
Reovirus Type 3 Dearing Variants Do Not Induce Necroptosis in RIPK3-Expressing Human Tumor Cell Lines


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
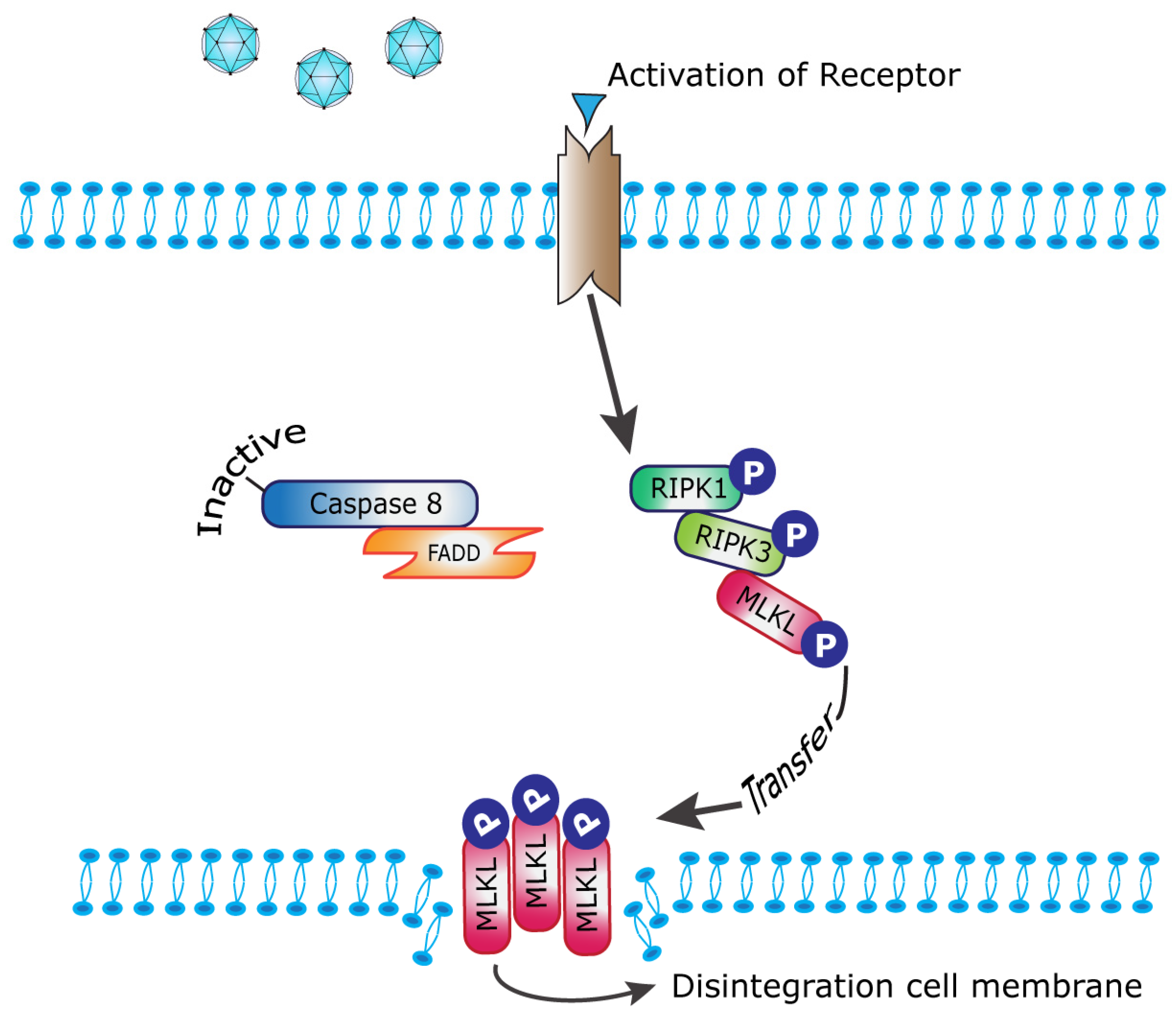
:1. Introduction
2. Results
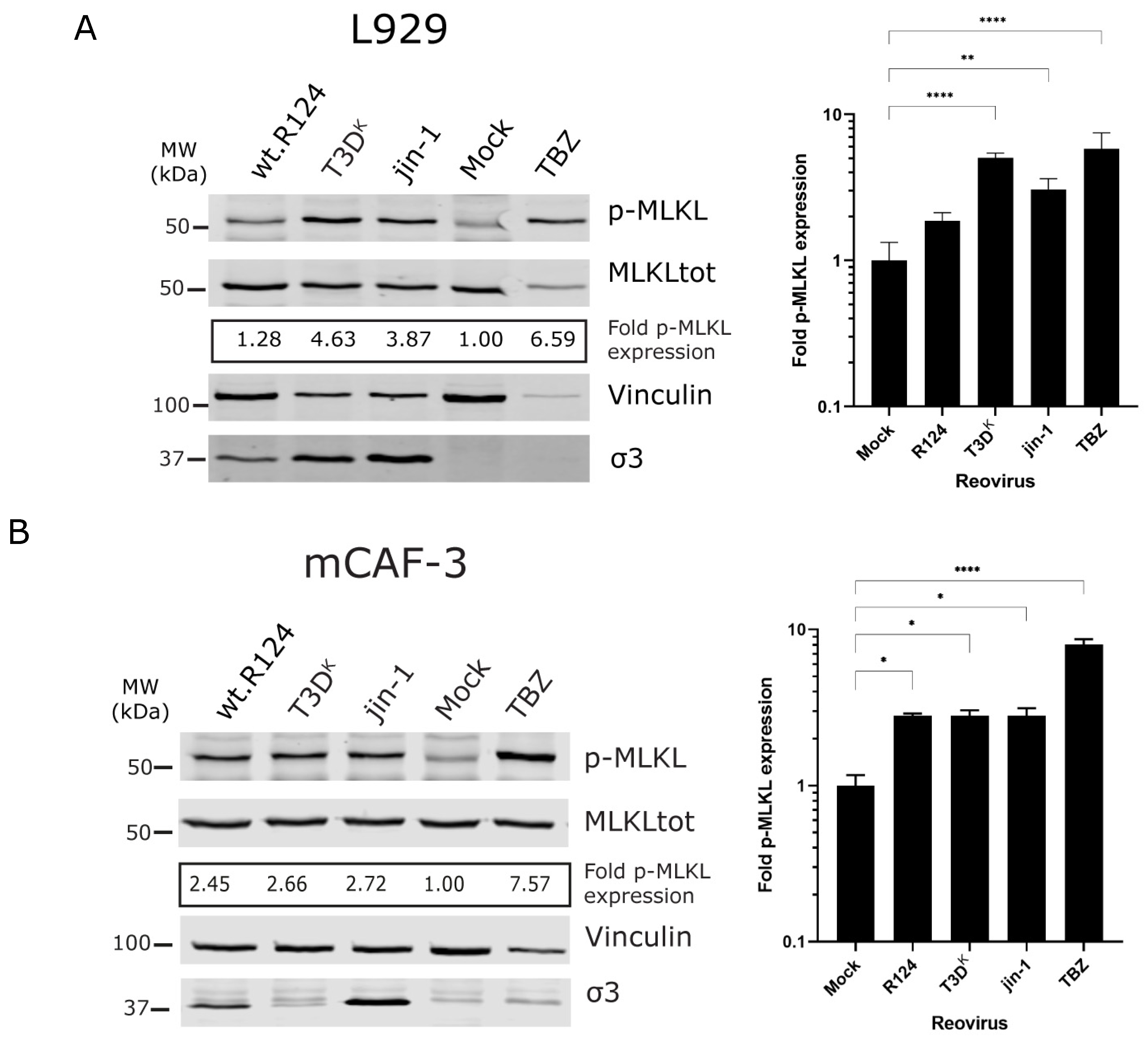
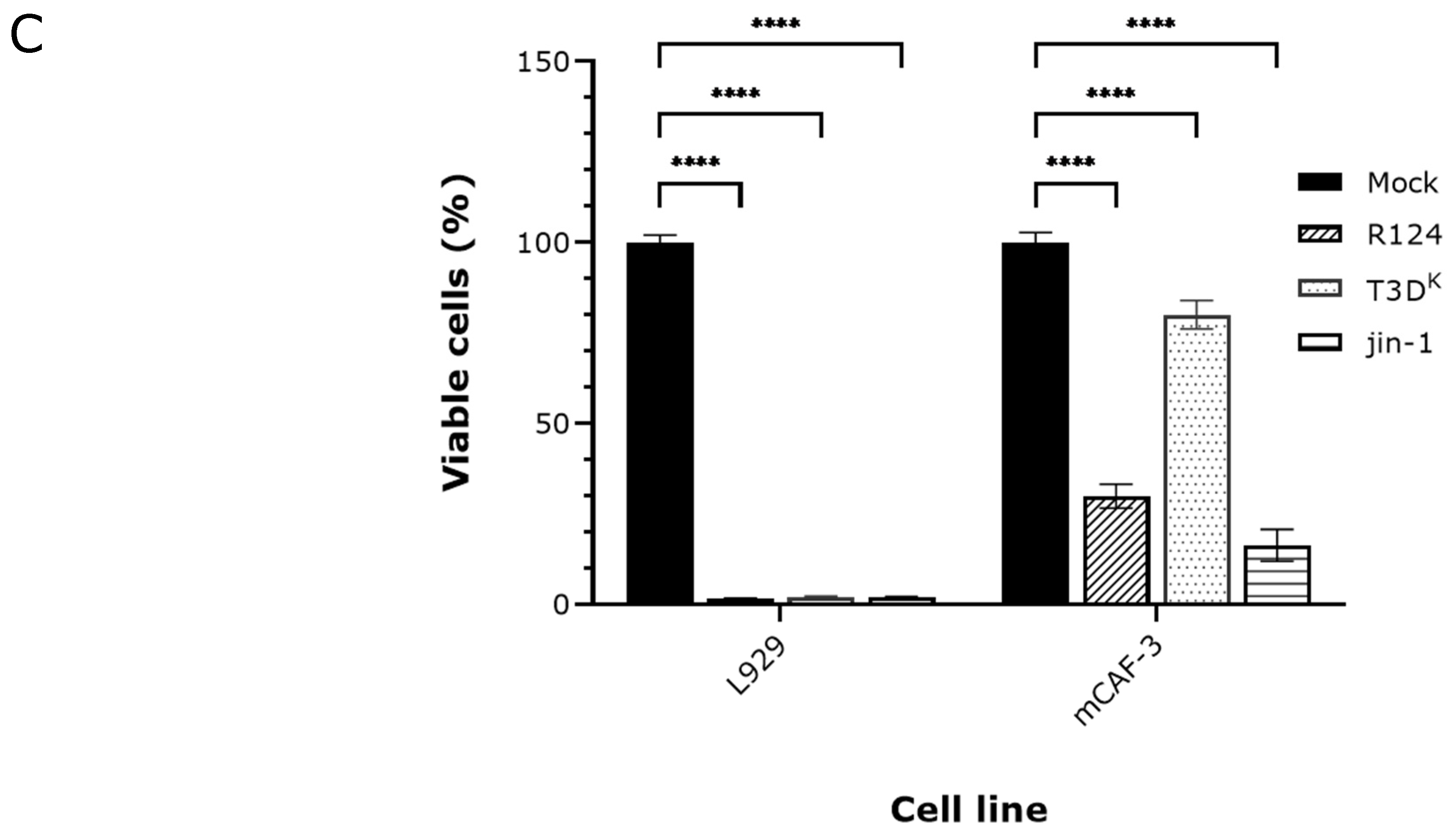
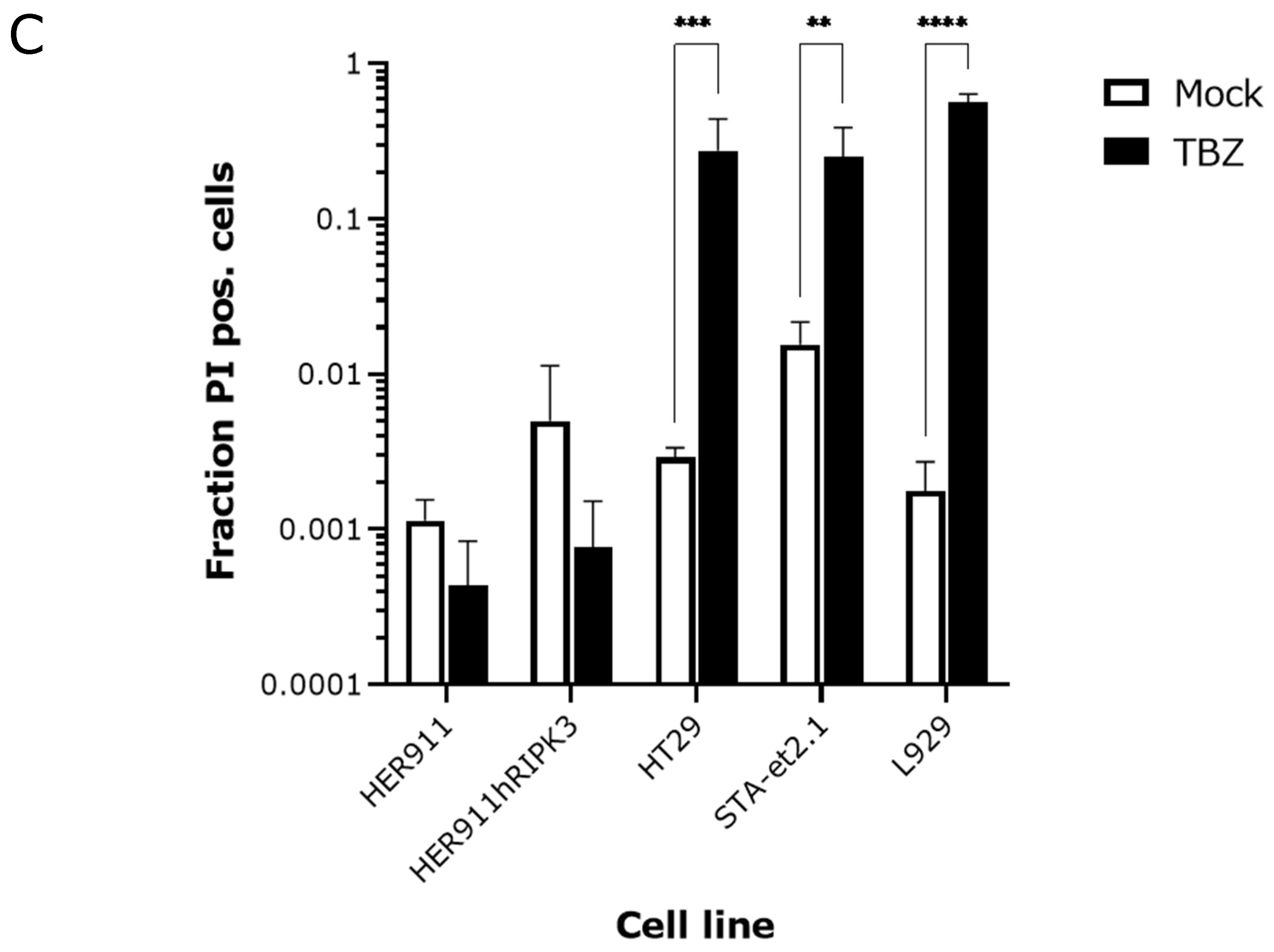
2.1. Detection of p-MLKL Induction in Reovirus-Infected Mouse Cells
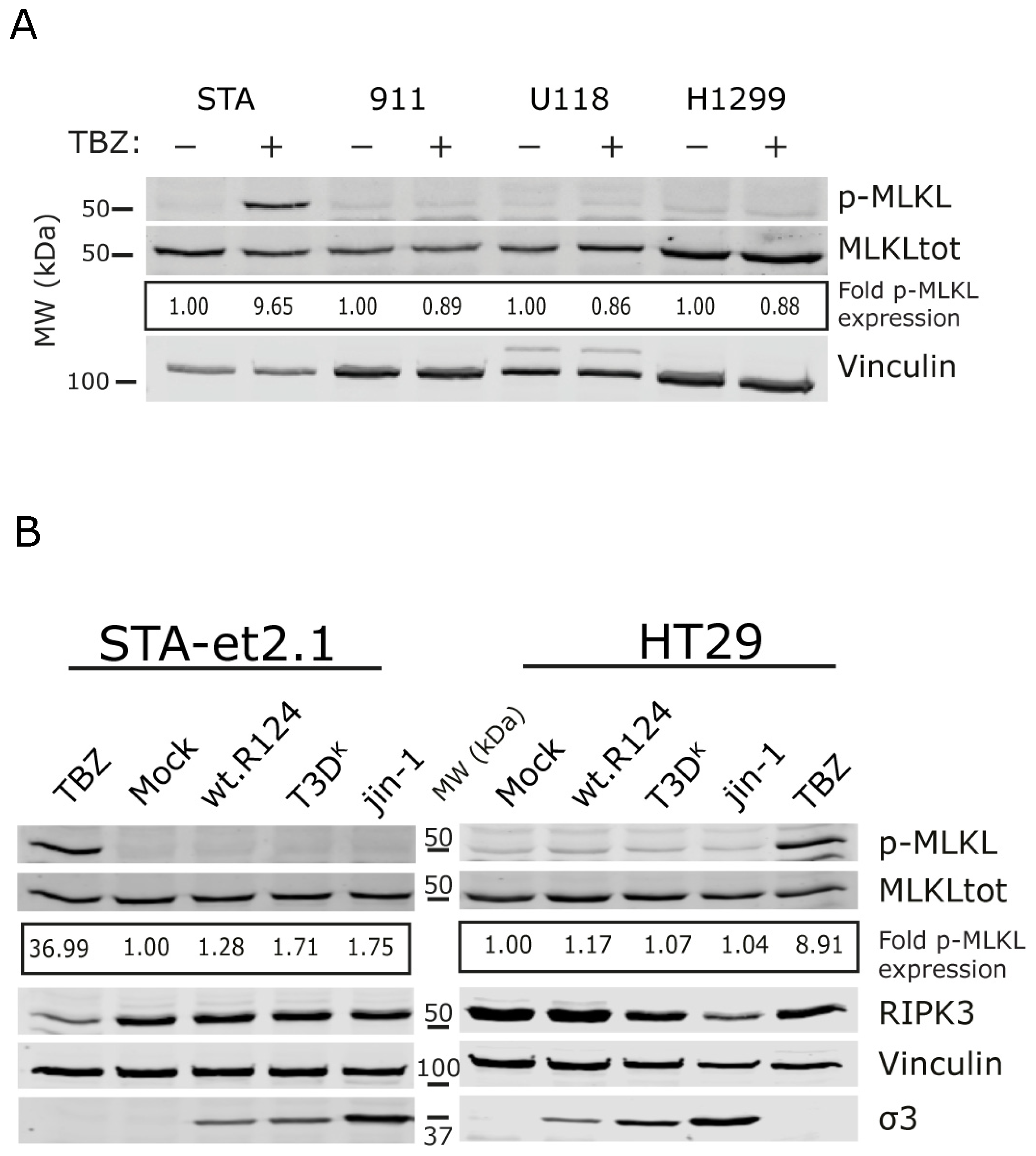
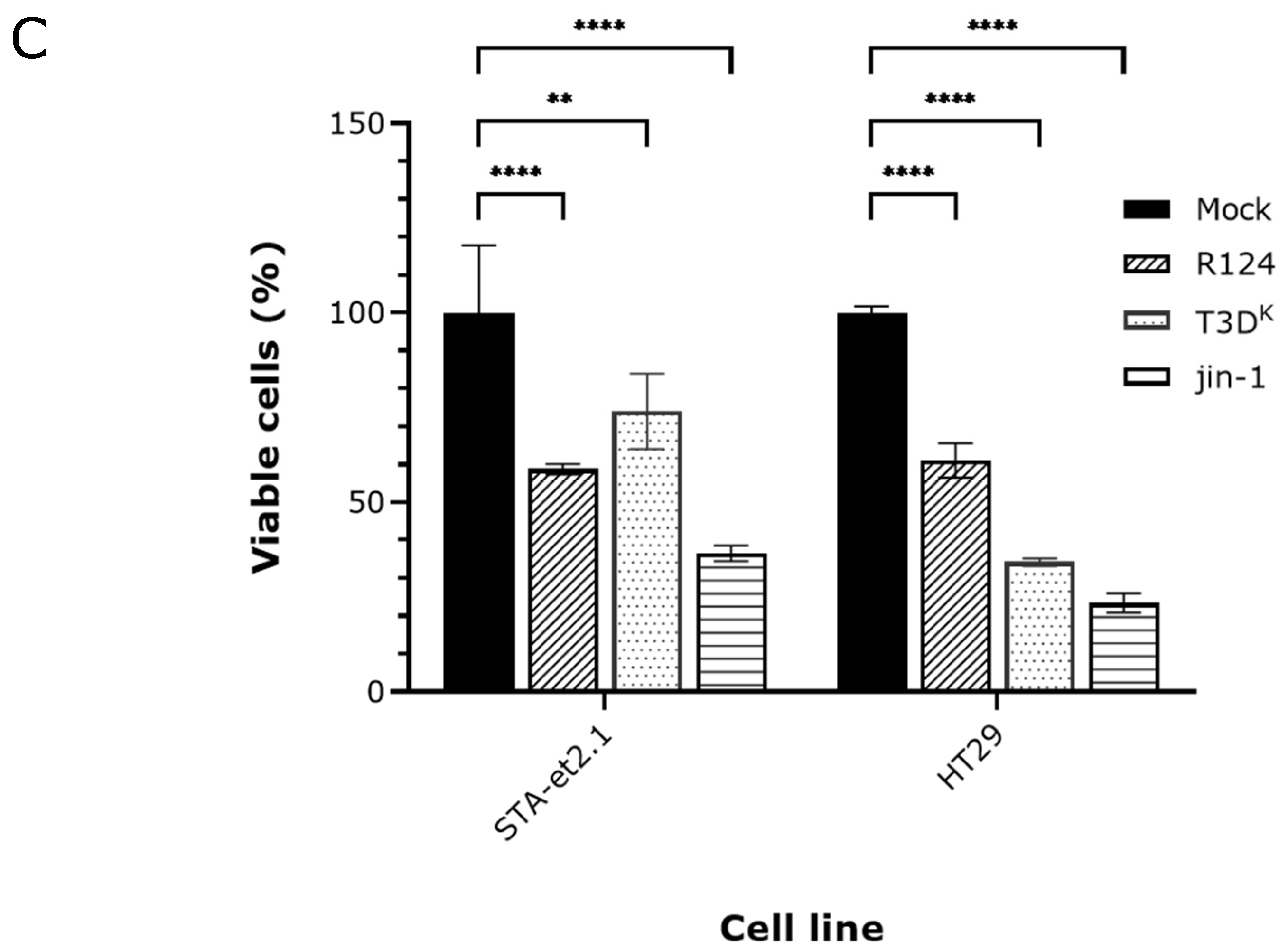
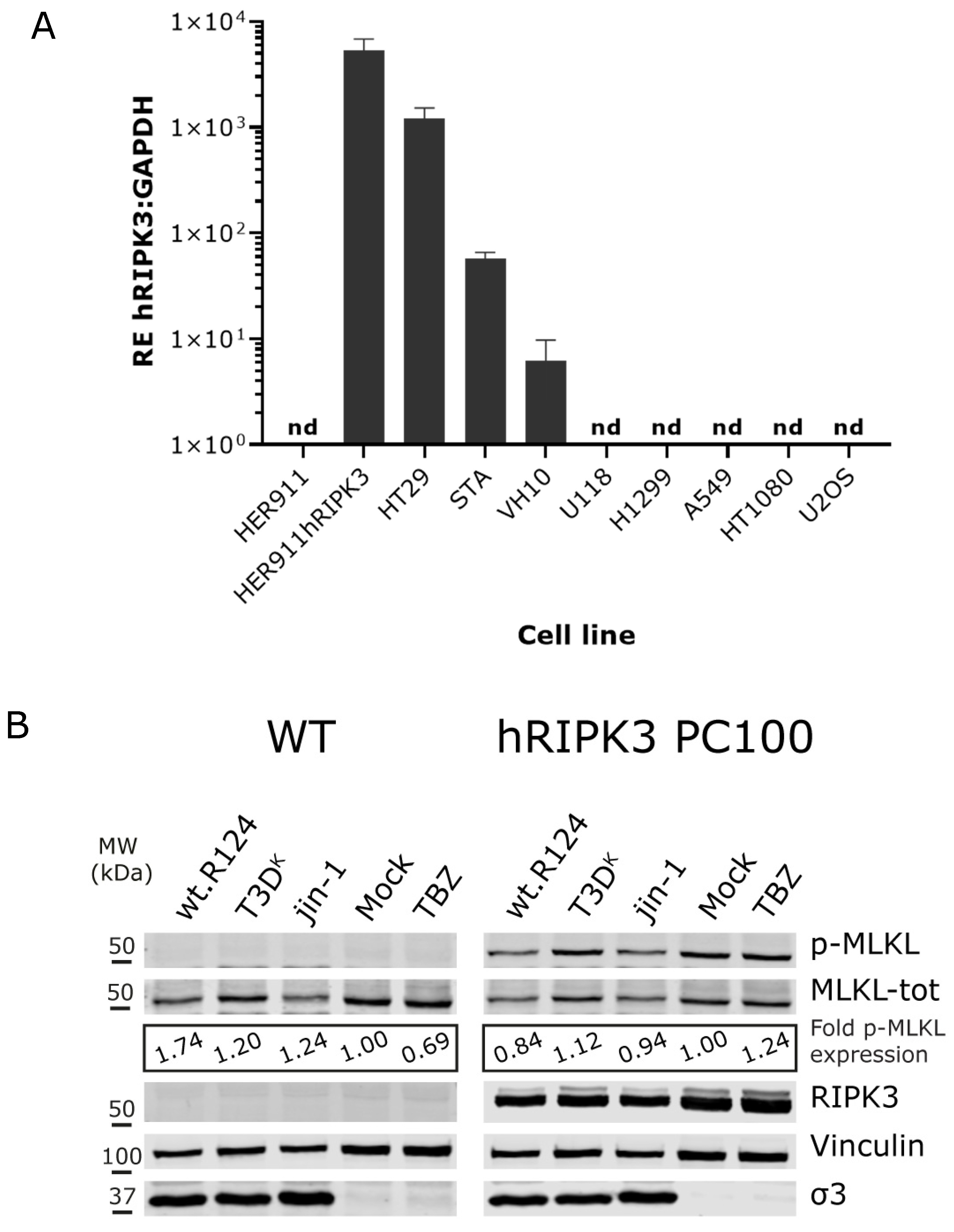
2.2. Induction of p-MLKL in Human Cell Lines
2.3. Overexpression of Human RIPK3 in HER911 Cells Does Not Lead to the Induction of p-MLKL by Reoviruses
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Viruses
4.2. Generation HER911 Cells Expressing Human RIPK3
4.3. Western Analysis
4.4. Cell Viability Detection
4.5. Human RIPK3 RT-qPCR
4.6. Cell Death Detection with Propidium Iodide
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [PubMed] [Green Version]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The clinical role of the TME in solid cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef]
- Gujar, S.; Pol, J.G.; Kroemer, G. Heating it up: Oncolytic viruses make tumors ‘hot’ and suitable for checkpoint blockade immunotherapies. Oncoimmunology 2018, 7, e1442169. [Google Scholar] [CrossRef] [PubMed]
- Moaven, O.; Mangieri, C.W.; Stauffer, J.A.; Anastasiadis, P.Z.; Borad, M.J. Evolving Role of Oncolytic Virotherapy: Challenges and Prospects in Clinical Practice. JCO Precis. Oncol. 2021, 5, 432–441. [Google Scholar] [CrossRef]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.; Bell, J.C.; Nelson, B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar]
