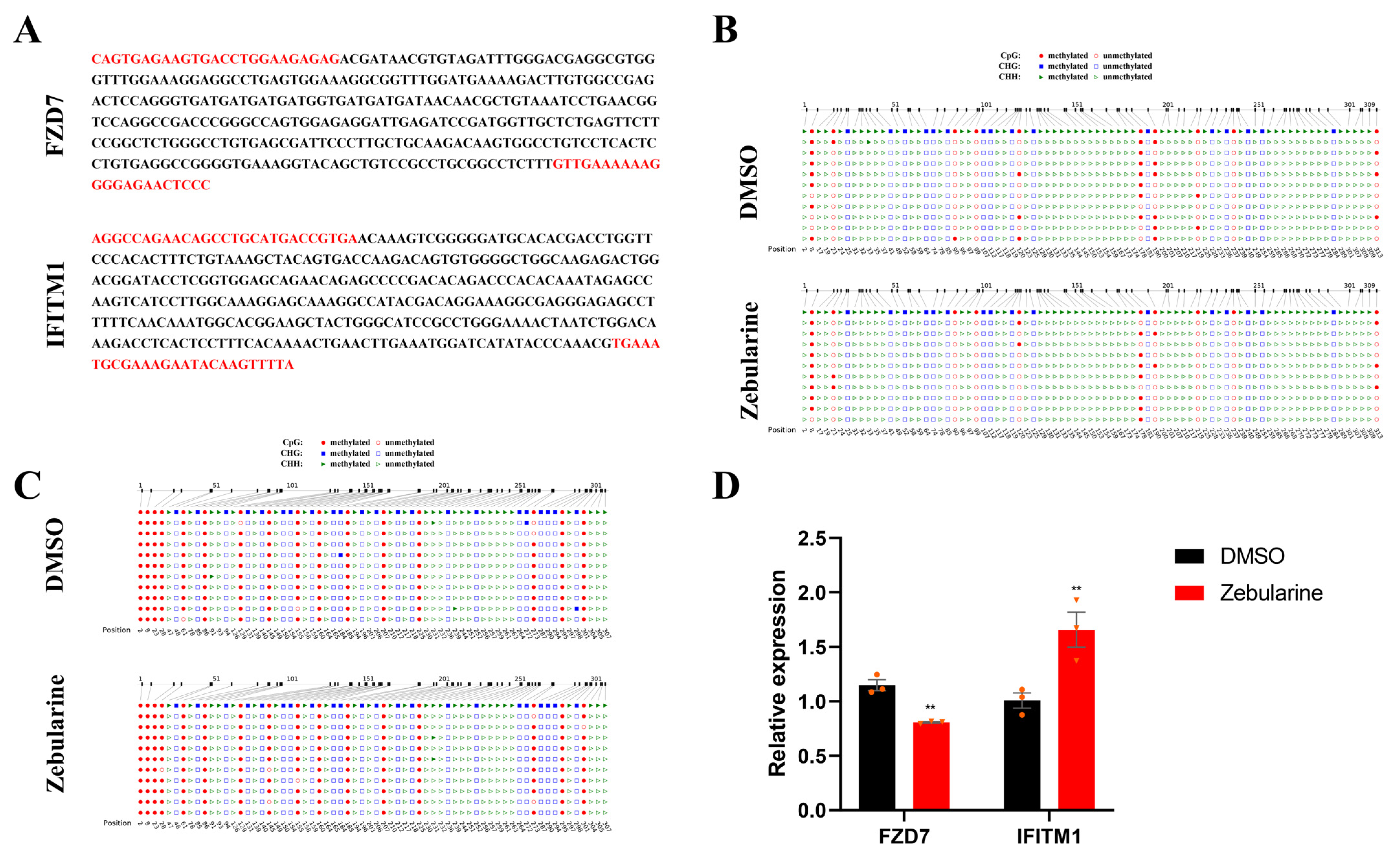
2.6. Zebularine Treatment
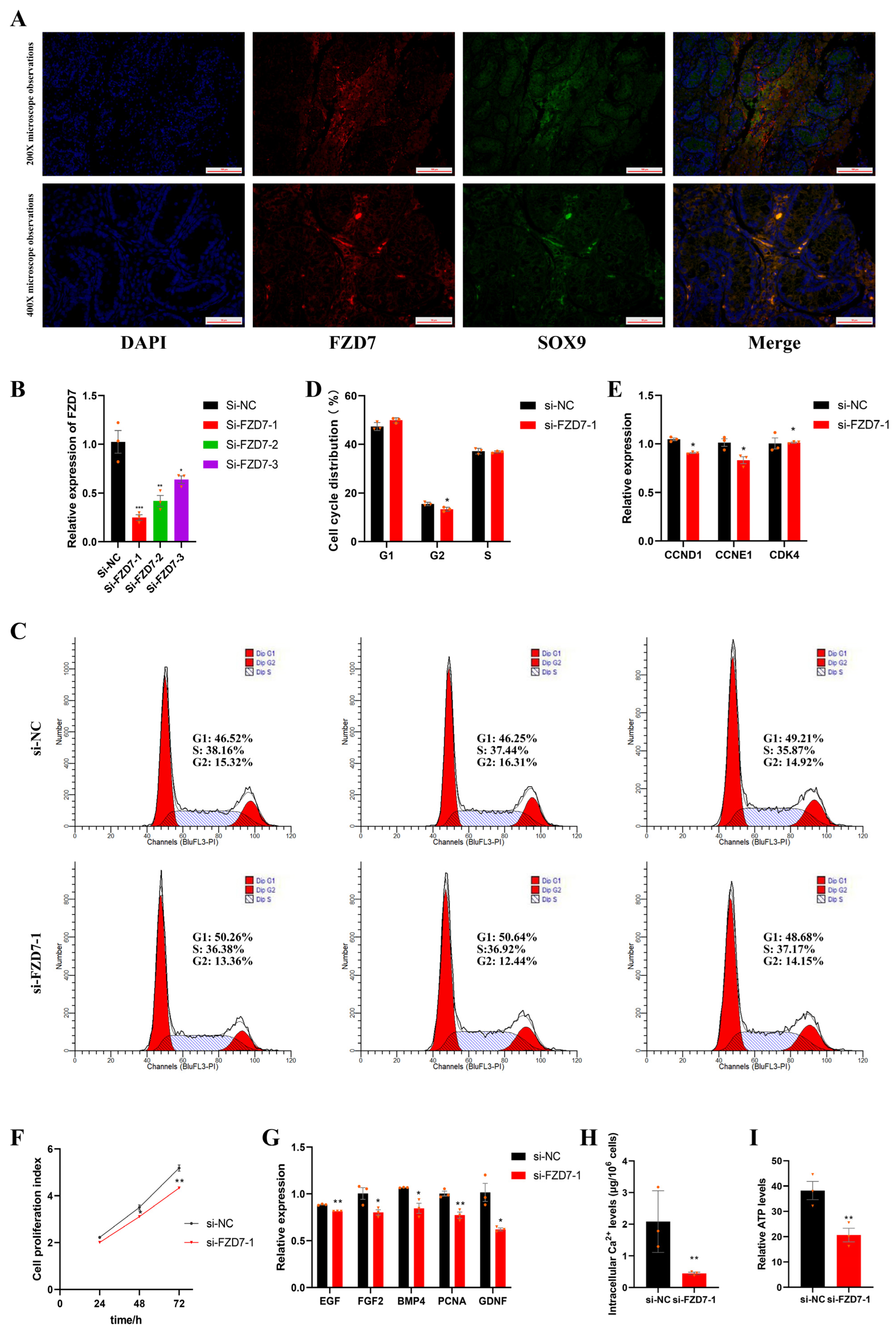
2.7. FZD7 Promotes Sertoli Cell Proliferation
2.8. FZD7 Increases Intracellular Ca2+ Concentrations and Adenosine Triphosphate (ATP) Levels
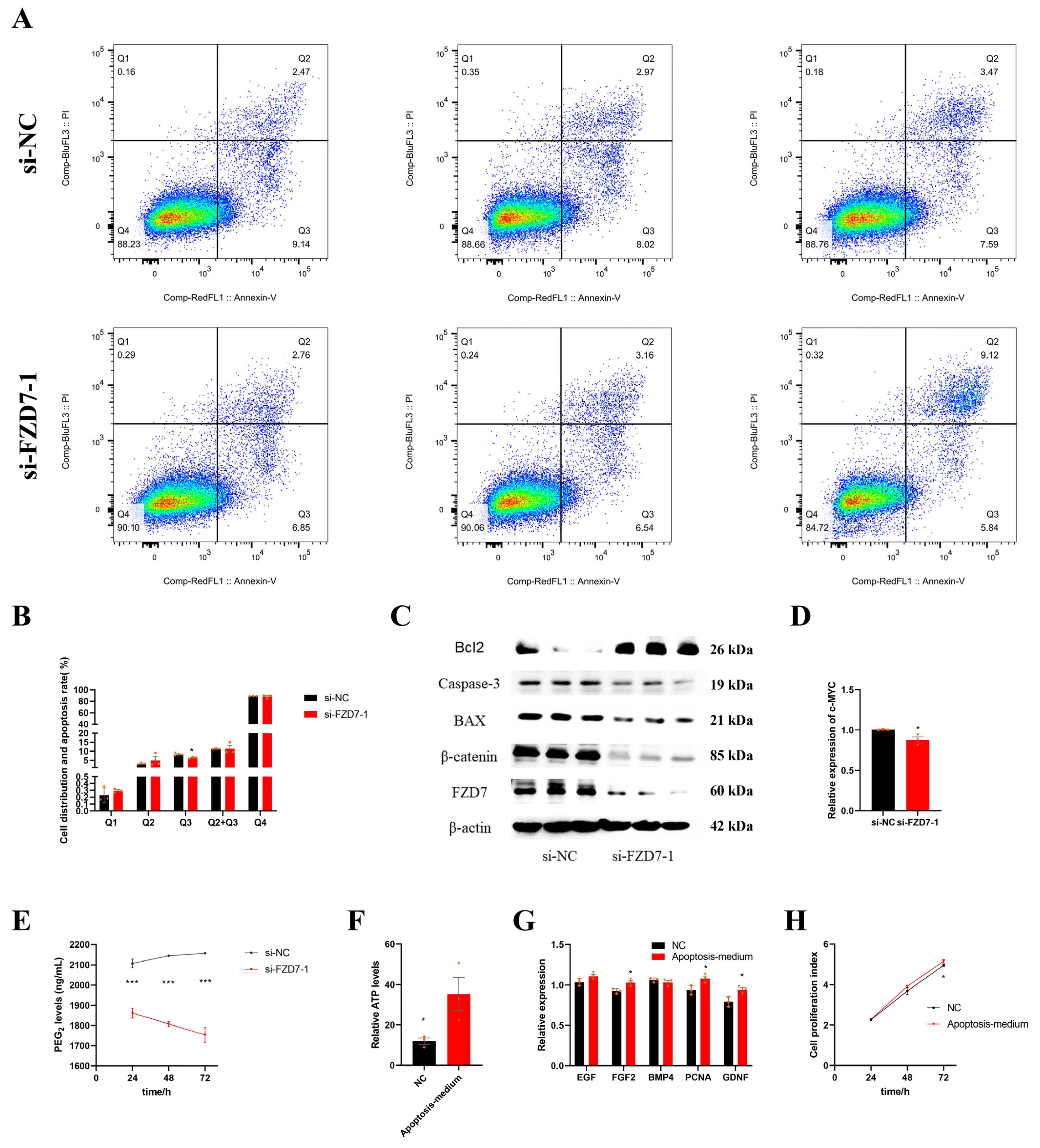
2.9. FZD7 Induces Early Apoptosis of Sertoli Cells
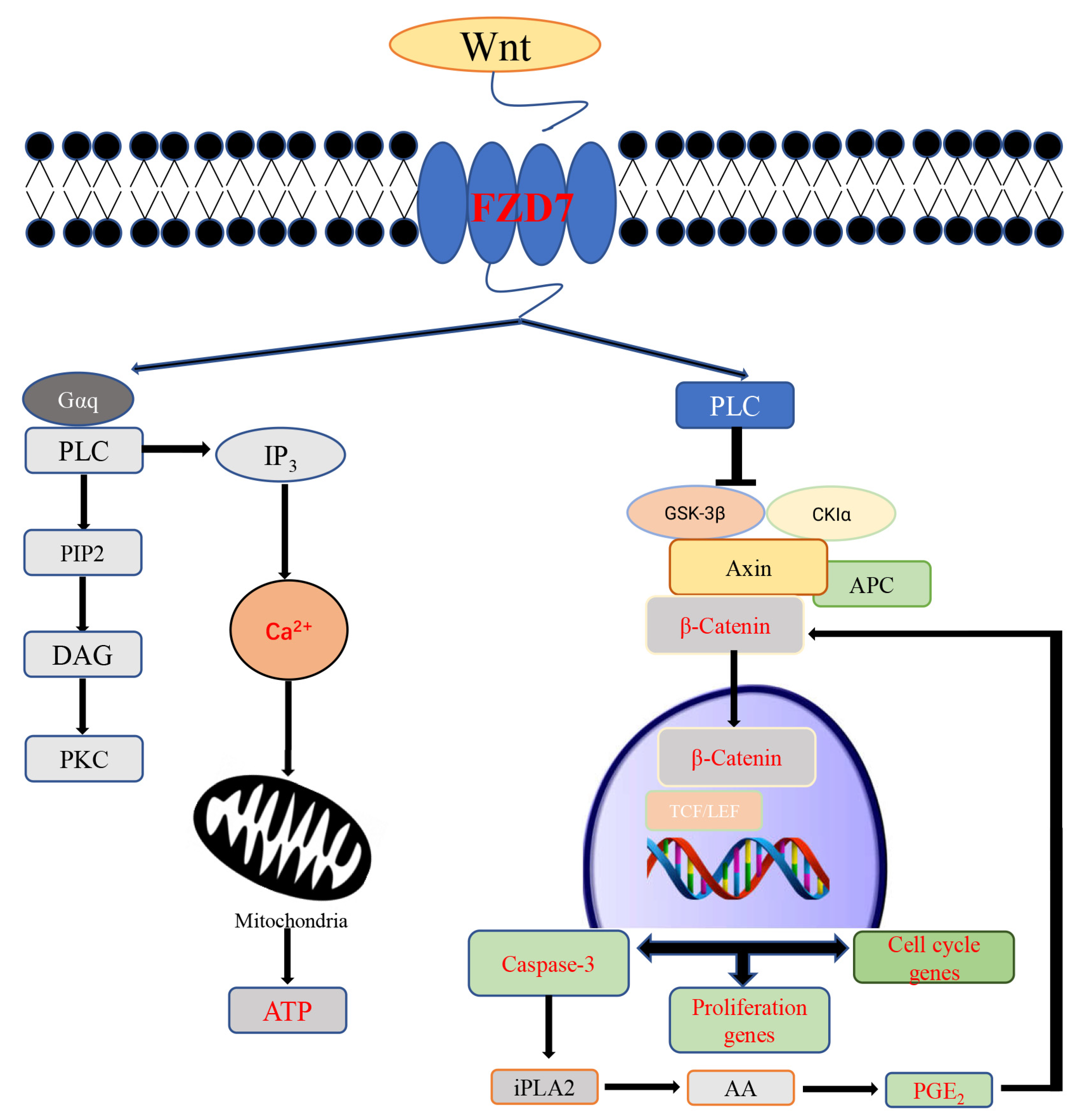
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Data Set from WGBS and RNA-Seq
4.3. Annotation of Gene and Genomic Features
4.4. Identification of DMRs and DMGs in Non-CpG Contexts
4.5. Correlation Analysis of DNA Methylation and Gene Expression
4.6. Enrichment Analysis
4.7. Construction and Module Analysis of the Protein–Protein Interaction (PPI) Network
4.8. Quantitative Real-Time PCR (qPCR)
4.9. Sertoli Cell Culture and Zebularine Treatment
4.10. Bisulfite Sequencing PCR (BSP)
4.11. RNA Interference (RNAi) of FZD7 and Transfection
4.12. Immunofluorescence Staining
4.13. Cell Cycle Assay
4.14. Cell Proliferation Assay
4.15. Cell Apoptosis Assay
4.16. Western Blotting
4.17. Measurement of the Intracellular Ca2+ Concentration
4.18. Adenosine Triphosphate (ATP) Assay
4.19. Prostaglandin E2 (PGE2) Assay
4.20. Apoptosis-Medium Treatment
4.21. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Griswold, M.D. 50 years of spermatogenesis: Sertoli cells and their interactions with germ cells. Biol. Reprod. 2018, 99, 87–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas-Herald, A.K.; Mitchell, R.T. Testicular Sertoli Cell Hormones in Differences in Sex Development. Front. Endocrinol. 2022, 13, 919670. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.J.; Khan, Y.S. Histology, Sertoli Cell. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Wu, Y.; Duan, P.; Wen, Y.; Zhang, J.; Wang, X.; Dong, J.; Zhao, Q.; Feng, S.; Lv, C.; Guo, Y.; et al. UHRF1 establishes crosstalk between somatic and germ cells in male reproduction. Cell Death Dis. 2022, 13, 377. [Google Scholar] [CrossRef]
