
PT320, a Sustained-Release GLP-1 Receptor Agonist, Ameliorates L-DOPA-Induced Dyskinesia in a Mouse Model of Parkinson’s Disease
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
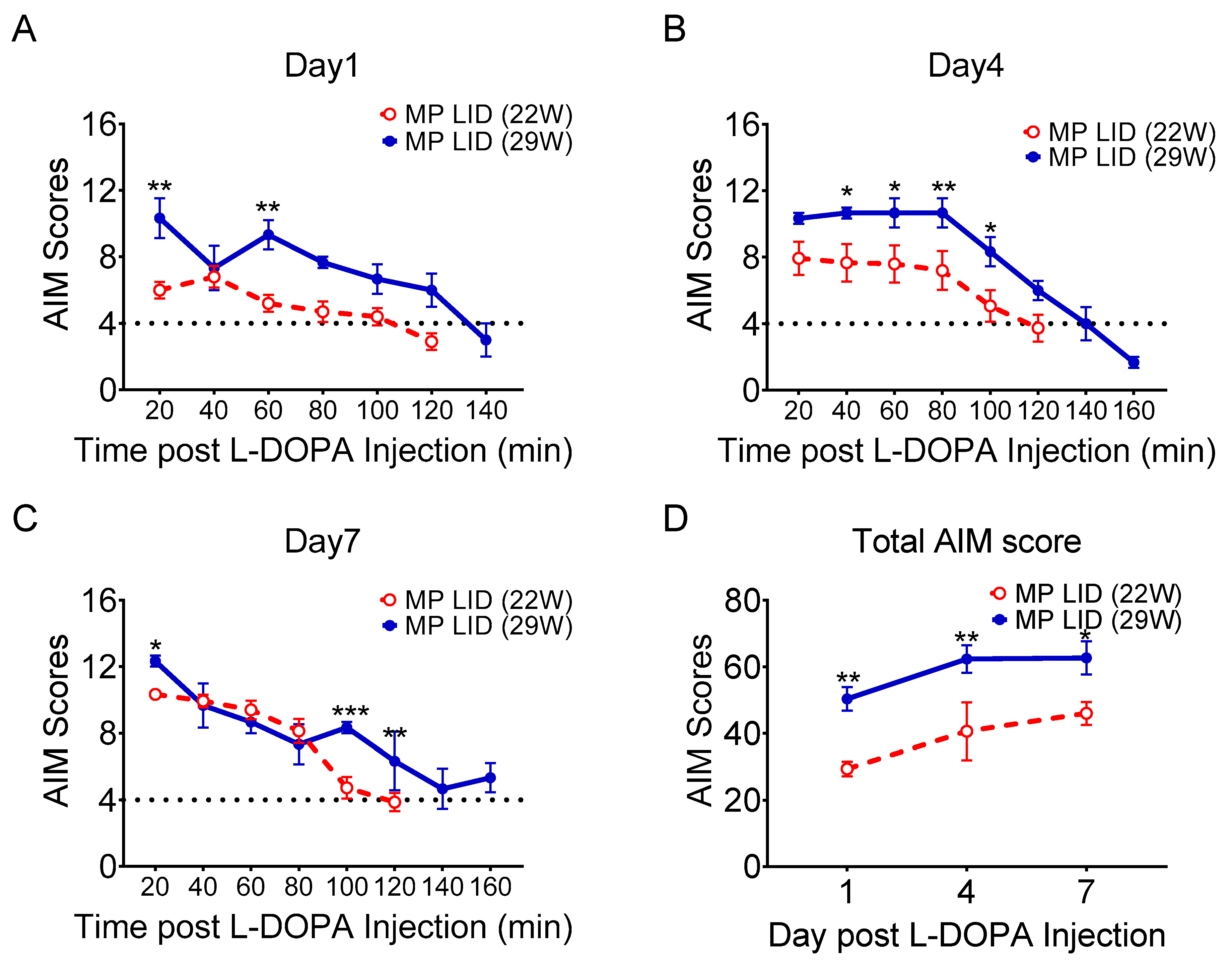
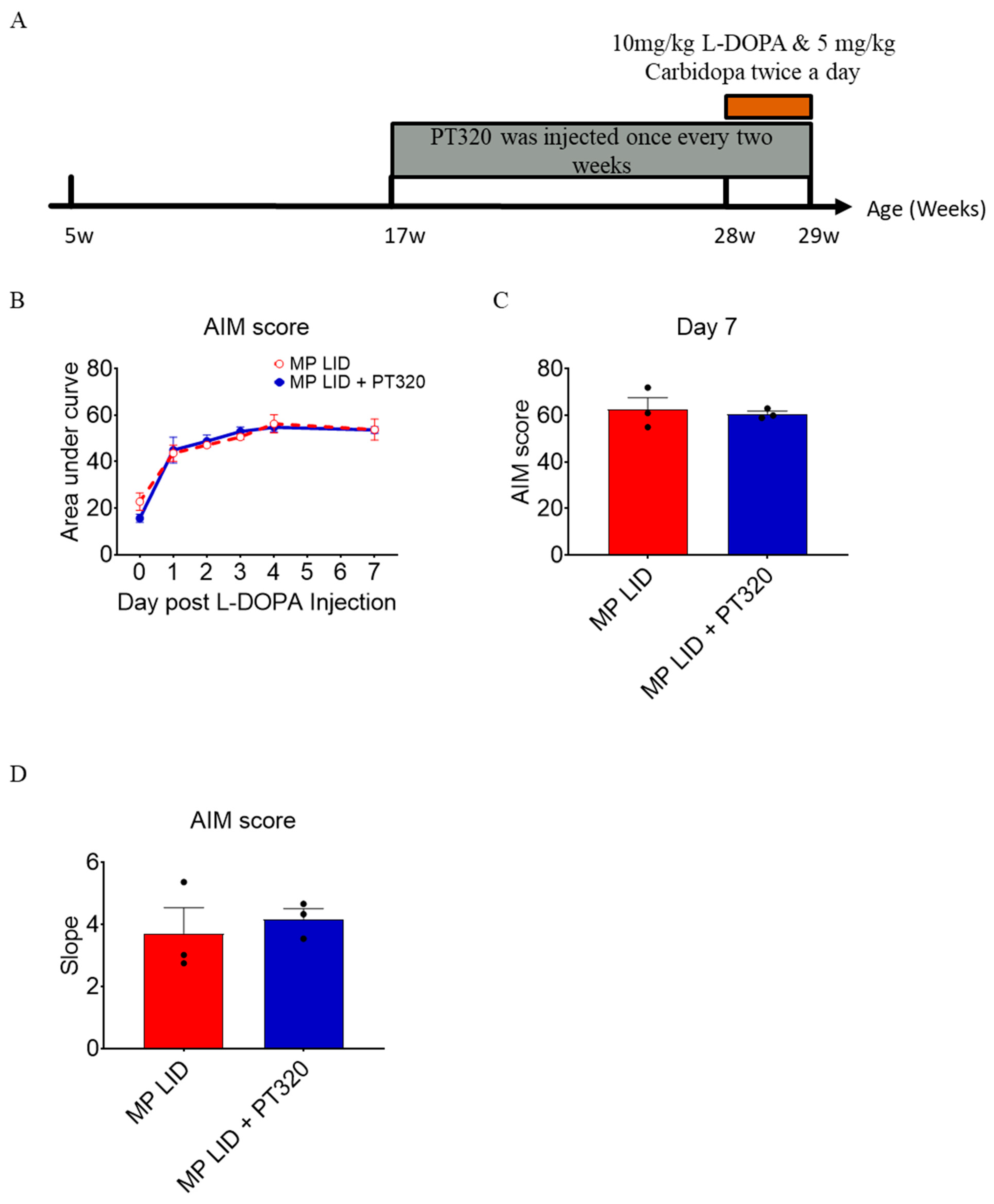
2.1. L-DOPA Priming
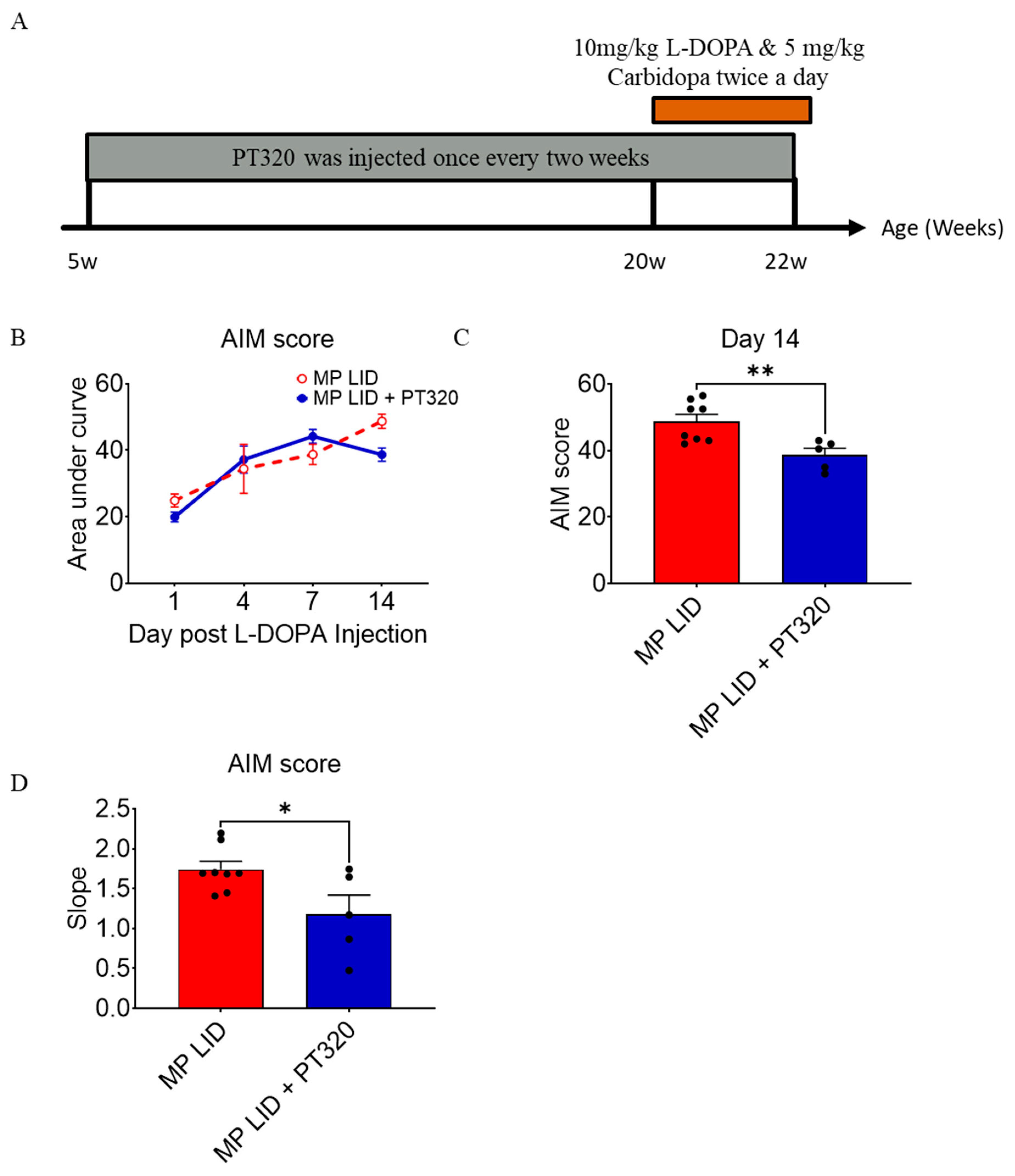
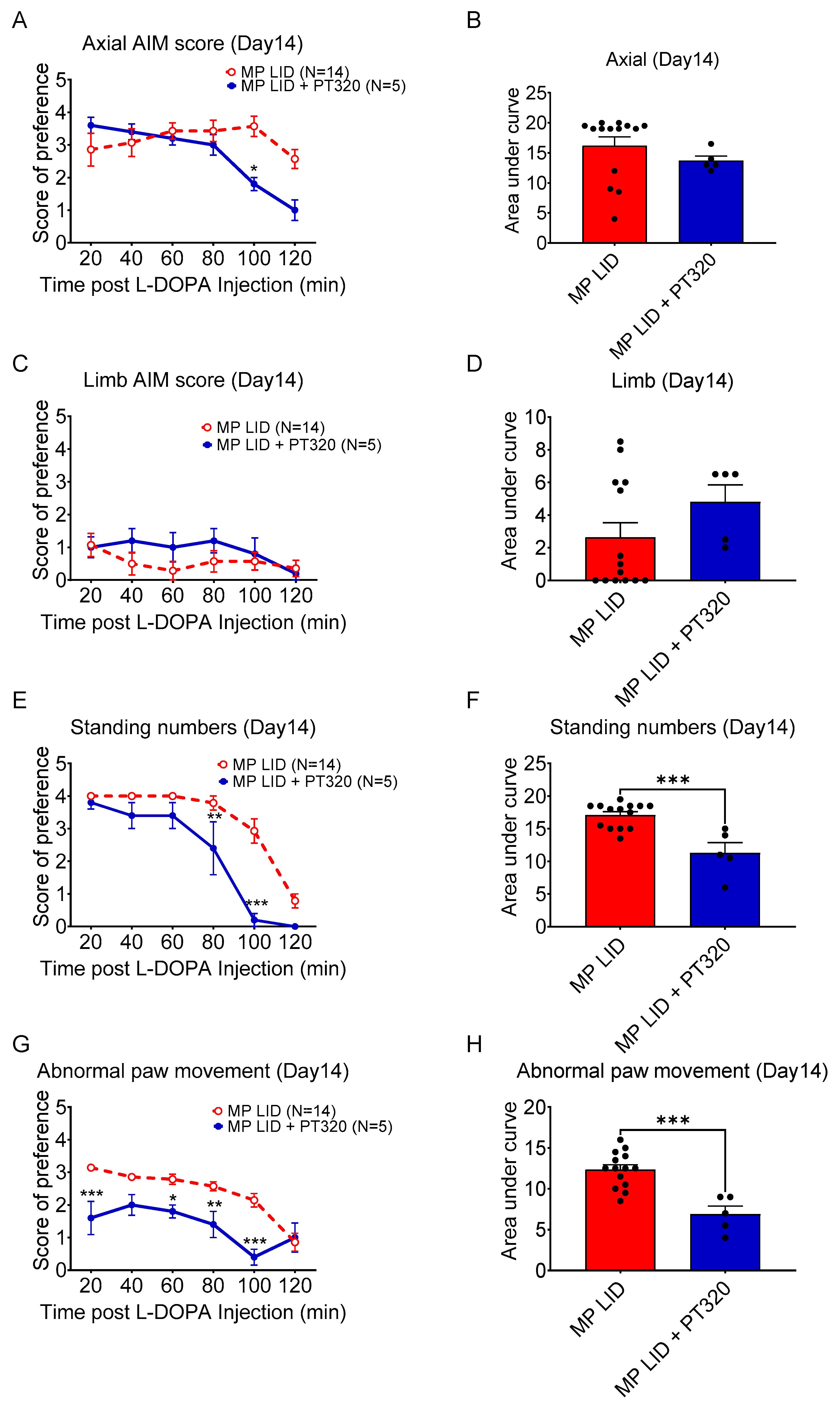
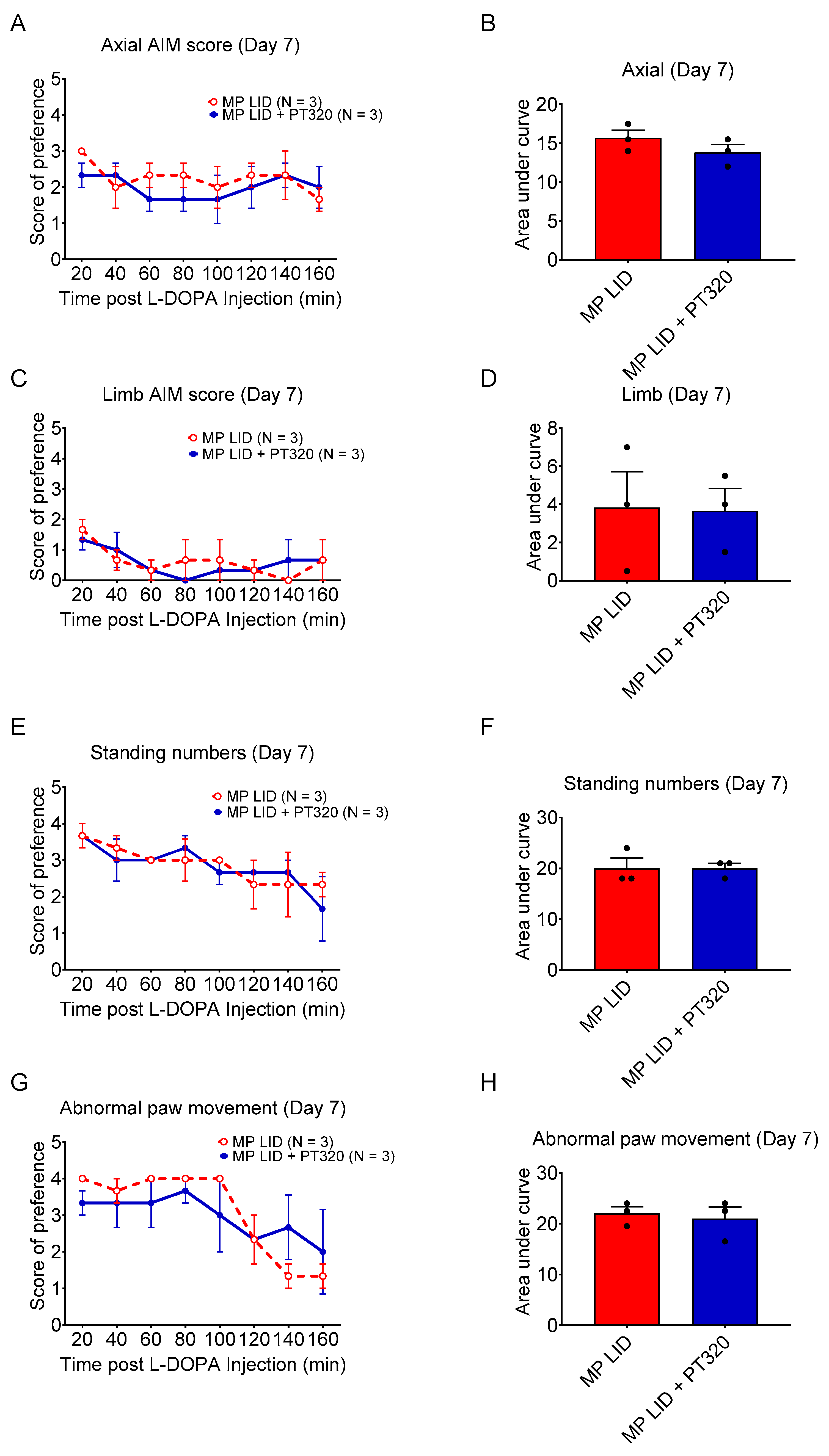
2.2. Early Administration of PT320 Reduced L-DOPA-Induced Dyskinesia
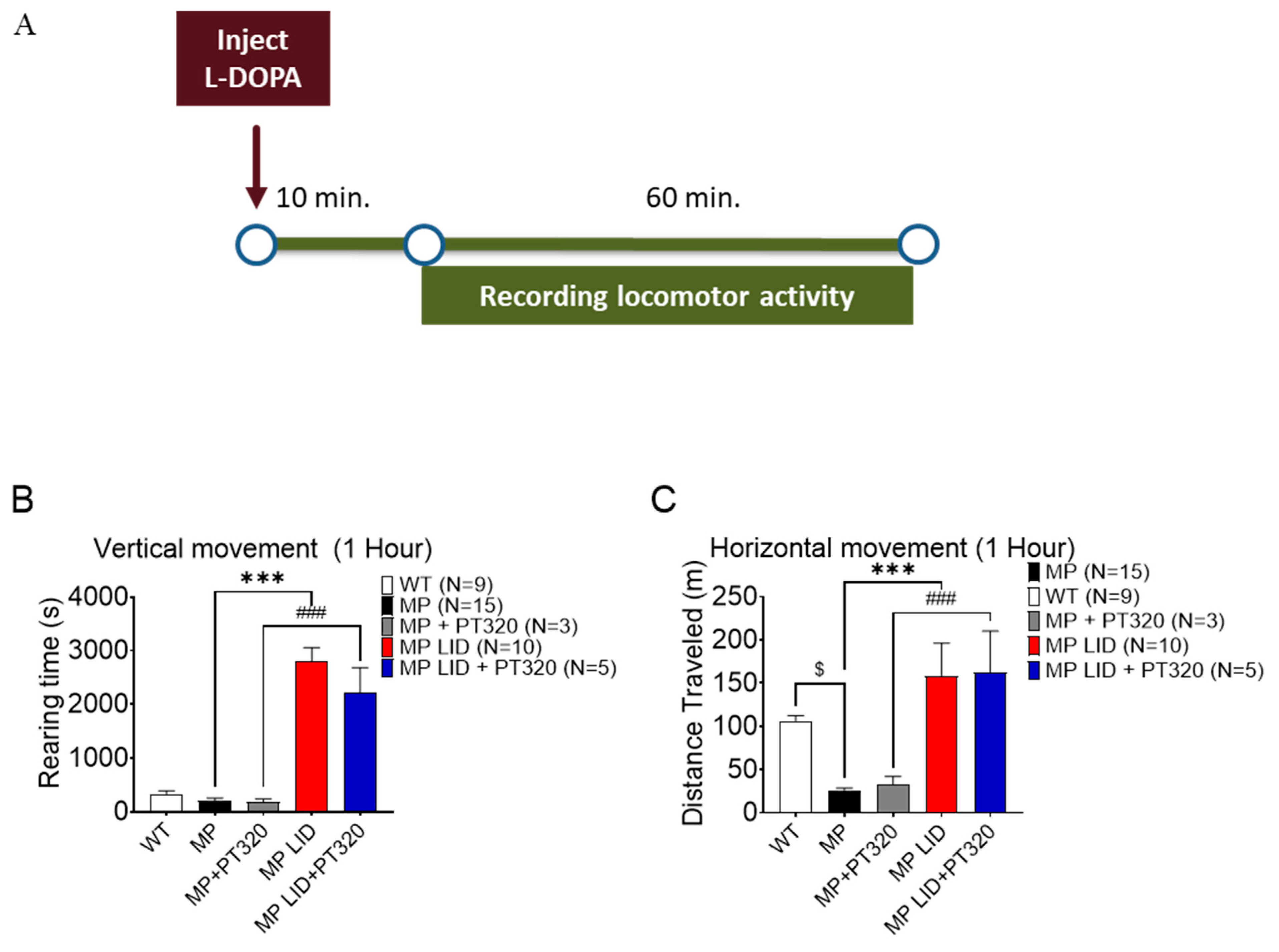
2.3. Early Administration of PT320 Did Not Alleviate L-DOPA-Induced Locomotor Hyperactivity
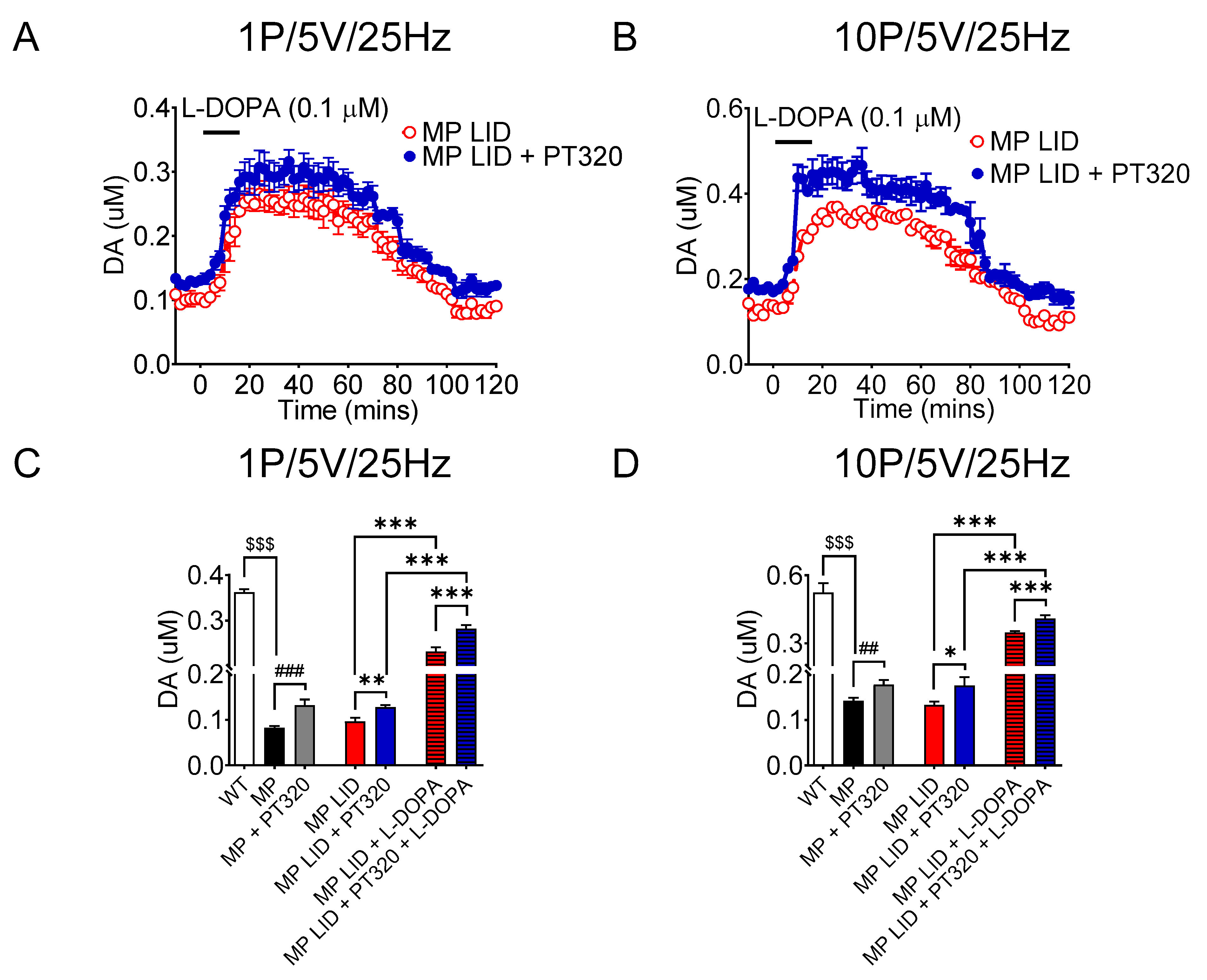
2.4. Early Administration of PT320 Increases Tonic and Phasic DA Release in Striatum of MitoPark LID Mice after L-DOPA Administration
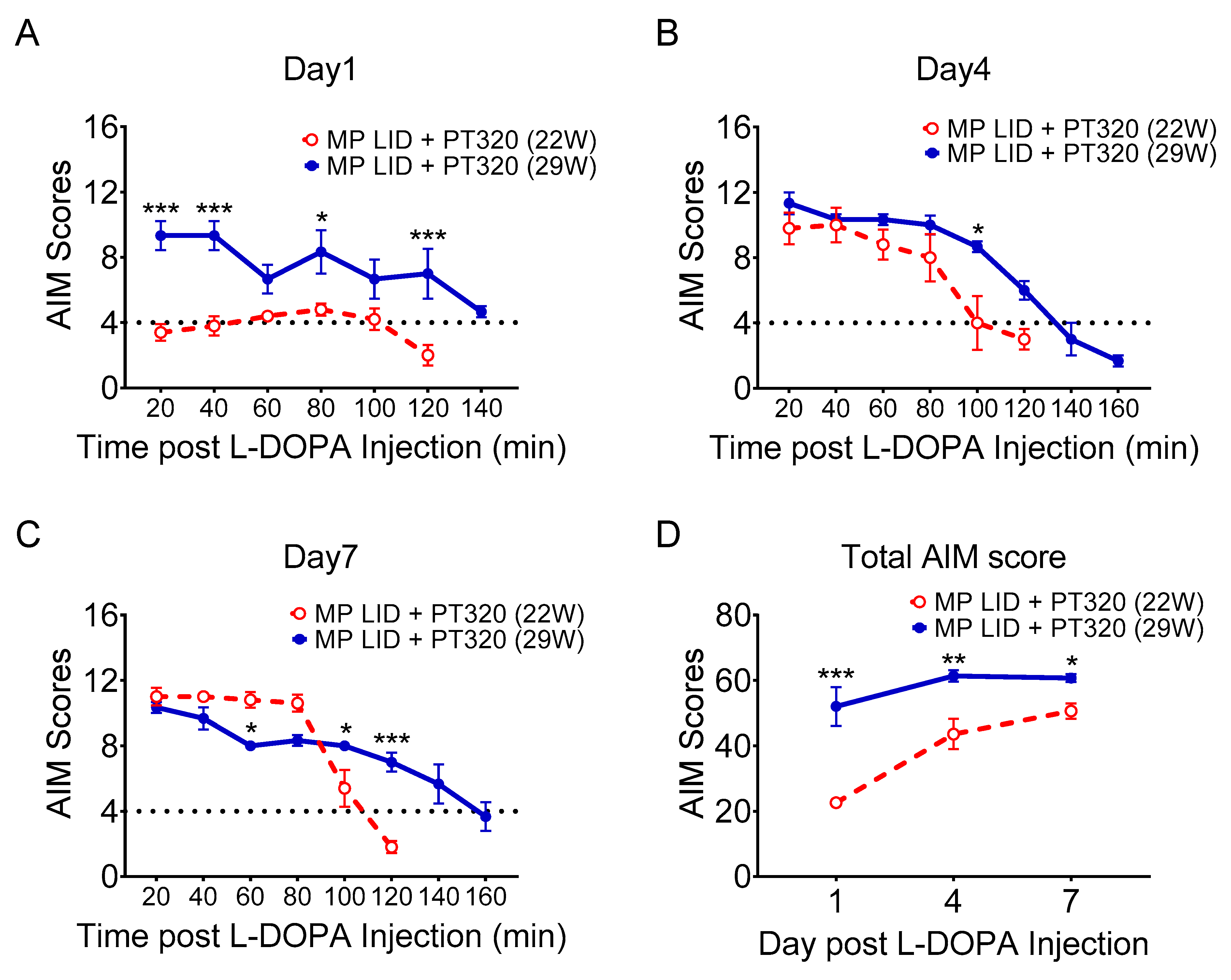
2.5. Late Administration of PT320 Did Not Mitigate L-DOPA-Induced Dyskinesia
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Treatments
4.3. Behavioral Tests
4.4. Fast Scan Cyclic Voltammetry (FSCV) for DA Dynamic Measurements in Brain Slices
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Calabresi, P.; Ghiglieri, V.; Mazzocchetti, P.; Corbelli, I.; Picconi, B. Levodopa-induced plasticity: A double-edged sword in Parkinson's disease? Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogetofte, H.; Alamyar, A.; Blaabjerg, M.; Meyer, M. Levodopa therapy for Parkinson’s disease: History, current status and perspectives. CNS Neurol. Disord. -Drug Targets (Former. Curr. Drug Targets-CNS Neurol. Disord.) 2020, 19, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Santini, E. Molecular Basis of L-DOPA-Induced Dyskinesia: Studies on Striatal Signaling; Institutionen för neurovetenskap/Department of Neuroscience: Stockholm, Sweden, 2009. [Google Scholar]