- Groeneveldt, C.; Kinderman, P.; van den Wollenberg, D.J.M.; van den Oever, R.L.; Middelburg, J.; Mustafa, D.A.M.; Hoeben, R.C.; van der Burg, S.H.; van Hall, T.; van Montfoort, N. Preconditioning of the tumor microenvironment with oncolytic reovirus converts CD3-bispecific antibody treatment into effective immunotherapy. J. Immunother. Cancer 2020, 8, e001191. [Google Scholar] [CrossRef]
- Kerkar, S.P.; Restifo, N.P. Cellular Constituents of Immune Escape within the Tumor Microenvironment. Cancer Res. 2012, 72, 3125–3130. [Google Scholar] [CrossRef] [Green Version]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar]
- Lee, P.; Clements, D.; Helson, E.; Gujar, S. Reovirus in cancer therapy: An evidence-based review. Oncolytic Virother. 2014, 3, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Müller, L.; Berkeley, R.; Barr, T.; Ilett, E.; Errington-Mais, F. Past, Present and Future of Oncolytic Reovirus. Cancers 2020, 12, 3219. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L. Oncolytic Immunotherapy: Dying the Right Way is a Key to Eliciting Potent Antitumor Immunity. Front. Oncol. 2014, 4, 74. [Google Scholar] [CrossRef] [Green Version]
- Phillips, M.B.; Stuart, J.D.; Rodriguez Stewart, R.M.; Berry, J.T.; Mainou, B.A.; Boehme, K.W. Current understanding of reovirus oncolysis mechanisms. Oncolytic Virother. 2018, 7, 53–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smakman, N.; van den Wollenberg, D.J.; Borel Rinkes, I.H.; Hoeben, R.C.; Kranenburg, O. Sensitization to Apoptosis Underlies KrasD12-Dependent Oncolysis of Murine C26 Colorectal Carcinoma Cells by Reovirus T3D. J. Virol. 