- Lucas, T.F.; Nascimento, A.R.; Pisolato, R.; Pimenta, M.T.; Lazari, M.F.; Porto, C.S. Receptors and signaling pathways involved in proliferation and differentiation of Sertoli cells. Spermatogenesis 2014, 4, e28138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orth, J.M.; Gunsalus, G.L.; Lamperti, A.A. Evidence from Sertoli cell-depleted rats indicates that spermatid number in adults depends on numbers of Sertoli cells produced during perinatal development. Endocrinology 1988, 122, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.; Chen, T. DNA Methylation Reprogramming during Mammalian Development. Genes 2019, 10, 257. [Google Scholar] [CrossRef] [Green Version]
- Gruenbaum, Y.; Stein, R.; Cedar, H.; Razin, A. Methylation of CpG sequences in eukaryotic DNA. FEBS Lett. 1981, 124, 67–71. [Google Scholar] [CrossRef] [Green Version]
- Ramsahoye, B.H.; Biniszkiewicz, D.; Lyko, F.; Clark, V.; Bird, A.P.; Jaenisch, R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. USA 2000, 97, 5237–5242. [Google Scholar] [CrossRef] [Green Version]
- Barres, R.; Osler, M.E.; Yan, J.; Rune, A.; Fritz, T.; Caidahl, K.; Krook, A.; Zierath, J.R. Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab. 2009, 10, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Monti, N.; Cavallaro, R.A.; Stoccoro, A.; Nicolia, V.; Scarpa, S.; Kovacs, G.G.; Fiorenza, M.T.; Lucarelli, M.; Aronica, E.; Ferrer, I.; et al. CpG and non-CpG Presenilin1 methylation pattern in course of neurodevelopment and neurodegeneration is associated with gene expression in human and murine brain. Epigenetics 2020, 15, 781–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichiyanagi, T.; Ichiyanagi, K.; Miyake, M.; Sasaki, H. Accumulation and loss of asymmetric non-CpG methylation during male germ-cell development. Nucleic Acids Res. 2013, 41, 738–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, N.; Toh, H.; Shirane, K.; Shirakawa, T.; Kobayashi, H.; Sato, T.; Sone, H.; Sato, Y.; Tomizawa, S.; Tsurusaki, Y.; et al. DNA methylation and gene expression dynamics during spermatogonial stem cell differentiation in the early postnatal mouse testis. BMC Genom. 