

(2-Hydroxy-3-Methoxybenzylidene)thiazolo[3,2-a]pyrimidines: Synthesis, Self-Assembly in the Crystalline Phase and Cytotoxic Activity
 , , , and
, , , and
Abstract
:1. Introduction
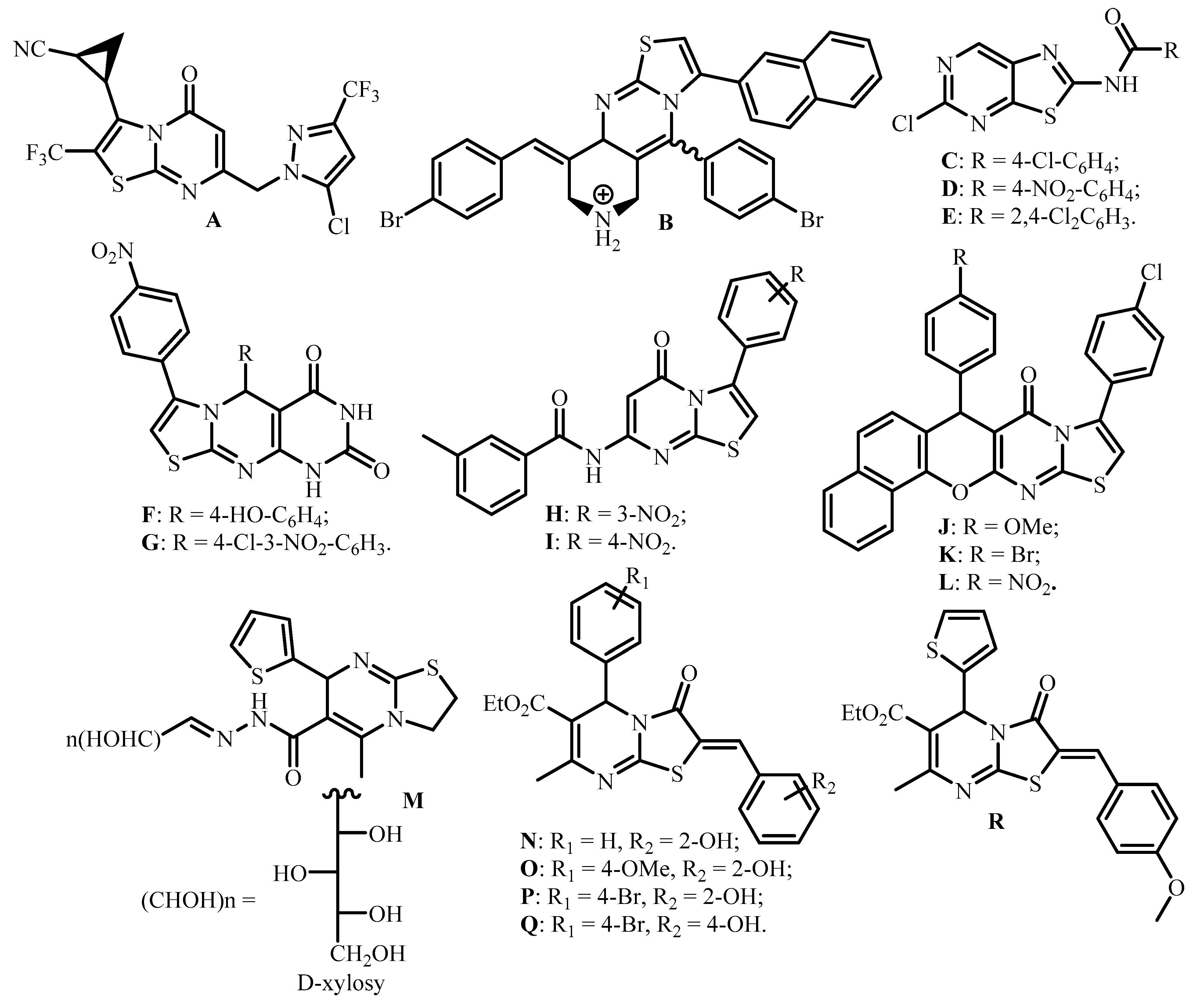
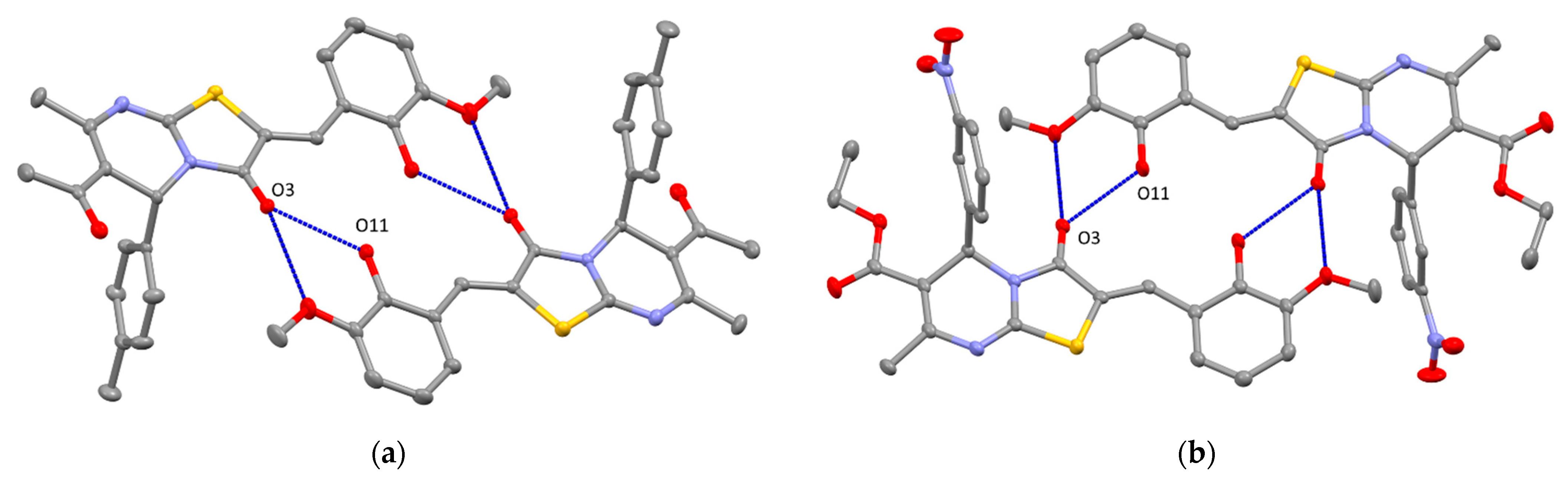