2005, 79, 14981–14985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thirukkumaran, C.M.; Shi, Z.Q.; Luider, J.; Kopciuk, K.; Gao, H.; Bahlis, N.; Neri, P.; Pho, M.; Stewart, D.; Mansoor, A.; et al. Reovirus as a viable therapeutic option for the treatment of multiple myeloma. Clin. Cancer Res. 2012, 18, 4962–4972. [Google Scholar] [CrossRef] [Green Version]
- Twigger, K.; Roulstone, V.; Kyula, J.; Karapanagiotou, E.M.; Syrigos, K.N.; Morgan, R.; White, C.; Bhide, S.; Nuovo, G.; Coffey, M.; et al. Reovirus exerts potent oncolytic effects in head and neck cancer cell lines that are independent of signalling in the EGFR pathway. BMC Cancer 2012, 12, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danthi, P.; Holm, G.H.; Stehle, T.; Dermody, T.S. Reovirus receptors, cell entry, and proapoptotic signaling. Adv. Exp. Med. Biol. 2013, 790, 42–71. [Google Scholar] [PubMed] [Green Version]
- Roebke, K.E.; Danthi, P. Cell Entry-Independent Role for the Reovirus μ1 Protein in Regulating Necroptosis and the Accumulation of Viral Gene Products. J. Virol. 2019, 93, e00199-19. [Google Scholar] [CrossRef] [Green Version]
- Berger, A.K.; Hiller, B.E.; Thete, D.; Snyder, A.J.; Perez, E., Jr.; Upton, J.W.; Danthi, P. Viral RNA at Two Stages of Reovirus Infection Is Required for the Induction of Necroptosis. J. Virol. 2017, 91, e02404-16. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, A.; Nomura, M.; Shuck, R.; Yustein, J. Cancer’s Achilles’ Heel: Apoptosis and Necroptosis to the Rescue. Int. J. Mol. Sci. 2016, 18, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Yu, J.; Zhang, L. Necroptosis: An alternative cell death program defending against cancer. Biochim. Biophys. Acta (BBA)–Rev. Cancer 2016, 1865, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Martens, S.; Bridelance, J.; Roelandt, R.; Vandenabeele, P.; Takahashi, N. MLKL in cancer: More than a necroptosis regulator. Cell Death Differ. 