2015, 16, 624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anqi, Y.; Saina, Y.; Chujie, C.; Yanfei, Y.; Xiangwei, T.; Jiajia, M.; Jiaojiao, X.; Maoliang, R.; Bin, C. Regulation of DNA methylation during the testicular development of Shaziling pigs. Genomics 2022, 114, 110450. [Google Scholar] [CrossRef]
- Gowher, H.; Jeltsch, A. Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: The enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J. Mol. Biol. 2001, 309, 1201–1208. [Google Scholar] [CrossRef]
- Xie, W.; Barr, C.L.; Kim, A.; Yue, F.; Lee, A.Y.; Eubanks, J.; Dempster, E.L.; Ren, B. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 2012, 148, 816–831. [Google Scholar] [CrossRef] [Green Version]
- Patil, V.; Ward, R.L.; Hesson, L.B. The evidence for functional non-CpG methylation in mammalian cells. Epigenetics 2014, 9, 823–828. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Chen, B.; Weng, B.; Tang, X.; Chen, Y.; Yang, A.; Chu, D.; Zeng, X.; Ran, M. miR-130a promotes immature porcine Sertoli cell growth by activating SMAD5 through the TGF-beta-PI3K/AKT signaling pathway. FASEB J. 2020, 34, 15164–15179. [Google Scholar] [CrossRef]
- Chapin, N.; Fernandez, J.; Poole, J.; Delatte, B. Anchor-based bisulfite sequencing determines genome-wide DNA methylation. Commun. Biol. 2022, 5, 596. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [Green Version]
- de Mendoza, A.; Poppe, D.; Buckberry, S.; Pflueger, J.; Albertin, C.B.; Daish, T.; Bertrand, S.; de la Calle-Mustienes, E.; Gomez-Skarmeta, J.L.; Nery, J.R.; et al. The emergence of the brain non-CpG methylation system in vertebrates. Nat. Ecol. Evol. 2021, 5, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Saito, Y.; Park, S.J.; Nakai, K. Existence and possible roles of independent non-CpG methylation in the mammalian brain. DNA Res. 2020, 27, dsaa020. [Google Scholar] [CrossRef] [PubMed]