- Bastide, M.F.; Meissner, W.G.; Picconi, B.; Fasano, S.; Fernagut, P.-O.; Feyder, M.; Francardo, V.; Alcacer, C.; Ding, Y.; Brambilla, R. Pathophysiology of L-dopa-induced motor and non-motor complications in Parkinson's disease. Prog. Neurobiol. 2015, 132, 96–168. [Google Scholar] [CrossRef]
- De la Fuente-Fernández, R.; Sossi, V.; Huang, Z.; Furtado, S.; Lu, J.-Q.; Calne, D.B.; Ruth, T.J.; Stoessl, A.J. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson's disease: Implications for dyskinesias. Brain 2004, 127, 2747–2754. [Google Scholar] [CrossRef] [Green Version]
- Lindgren, H.S.; Andersson, D.R.; Lagerkvist, S.; Nissbrandt, H.; Cenci, M.A. l-DOPA-induced dopamine efflux in the striatum and the substantia nigra in a rat model of Parkinson’s disease: Temporal and quantitative relationship to the expression of dyskinesia. J. Neurochem. 2010, 112, 1465–1476. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P. Molecular mechanisms of L-DOPA-induced dyskinesia. Nat. Rev. Neurosci. 2008, 9, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Tseng, K.Y.; Kuo, T.T.; Wang, V.; Huang, E.Y.; Ma, K.H.; Olson, L.; Hoffer, B.J.; Chen, Y.H. Tetrabenazine Mitigates Aberrant Release and Clearance of Dopamine in the Nigrostriatal System, and Alleviates L-DOPA-Induced Dyskinesia in a Mouse Model of Parkinson’s Disease. J. Park. Dis. 2022, 12, 1545–1565. [Google Scholar] [CrossRef]
- Werner, F.-M.; Coveñas, R. Classical Neurotransmitters and Neuropeptides Involved in Parkinson’s Disease: Focus on Anti-Parkinsonian Drugs. Curr. Drug Ther. 2015, 10, 66–81. [Google Scholar] [CrossRef]
- Gonzalez-Latapi, P.; Bhowmick, S.S.; Saranza, G.; Fox, S.H. Non-dopaminergic treatments for motor control in Parkinson’s disease: An update. CNS Drugs 2020, 34, 1025–1044. [Google Scholar] [CrossRef]
- Zheng, C.Q.; Fan, H.X.; Li, X.X.; Li, J.J.; Sheng, S.; Zhang, F. Resveratrol Alleviates Levodopa-Induced Dyskinesia in Rats. Front Immunol. 2021, 12, 683577. [Google Scholar] [CrossRef]