2021, 28, 1757–1772. [Google Scholar] [CrossRef]
- Moujalled, D.M.; Cook, W.D.; Murphy, J.M.; Vaux, D.L. Necroptosis induced by RIPK3 requires MLKL but not Drp1. Cell Death Dis. 2014, 5, e1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouan-Lanhouet, S.; Arshad, M.I.; Piquet-Pellorce, C.; Martin-Chouly, C.; Le Moigne-Muller, G.; Van Herreweghe, F.; Takahashi, N.; Sergent, O.; Lagadic-Gossmann, D.; Vandenabeele, P.; et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 2012, 19, 2003–2014. [Google Scholar] [PubMed] [Green Version]
- Berger, A.K.; Danthi, P. Reovirus activates a caspase-independent cell death pathway. MBio 2013, 4, e00178-13. [Google Scholar] [CrossRef] [Green Version]
- Koo, G.B.; Morgan, M.J.; Lee, D.G.; Kim, W.J.; Yoon, J.H.; Koo, J.S.; Kim, S.I.; Kim, S.J.; Son, M.K.; Hong, S.S.; et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015, 25, 707–725. [Google Scholar] [CrossRef]
- Van den Wollenberg, D.J.M.; Dautzenberg, I.J.C.; van den Hengel, S.K.; Cramer, S.J.; de Groot, R.J.; Hoeben, R.C. Isolation of reovirus T3D mutants capable of infecting human tumor cells independent of junction adhesion molecule-A. PLoS ONE 2012, 7, e48064. [Google Scholar] [CrossRef] [Green Version]
- DeAntoneo, C.; Danthi, P.; Balachandran, S. Reovirus Activated Cell Death Pathways. Cells 2022, 11, 1757. [Google Scholar] [CrossRef]