- Malone, C.S.; Miner, M.D.; Doerr, J.R.; Jackson, J.P.; Jacobsen, S.E.; Wall, R.; Teitell, M. CmC(A/T)GG DNA methylation in mature B cell lymphoma gene silencing. Proc. Natl. Acad. Sci. USA 2001, 98, 10404–10409. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Xiong, W.; Liu, X.; Xiao, W.; Guo, Y.; Tan, J.; Li, Y. Hypomethylation at non-CpG/CpG sites in the promoter of HIF-1alpha gene combined with enhanced H3K9Ac modification contribute to maintain higher HIF-1alpha expression in breast cancer. Oncogenesis 2019, 8, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucarelli, M.; Ferraguti, G.; Fuso, A. Active Demethylation of Non-CpG Moieties in Animals: A Neglected Research Area. Int. J. Mol. Sci. 2019, 20, 6272. [Google Scholar] [CrossRef] [Green Version]
- Sharma, G.; Upadhyay, S.; Srilalitha, M.; Nandicoori, V.K.; Khosla, S. The interaction of mycobacterial protein Rv2966c with host chromatin is mediated through non-CpG methylation and histone H3/H4 binding. Nucleic Acids Res. 2015, 43, 3922–3937. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Velasco, G.; Francastel, C. Genetics meets DNA methylation in rare diseases. Clin. Genet 2019, 95, 210–220. [Google Scholar] [CrossRef]
- Luo, C.; Hajkova, P.; Ecker, J.R. Dynamic DNA methylation: In the right place at the right time. Science 2018, 361, 1336–1340. [Google Scholar] [CrossRef] [Green Version]
- Weng, B.; Ran, M.; Chen, B.; He, C.; Dong, L.; Peng, F. Genome-wide analysis of long non-coding RNAs and their role in postnatal porcine testis development. Genomics 2017, 109, 446–456. [Google Scholar] [CrossRef]
- Li, T.; Luo, R.; Wang, X.; Wang, H.; Zhao, X.; Guo, Y.; Jiang, H.; Ma, Y. Unraveling Stage-Dependent Expression Patterns of Circular RNAs and Their Related ceRNA Modulation in Ovine Postnatal Testis Development. Front. Cell Dev. Biol. 2021, 9, 627439. [Google Scholar] [CrossRef] [PubMed]