- Good, C.H.; Hoffman, A.F.; Hoffer, B.J.; Chefer, V.I.; Shippenberg, T.S.; Backman, C.M.; Larsson, N.G.; Olson, L.; Gellhaar, S.; Galter, D.; et al. Impaired nigrostriatal function precedes behavioral deficits in a genetic mitochondrial model of Parkinson's disease. FASEB J. 2011, 25, 1333–1344. [Google Scholar] [CrossRef] [Green Version]
- Galter, D.; Pernold, K.; Yoshitake, T.; Lindqvist, E.; Hoffer, B.; Kehr, J.; Larsson, N.G.; Olson, L. MitoPark mice mirror the slow progression of key symptoms and L-DOPA response in Parkinson's disease. Genes Brain Behav. 2010, 9, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Ekstrand, M.I.; Galter, D. The MitoPark Mouse–An animal model of Parkinson's disease with impaired respiratory chain function in dopamine neurons. Park. Relat. Disord. 2009, 15, S185–S188. [Google Scholar] [CrossRef]
- Glotfelty, E.J.; Olson, L.; Karlsson, T.E.; Li, Y.; Greig, N.H. Glucagon-like peptide-1 (GLP-1)-based receptor agonists as a treatment for Parkinson’s disease. Expert Opin. Investig. Drugs 2020, 29, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Reich, N.; Hölscher, C. The neuroprotective effects of glucagon-like peptide 1 in Alzheimer’s and Parkinson’s disease: An in-depth review. Front. Neurosci. 2022, 16, 970925. [Google Scholar] [CrossRef] [PubMed]
- Salameh, T.S.; Rhea, E.M.; Talbot, K.; Banks, W.A. Brain uptake pharmacokinetics of incretin receptor agonists showing promise as Alzheimer’s and Parkinson’s disease therapeutics. Biochem. Pharmacol. 2020, 180, 114187. [Google Scholar] [CrossRef] [PubMed]
- Athauda, D.; Maclagan, K.; Skene, S.S.; Bajwa-Joseph, M.; Letchford, D.; Chowdhury, K.; Hibbert, S.; Budnik, N.; Zampedri, L.; Dickson, J. Exenatide once weekly versus placebo in Parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1664–1675. [Google Scholar] [CrossRef]