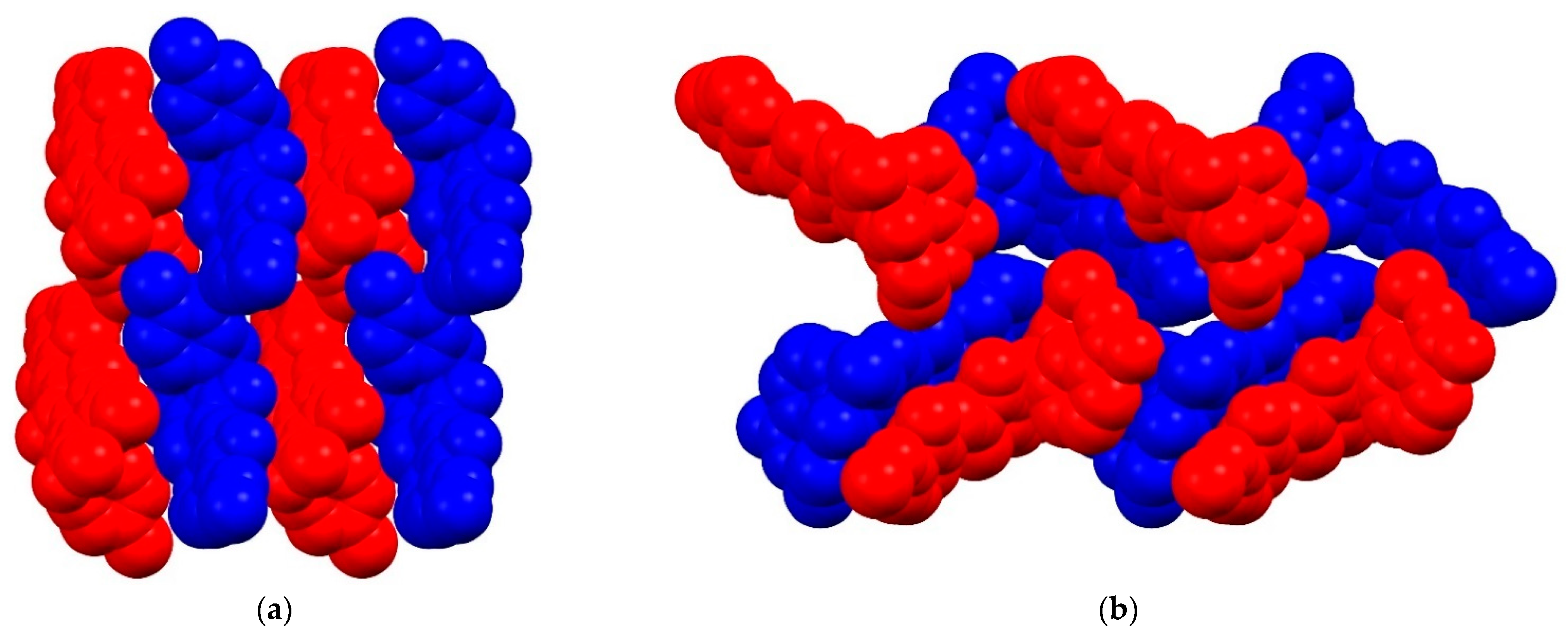
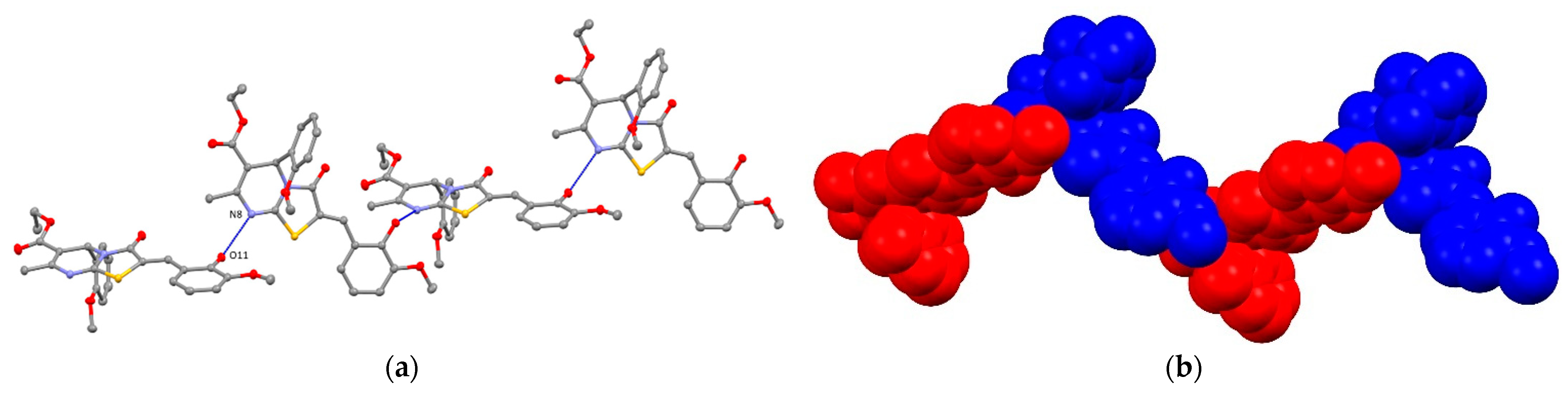