- Mohamed, A.; Konda, P.; Eaton, H.E.; Gujar, S.; Smiley, J.R.; Shmulevitz, M. Closely related reovirus lab strains induce opposite expression of RIG-I/IFN-dependent versus -independent host genes, via mechanisms of slow replication versus polymorphisms in dsRNA binding σ3 respectively. PLoS Pathog. 2020, 16, e1008803. [Google Scholar] [CrossRef]
- Hiller, B.E.; Berger, A.K.; Danthi, P. Viral gene expression potentiates reovirus-induced necrosis. Virology 2015, 484, 386–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahalingam, D.; Goel, S.; Aparo, S.; Patel Arora, S.; Noronha, N.; Tran, H.; Chakrabarty, R.; Selvaggi, G.; Gutierrez, A.; Coffey, M.; et al. A Phase II Study of Pelareorep (REOLYSIN®) in Combination with Gemcitabine for Patients with Advanced Pancreatic Adenocarcinoma. Cancers 2018, 10, 160. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Smiley, J.R.; Shmulevitz, M. Polymorphisms in the Most Oncolytic Reovirus Strain Confer Enhanced Cell Attachment, Transcription, and Single-Step Replication Kinetics. J. Virol. 2020, 94, e01937-19. [Google Scholar] [CrossRef] [PubMed]
- Boudreault, S.; Lemay, G.; Bisaillon, M. U5 snRNP Core Proteins Are Key Components of the Defense Response against Viral Infection through Their Roles in Programmed Cell Death and Interferon Induction. Viruses 2022, 14, 2710. [Google Scholar]
- Boudreault, S.; Durand, M.; Martineau, C.A.; Perreault, J.P.; Lemay, G.; Bisaillon, M. Reovirus μ2 protein modulates host cell alternative splicing by reducing protein levels of U5 snRNP core components. Nucleic Acids Res. 2022, 50, 5263–5281. [Google Scholar] [CrossRef]
- Lanoie, D.; Lemay, G. Multiple proteins differing between laboratory stocks of mammalian orthoreoviruses affect both virus sensitivity to interferon and induction of interferon production during infection. Virus Res. 2018, 247, 40–46. [Google Scholar] [CrossRef]