- Li, Y.; Vaughan, K.L.; Tweedie, D.; Jung, J.; Kim, H.K.; Choi, H.-I.; Kim, D.S.; Mattison, J.A.; Greig, N.H. Pharmacokinetics of Exenatide in nonhuman primates following its administration in the form of sustained-release PT320 and Bydureon. Sci. Rep. 2019, 9, 17208. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Yu, S.-J.; Li, Y.; Lecca, D.; Glotfelty, E.; Kim, H.K.; Choi, H.-I.; Hoffer, B.J.; Greig, N.H.; Kim, D.S. Post-treatment with PT302, a long-acting Exendin-4 sustained release formulation, reduces dopaminergic neurodegeneration in a 6-Hydroxydopamine rat model of Parkinson’s disease. Sci. Rep. 2018, 8, 10722. [Google Scholar]
- Bader, M.; Li, Y.; Lecca, D.; Rubovitch, V.; Tweedie, D.; Glotfelty, E.; Rachmany, L.; Kim, H.K.; Choi, H.-I.; Hoffer, B.J. Pharmacokinetics and efficacy of PT302, a sustained-release Exenatide formulation, in a murine model of mild traumatic brain injury. Neurobiol. Dis. 2019, 124, 439–453. [Google Scholar] [CrossRef]
- Wang, V.; Kuo, T.-T.; Huang, E.Y.-K.; Ma, K.-H.; Chou, Y.-C.; Fu, Z.-Y.; Lai, L.-W.; Jung, J.; Choi, H., II.; Choi, D.-S. Sustained release GLP-1 Agonist PT320 delays disease progression in a mouse model of Parkinson’s disease. ACS Pharmacol. Transl. Sci. 2021, 4, 858–869. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Terzioglu, M.; Galter, D.; Zhu, S.; Hofstetter, C.; Lindqvist, E.; Thams, S.; Bergstrand, A.; Hansson, F.S.; Trifunovic, A.; et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 1325–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gellhaar, S.; Marcellino, D.; Abrams, M.B.; Galter, D. Chronic L-DOPA induces hyperactivity, normalization of gait and dyskinetic behavior in MitoPark mice. Genes Brain Behav. 2015, 14, 260–270. [Google Scholar] [CrossRef] [Green Version]