- Zirkin, B.R.; Santulli, R.; Awoniyi, C.A.; Ewing, L.L. Maintenance of advanced spermatogenic cells in the adult rat testis: Quantitative relationship to testosterone concentration within the testis. Endocrinology 1989, 124, 3043–3049. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Li, X.; Li, K.; Zhang, J.; Li, Y.; Gong, D.; Zhao, G.; Zheng, Q.; Yuan, M.; Lin, P.; et al. Linoleic acid promotes testosterone production by activating Leydig cell GPR120/ERK pathway and restores BPA-impaired testicular toxicity. Steroids 2020, 163, 108677. [Google Scholar] [CrossRef]
- Sargent, K.M.; McFee, R.M.; Spuri Gomes, R.; Cupp, A.S. Vascular endothelial growth factor A: Just one of multiple mechanisms for sex-specific vascular development within the testis? J. Endocrinol. 2015, 227, R31–R50. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Li, S.Y.; DeFalco, T. Immune and vascular contributions to organogenesis of the testis and ovary. FEBS J. 2022, 289, 2386–2408. [Google Scholar] [CrossRef]
- Bott, R.C.; McFee, R.M.; Clopton, D.T.; Toombs, C.; Cupp, A.S. Vascular endothelial growth factor and kinase domain region receptor are involved in both seminiferous cord formation and vascular development during testis morphogenesis in the rat. Biol. Reprod. 2006, 75, 56–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruso, M.; Ferranti, F.; Corano Scheri, K.; Dobrowolny, G.; Ciccarone, F.; Grammatico, P.; Catizone, A.; Ricci, G. R-spondin 1/dickkopf-1/beta-catenin machinery is involved in testicular embryonic angiogenesis. PLoS ONE 2015, 10, e0124213. [Google Scholar] [CrossRef] [Green Version]
- Taciak, B.; Pruszynska, I.; Kiraga, L.; Bialasek, M.; Krol, M. Wnt signaling pathway in development and cancer. J. Physiol. Pharmacol. 2018, 69, 185–196. [Google Scholar] [CrossRef]
- Dong, W.L.; Tan, F.Q.; Yang, W.X. Wnt signaling in testis development: Unnecessary or essential? Gene 2015, 565, 155–165. [Google Scholar] [CrossRef]
- Lau, Y.F.; Li, Y. The human and mouse sex-determining SRY genes repress the Rspol/beta-catenin signaling. J. Genet. Genom. 2009, 36, 193–202. [Google Scholar] [CrossRef]