- Meng, Y.; Davies, K.A.; Fitzgibbon, C.; Young, S.N.; Garnish, S.E.; Horne, C.R.; Luo, C.; Garnier, J.-M.; Liang, L.-Y.; Cowan, A.D.; et al. Human RIPK3 maintains MLKL in an inactive conformation prior to cell death by necroptosis. Nat. Common. 2021, 12, 6783. [Google Scholar] [CrossRef]
- Petrie, E.J.; Birkinshaw, R.W.; Koide, A.; Denbaum, E.; Hildebrand, J.M.; Garnish, S.E.; Davies, K.A.; Sandow, J.J.; Samson, A.L.; Gavin, X.; et al. Identification of MLKL membrane translocation as a checkpoint in necroptotic cell death using Monobodies. Proc. Natl. Acad. Sci. USA 2020, 117, 8468–8475. [Google Scholar] [CrossRef]
- Petrie, E.J.; Czabotar, P.E.; Murphy, J.M. The Structural Basis of Necroptotic Cell Death Signaling. Trends Biochem. Sci. 2019, 44, 53–63. [Google Scholar] [CrossRef]
- Omoto, S.; Guo, H.; Talekar, G.R.; Roback, L.; Kaiser, W.J.; Mocarski, E.S. Suppression of RIP3-dependent Necroptosis by Human Cytomegalovirus. J. Biol. Chem. 2015, 290, 11635–11648. [Google Scholar] [CrossRef] [Green Version]
- Petrie, E.J.; Sandow, J.J.; Lehmann, W.I.L.; Liang, L.Y.; Coursier, D.; Young, S.N.; Kersten, W.J.A.; Fitzgibbon, C.; Samson, A.L.; Jacobsen, A.V.; et al. Viral MLKL Homologs Subvert Necroptotic Cell Death by Sequestering Cellular RIPK3. Cell Rep. 2019, 28, 3309–3319.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.K.; Kim, W.-J.; Yoon, J.-H.; Ji, J.-H.; Morgan, M.J.; Cho, H.; Kim, Y.C.; Kim, Y.-S. Upregulated RIP3 Expression Potentiates MLKL Phosphorylation–Mediated Programmed Necrosis in Toxic Epidermal Necrolysis. J. Investig. Dermatol. 