- Shan, L.; Diaz, O.; Zhang, Y.; Ladenheim, B.; Cadet, J.-L.; Chiang, Y.-H.; Olson, L.; Hoffer, B.J.; Bäckman, C.M. L-Dopa induced dyskinesias in Parkinsonian mice: Disease severity or L-Dopa history. Brain Res. 2015, 1618, 261–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, T.; Lahiri, D.K.; Chen, D.; Zhou, J.I.E.; Shaw, K.T.Y.; Egan, J.M.; Greig, N.H. A novel neurotrophic property of glucagon-like peptide 1: A promoter of nerve growth factor-mediated differentiation in PC12 cells. J. Pharmacol. Exp. Ther. 2002, 300, 958–966. [Google Scholar] [CrossRef] [Green Version]
- Perry, T.; Lahiri, D.K.; Sambamurti, K.; Chen, D.; Mattson, M.P.; Egan, J.M.; Greig, N.H. Glucagon-like peptide-1 decreases endogenous amyloid-β peptide (Aβ) levels and protects hippocampal neurons from death induced by Aβ and iron. J. Neurosci. Res. 2003, 72, 603–612. [Google Scholar] [CrossRef]
- Li, Y.; Perry, T.; Kindy, M.S.; Harvey, B.K.; Tweedie, D.; Holloway, H.W.; Powers, K.; Shen, H.; Egan, J.M.; Sambamurti, K. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc. Natl. Acad. Sci. USA 2009, 106, 1285–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Duffy, K.B.; Ottinger, M.A.; Ray, B.; Bailey, J.A.; Holloway, H.W.; Tweedie, D.; Perry, T.; Mattson, M.P.; Kapogiannis, D. GLP-1 receptor stimulation reduces amyloid-β peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer's disease. J. Alzheimer's Dis. 2010, 19, 1205–1219. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Tweedie, D.; Mattson, M.P.; Holloway, H.W.; Greig, N.H. Enhancing the GLP-1 receptor signaling pathway leads to proliferation and neuroprotection in human neuroblastoma cells. J. Neurochem. 2010, 113, 1621–1631. [Google Scholar] [CrossRef] [Green Version]
- Botfield, H.F.; Uldall, M.S.; Westgate, C.S.J.; Mitchell, J.L.; Hagen, S.M.; Gonzalez, A.M.; Hodson, D.J.; Jensen, R.H.; Sinclair, A.J. A glucagon-like peptide-1 receptor agonist reduces intracranial pressure in a rat model of hydrocephalus. Sci. Transl. Med. 2017, 9, eaan0972. [Google Scholar] [CrossRef] [Green Version]