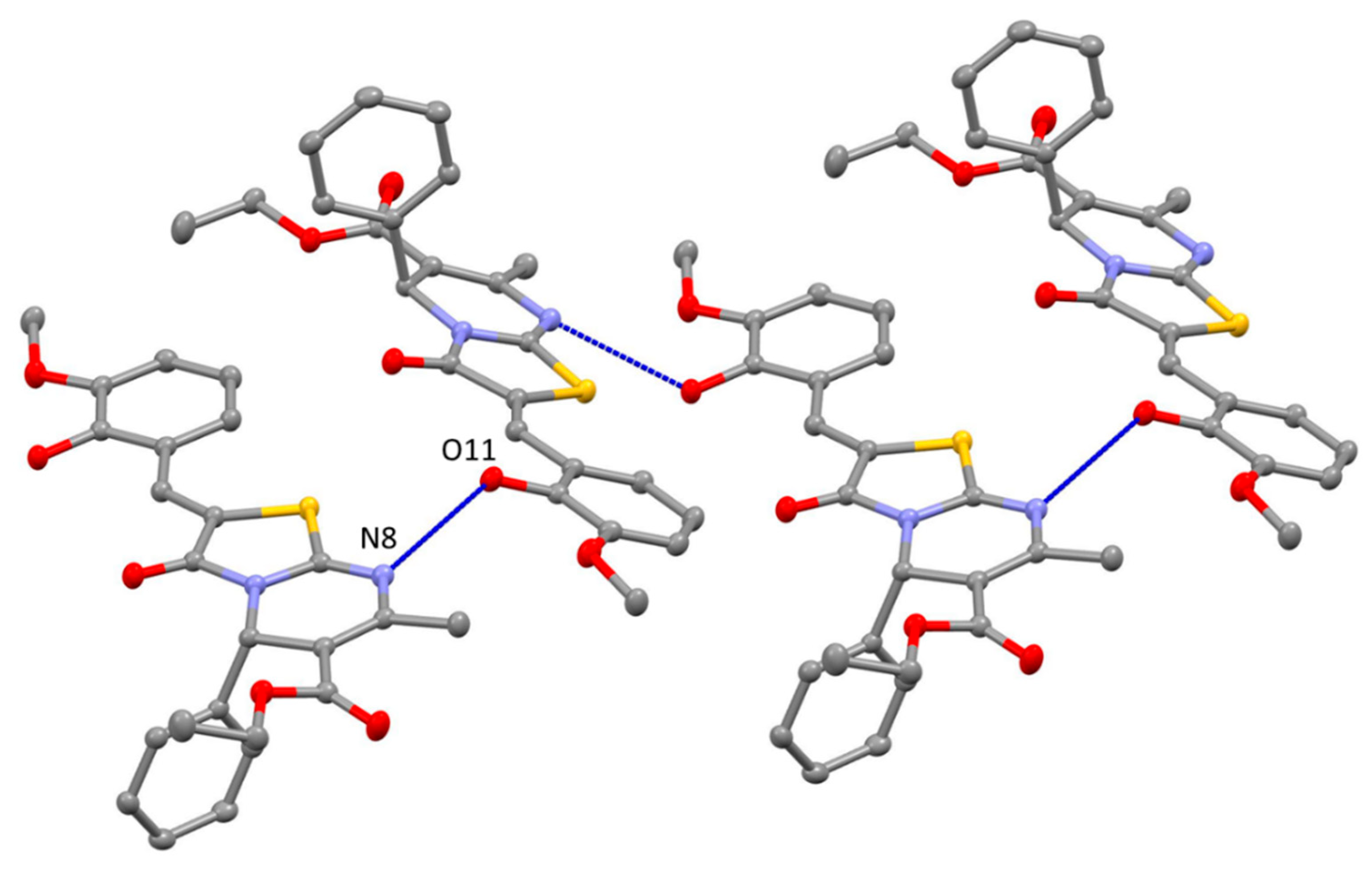
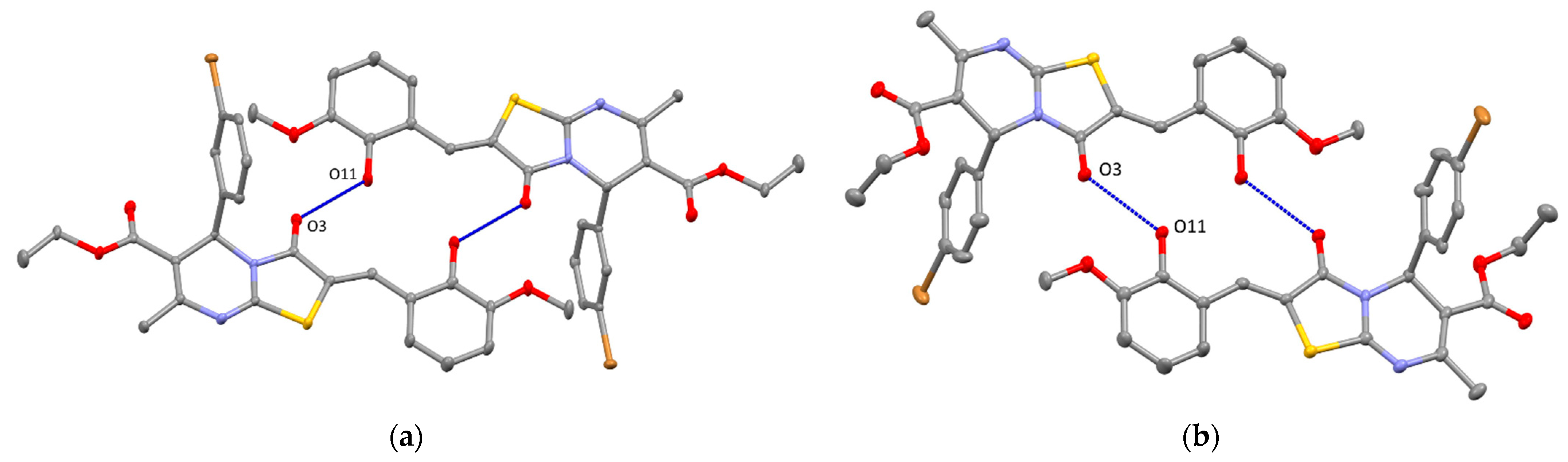