2015, 135, 2021–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallaux, F.J.; Kranenburg, O.; Cramer, S.J.; Houweling, A.; van Ormondt, H.; Hoeben, R.C.; van Der Eb, A.J. Characterization of 911: A new helper cell line for the titration and propagation of early region 1-deleted adenoviral vectors. Hum. Gene Ther. 1996, 7, 215–222. [Google Scholar] [CrossRef]
- Klein, B.; Pastink, A.; Odijk, H.; Westerveld, A.; van der Eb, A.J. Transformation and immortalization of diploid xeroderma pigmentosum fibroblasts. Exp. Cell Res. 1990, 191, 256–262. [Google Scholar] [CrossRef]
- Kovar, H.; Jug, G.; Aryee, D.N.; Zoubek, A.; Ambros, P.; Gruber, B.; Windhager, R.; Gadner, H. Among genes involved in the RB dependent cell cycle regulatory cascade, the p16 tumor suppressor gene is frequently lost in the Ewing family of tumors. Oncogene 1997, 15, 2225–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchholz, U.J.; Finke, S.; Conzelmann, K.-K. Generation of Bovine Respiratory Syncytial Virus (BRSV) from cDNA: BRSV NS2 Is Not Essential for Virus Replication in Tissue Culture, and the Human RSV Leader Region Acts as a Functional BRSV Genome Promoter. J. Virol. 1999, 73, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Ooms, L.S.; Ikizler, M.; Chappell, J.D.; Dermody, T.S. An improved reverse genetics system for mammalian orthoreoviruses. Virology 2010, 398, 194–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Wollenberg, D.J.; Dautzenberg, I.J.; Ros, W.; Lipinska, A.D.; van den Hengel, S.K.; Hoeben, R.C. Replicating reoviruses with a transgene replacing the codons for the head domain of the viral spike. Gene Ther. 2015, 22, 51–63. [Google Scholar]