- Athauda, D.; Foltynie, T. Protective effects of the GLP-1 mimetic exendin-4 in Parkinson's disease. Neuropharmacology 2018, 136, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Glotfelty, E.J.; Delgado, T.E.; Tovar-y-Romo, L.B.; Luo, Y.; Hoffer, B.J.; Olson, L.; Karlsson, T.E.; Mattson, M.P.; Harvey, B.K.; Tweedie, D. Incretin mimetics as rational candidates for the treatment of traumatic brain injury. ACS Pharmacol. Transl. Sci. 2019, 2, 66–91. [Google Scholar] [CrossRef] [PubMed]
- Tovar-y-Romo, L.B.; Ramírez-Jarquín, U.N.; Lazo-Gómez, R.; Tapia, R. Trophic factors as modulators of motor neuron physiology and survival: Implications for ALS therapy. Front. Cell. Neurosci. 2014, 8, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Z.; Chen, H.; Xia, M.; Chang, J.; Li, X.; Ye, S.; Wu, S.; Jiang, S.; Bao, J.; Wang, B. Activation of glucagon-like peptide-1 receptor in microglia attenuates neuroinflammation-induced glial scarring via rescuing Arf and Rho GAP adapter protein 3 expressions after nerve injury. Int. J. Biol. Sci. 2022, 18, 1328. [Google Scholar] [CrossRef]
- Xie, Y.; Zheng, J.; Li, S.; Li, H.; Zhou, Y.; Zheng, W.; Zhang, M.; Liu, L.; Chen, Z. GLP-1 improves the neuronal supportive ability of astrocytes in Alzheimer’s disease by regulating mitochondrial dysfunction via the cAMP/PKA pathway. Biochem. Pharmacol. 2021, 188, 114578. [Google Scholar] [CrossRef]
- Jankovic, J.; Rajput, A.H.; McDermott, M.P.; Perl, D.P.; Parkinson Study, G. The evolution of diagnosis in early Parkinson disease. Arch. Neurol. 2000, 57, 369–372. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Van Gerpen, J.A.; Bower, J.H.; Ahlskog, J.E. Levodopa-dyskinesia incidence by age of Parkinson’s disease onset. Mov. Disord. 2005, 20, 342–344. [Google Scholar] [CrossRef]