- Wang, X.; Adegoke, E.O.; Ma, M.; Huang, F.; Zhang, H.; Adeniran, S.O.; Zheng, P.; Zhang, G. Influence of Wilms’ tumor suppressor gene WT1 on bovine Sertoli cells polarity and tight junctions via non-canonical WNT signaling pathway. Theriogenology 2019, 138, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Sheldahl, L.C.; Park, M.; Malbon, C.C.; Moon, R.T. Protein kinase C is differentially stimulated by Wnt and Frizzled homologs in a G-protein-dependent manner. Curr. Biol. 1999, 9, 695–698. [Google Scholar] [CrossRef] [Green Version]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium-cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J. Calcium signalling and cell proliferation. Bioessays 1995, 17, 491–500. [Google Scholar] [CrossRef]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef] [Green Version]
- Harr, M.W.; Distelhorst, C.W. Apoptosis and autophagy: Decoding calcium signals that mediate life or death. Cold Spring Harb. Perspect. Biol. 2010, 2, a005579. [Google Scholar] [CrossRef] [PubMed]
- Morata, G.; Ripoll, P. Minutes: Mutants of drosophila autonomously affecting cell division rate. Dev. Biol. 1975, 42, 211–221. [Google Scholar] [CrossRef]
- Gauron, C.; Rampon, C.; Bouzaffour, M.; Ipendey, E.; Teillon, J.; Volovitch, M.; Vriz, S. Sustained production of ROS triggers compensatory proliferation and is required for regeneration to proceed. Sci. Rep. 2013, 3, 2084. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Wang, B.; Chen, P.P.; Wang, Y.Z.; Miao, N.J.; Yin, F.; Cheng, Q.; Zhou, Z.L.; Xie, H.Y.; Zhou, L.; et al. c-Myc promotes tubular cell apoptosis in ischemia-reperfusion-induced renal injury by negatively regulating c-FLIP and enhancing FasL/Fas-mediated apoptosis pathway. Acta. Pharmacol. Sin. 2019, 40, 1058–1066. [Google Scholar] [CrossRef]