- Berard, A.; Coombs, K.M. Mammalian reoviruses: Propagation, quantification, and storage. Curr. Protoc. Microbiol. 2009, 14, 15C. 1.1–15C. 1.18. [Google Scholar]
- Uil, T.G.; de Vrij, J.; Vellinga, J.; Rabelink, M.J.; Cramer, S.J.; Chan, O.Y.; Pugnali, M.; Magnusson, M.; Lindholm, L.; Boulanger, P.; et al. A lentiviral vector-based adenovirus fiber-pseudotyping approach for expedited functional assessment of candidate retargeted fibers. J. Gene Med. 2009, 11, 990–1004. [Google Scholar] [CrossRef]
- Vellinga, J.; Uil, T.G.; De, V.J.; Rabelink, M.J.; Lindholm, L.; Hoeben, R.C. A system for efficient generation of adenovirus protein IX-producing helper cell lines. J. Gene Med. 2006, 8, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Bogdanov, K.; Kovalenko, A.; Wallach, D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. 2016, 23, 253–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van den Wollenberg, D.J.M.; Kemp, V.; Rabelink, M.J.W.E.; Hoeben, R.C. Reovirus Type 3 Dearing Variants Do Not Induce Necroptosis in RIPK3-Expressing Human Tumor Cell Lines. Int. J. Mol. Sci. 2023, 24, 2320. https://doi.org/10.3390/ijms24032320
van den Wollenberg DJM, Kemp V, Rabelink MJWE, Hoeben RC. Reovirus Type 3 Dearing Variants Do Not Induce Necroptosis in RIPK3-Expressing Human Tumor Cell Lines. International Journal of Molecular Sciences. 2023; 24(3):2320. https://doi.org/10.3390/ijms24032320
Chicago/Turabian Stylevan den Wollenberg, Diana J. M., Vera Kemp, Martijn J. W. E. Rabelink, and Rob C. Hoeben. 2023. "Reovirus Type 3 Dearing Variants Do Not Induce Necroptosis in RIPK3-Expressing Human Tumor Cell Lines" International Journal of Molecular Sciences 24, no. 3: 2320. https://doi.org/10.3390/ijms24032320