- Schrag, A.; Quinn, N. Dyskinesias and motor fluctuations in Parkinson's disease: A community-based study. Brain 2000, 123, 2297–2305. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.-J.; Chen, S.; Yang, Y.-Y.; Glotfelty, E.J.; Jung, J.; Kim, H.K.; Choi, H.-I.; Choi, D.-S.; Hoffer, B.J.; Greig, N.H. PT320, sustained-release exendin-4, mitigates L-DOPA-induced dyskinesia in a rat 6-hydroxydopamine model of Parkinson’s disease. Front. Neurosci. 2020, 14, 785. [Google Scholar] [CrossRef]
- Ding, Y.; Restrepo, J.; Won, L.; Hwang, D.-Y.; Kim, K.-S.; Kang, U.J. Chronic 3, 4-dihydroxyphenylalanine treatment induces dyskinesia in aphakia mice, a novel genetic model of Parkinson’s disease. Neurobiol. Dis. 2007, 27, 11–23. [Google Scholar] [CrossRef] [Green Version]
- Sebastianutto, I.; Maslava, N.; Hopkins, C.R.; Cenci, M.A. Validation of an improved scale for rating l-DOPA-induced dyskinesia in the mouse and effects of specific dopamine receptor antagonists. Neurobiol. Dis. 2016, 96, 156–170. [Google Scholar] [CrossRef]
- Lundblad, M.; Picconi, B.; Lindgren, H.; Cenci, M.A. A model of L-DOPA-induced dyskinesia in 6-hydroxydopamine lesioned mice: Relation to motor and cellular parameters of nigrostriatal function. Neurobiol. Dis. 2004, 16, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Harvey, B.K.; Hoffman, A.F.; Wang, Y.; Chiang, Y.H.; Lupica, C.R. MPTP-induced deficits in striatal synaptic plasticity are prevented by glial cell line-derived neurotrophic factor expressed via an adeno-associated viral vector. FASEB J. 2008, 22, 261–275. [Google Scholar] [CrossRef] [Green Version]
- Good, C.H.; Wang, H.; Chen, Y.-H.; Mejias-Aponte, C.A.; Hoffman, A.F.; Lupica, C.R. Dopamine D4 receptor excitation of lateral habenula neurons via multiple cellular mechanisms. J. Neurosci. 2013, 33, 16853–16864. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.-Y.; Reddy, S.P.; Kleeberger, S.R. Nrf2 defends the lung from oxidative stress. Antioxid. Redox Signal. 2006, 8, 76–87. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Huang, E.Y.-K.; Kuo, T.