- Geske, F.J.; Lieberman, R.; Strange, R.; Gerschenson, L.E. Early stages of p53-induced apoptosis are reversible. Cell Death Differ. 2001, 8, 182–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Wu, X.; Wang, Y.; Zhang, K.; Wu, J.; Yuan, Y.C.; Deng, X.; Chen, L.; Kim, C.C.; Lau, S.; et al. FZD7 has a critical role in cell proliferation in triple negative breast cancer. Oncogene 2011, 30, 4437–4446. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Hirasawa, K.; Bostwick, D.G.; Bergstralh, E.J.; Slezak, J.M.; Anderl, K.L.; Borell, T.J.; Lieber, M.M.; Jenkins, R.B. Loss of p53 and c-myc overrepresentation in stage T(2-3)N(1-3)M(0) prostate cancer are potential markers for cancer progression. Mod. Pathol. 2002, 15, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Wang, D.; Zhao, Z.; Xiao, Y.; Sengupta, S.; Xiao, Y.; Zhang, R.; Lauber, K.; Wesselborg, S.; Feng, L.; et al. Caspase-3-dependent activation of calcium-independent phospholipase A2 enhances cell migration in non-apoptotic ovarian cancer cells. J. Biol. Chem. 2006, 281, 29357–29368. [Google Scholar] [CrossRef] [Green Version]
- Bell, E.; Ponthan, F.; Whitworth, C.; Westermann, F.; Thomas, H.; Redfern, C.P. Cell survival signalling through PPARdelta and arachidonic acid metabolites in neuroblastoma. PLoS ONE 2013, 8, e68859. [Google Scholar] [CrossRef] [Green Version]
- Chera, S.; Ghila, L.; Dobretz, K.; Wenger, Y.; Bauer, C.; Buzgariu, W.; Martinou, J.C.; Galliot, B. Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev. Cell 2009, 17, 279–289. [Google Scholar] [CrossRef]
- Goessling, W.; North, T.E.; Loewer, S.; Lord, A.M.; Lee, S.; Stoick-Cooper, C.L.; Weidinger, G.; Puder, M.; Daley, G.Q.; Moon, R.T.; et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell 2009, 136, 1136–1147. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Chen, X.; Zhang, S.; Zhu, J.; Tang, B.; Wang, A.; Dong, L.; Zhang, Z.; Yu, C.; Sun, Y.; et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genom. Proteom. Bioinform. 2021, 19, 578–583. [Google Scholar] [CrossRef]
- CNCB-NGDC Members and Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 2022, 50, D27–D38. [Google Scholar] [CrossRef]