-T.; Hoffer, B.J.; Miller, J.; Chou, Y.-C.; Chiang, Y.-H. Dopamine release in the nucleus accumbens is altered following traumatic brain injury. Neuroscience 2017, 348, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Kawagoe, K.T.; Zimmerman, J.B.; Wightman, R.M. Principles of voltammetry and microelectrode surface states. J. Neurosci. Methods 1993, 48, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Thanvi, B.; Lo, N.; Robinson, T. Levodopa-induced dyskinesia in Parkinson’s disease: Clinical features, pathogenesis, prevention and treatment. Postgrad. Med. J. 2007, 83, 384–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cenci, M.A. Presynaptic mechanisms of l-DOPA-induced dyskinesia: The findings, the debate, and the therapeutic implications. Front. Neurol. 2014, 5, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, T.-T.; Chen, Y.-H.; Wang, V.; Huang, E.Y.-K.; Ma, K.-H.; Greig, N.H.; Jung, J.; Choi, H.-I.; Olson, L.; Hoffer, B.J.; et al. PT320, a Sustained-Release GLP-1 Receptor Agonist, Ameliorates L-DOPA-Induced Dyskinesia in a Mouse Model of Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 4687. https://doi.org/10.3390/ijms24054687
Kuo T-T, Chen Y-H, Wang V, Huang EY-K, Ma K-H, Greig NH, Jung J, Choi H-I, Olson L, Hoffer BJ, et al. PT320, a Sustained-Release GLP-1 Receptor Agonist, Ameliorates L-DOPA-Induced Dyskinesia in a Mouse Model of Parkinson’s Disease. International Journal of Molecular Sciences. 2023; 24(5):4687. https://doi.org/10.3390/ijms24054687
Chicago/Turabian StyleKuo, Tung-Tai, Yuan-Hao Chen, Vicki Wang, Eagle Yi-Kung Huang, Kuo-Hsing Ma, Nigel H. Greig, Jin Jung, Ho-II Choi, Lars Olson, Barry J. Hoffer, and et al. 2023. "PT320, a Sustained-Release GLP-1 Receptor Agonist, Ameliorates L-DOPA-Induced Dyskinesia in a Mouse Model of Parkinson’s Disease" International Journal of Molecular Sciences 24, no. 5: 4687. https://doi.org/10.3390/ijms24054687