- Haeussler, M.; Zweig, A.S.; Tyner, C.; Speir, M.L.; Rosenbloom, K.R.; Raney, B.J.; Lee, C.M.; Lee, B.T.; Hinrichs, A.S.; Gonzalez, J.N.; et al. The UCSC Genome Browser database: 2019 update. Nucleic Acids Res. 2019, 47, D853–D858. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Conneely, K.N.; Wu, H. A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic Acids Res. 2014, 42, e69. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Wu, H. Differential methylation analysis for BS-seq data under general experimental design. Bioinformatics 2016, 32, 1446–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. S4), S11. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Gene Symbol | Score |
|---|---|---|
| 1 | VEGFA | 36 |
| 2 | PECAM1 | 21 |
| 3 | FZD7 | 17 |
| 4 | CXCL12 | 16 |
| 5 | VANGL1 | 15 |
| 6 | WNT9A | 14 |
| 6 | GRAP | 14 |
| 8 | FGF18 | 12 |
| 8 | ITGB6 | 12 |
| 10 | RYK | 10 |
| Genes | Zebularine-Treated Group | DMSO-Treated Group | ||||
|---|---|---|---|---|---|---|
| CpG | CHG | CHH | CpG | CHG | CHH | |
| FZD7 | 95.62 | 0 | 0.71 | 96.88 | 1.36 | 1.07 |
| IFITM1 | 25 | 0 | 0 | 30 | 0 | 0.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, A.; Yan, S.; Yin, Y.; Chen, C.; Tang, X.; Ran, M.; Chen, B. FZD7, Regulated by Non-CpG Methylation, Plays an Important Role in Immature Porcine Sertoli Cell Proliferation. Int. J. Mol. Sci. 2023, 24, 6179. https://doi.org/10.3390/ijms24076179
Yang A, Yan S, Yin Y, Chen C, Tang X, Ran M, Chen B. FZD7, Regulated by Non-CpG Methylation, Plays an Important Role in Immature Porcine Sertoli Cell Proliferation. International Journal of Molecular Sciences. 2023; 24(7):6179. https://doi.org/10.3390/ijms24076179
Chicago/Turabian StyleYang, Anqi, Saina Yan, Yanfei Yin, Chujie Chen, Xiangwei Tang, Maoliang Ran, and Bin Chen. 2023. "FZD7, Regulated by Non-CpG Methylation, Plays an Important Role in Immature Porcine Sertoli Cell Proliferation" International Journal of Molecular Sciences 24, no. 7: 6179. https://doi.org/10.3390/ijms24076179