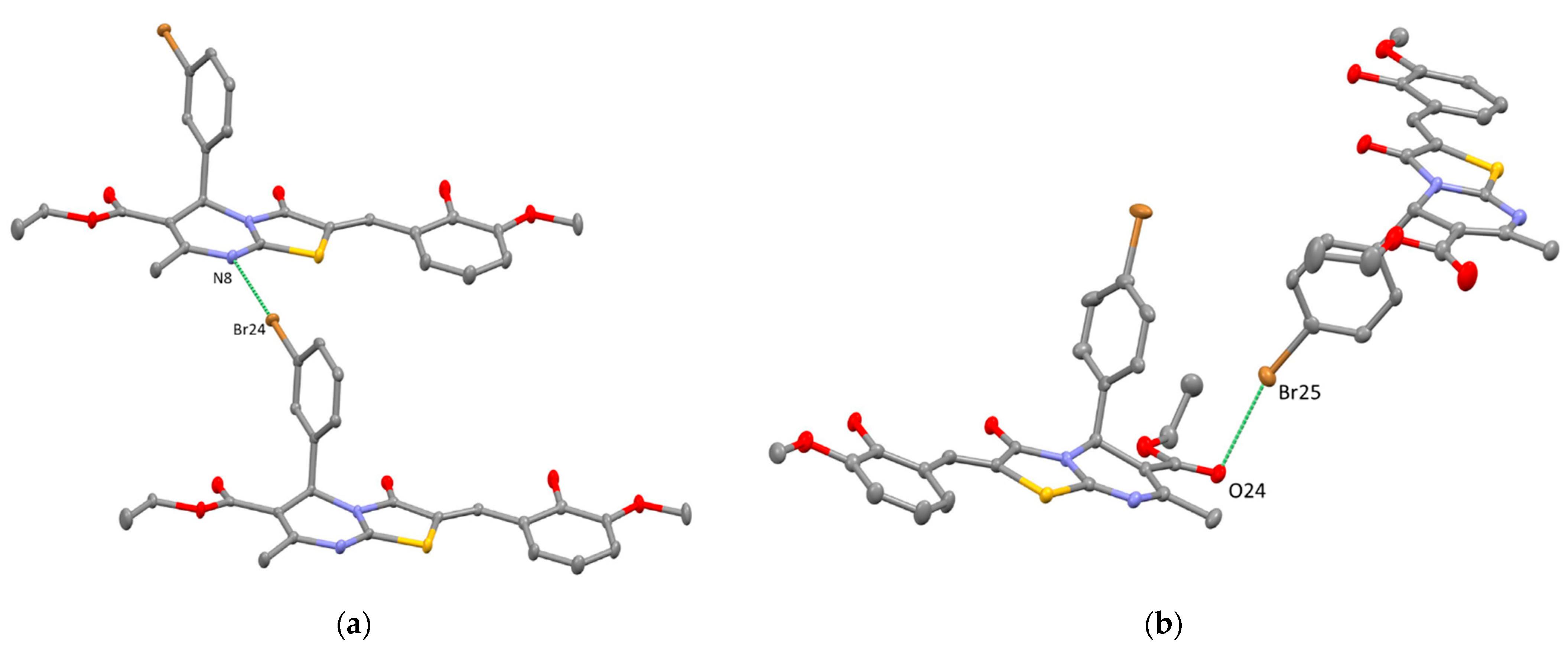
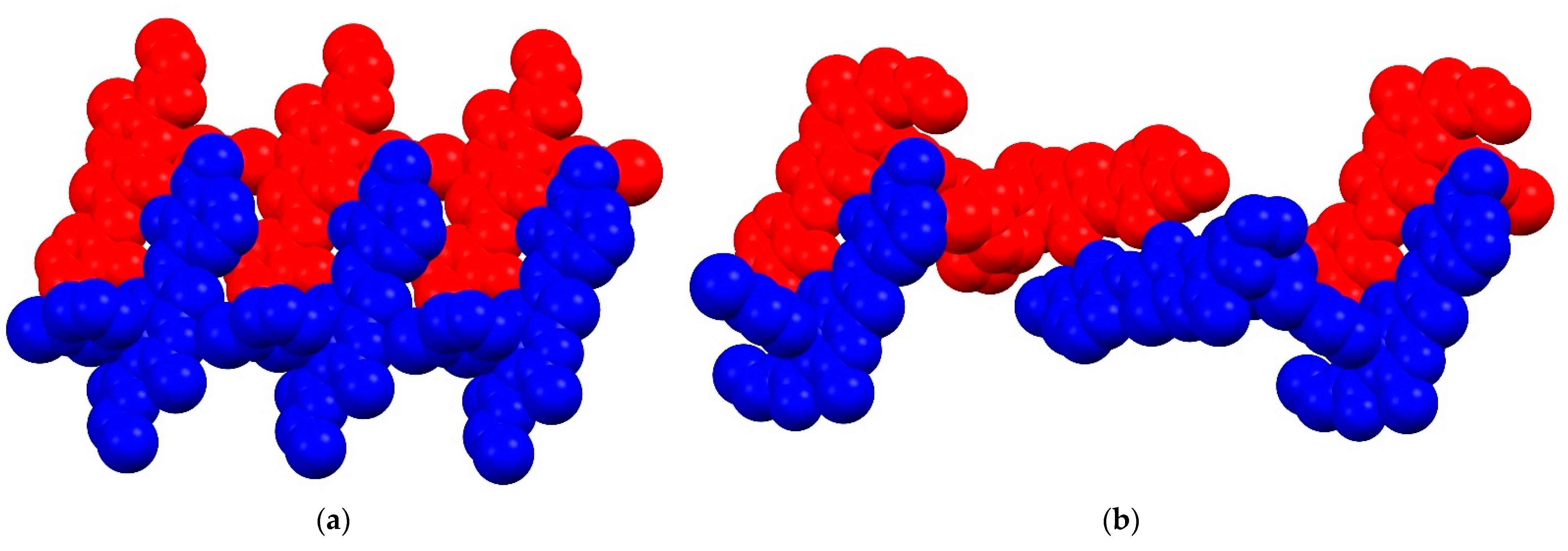
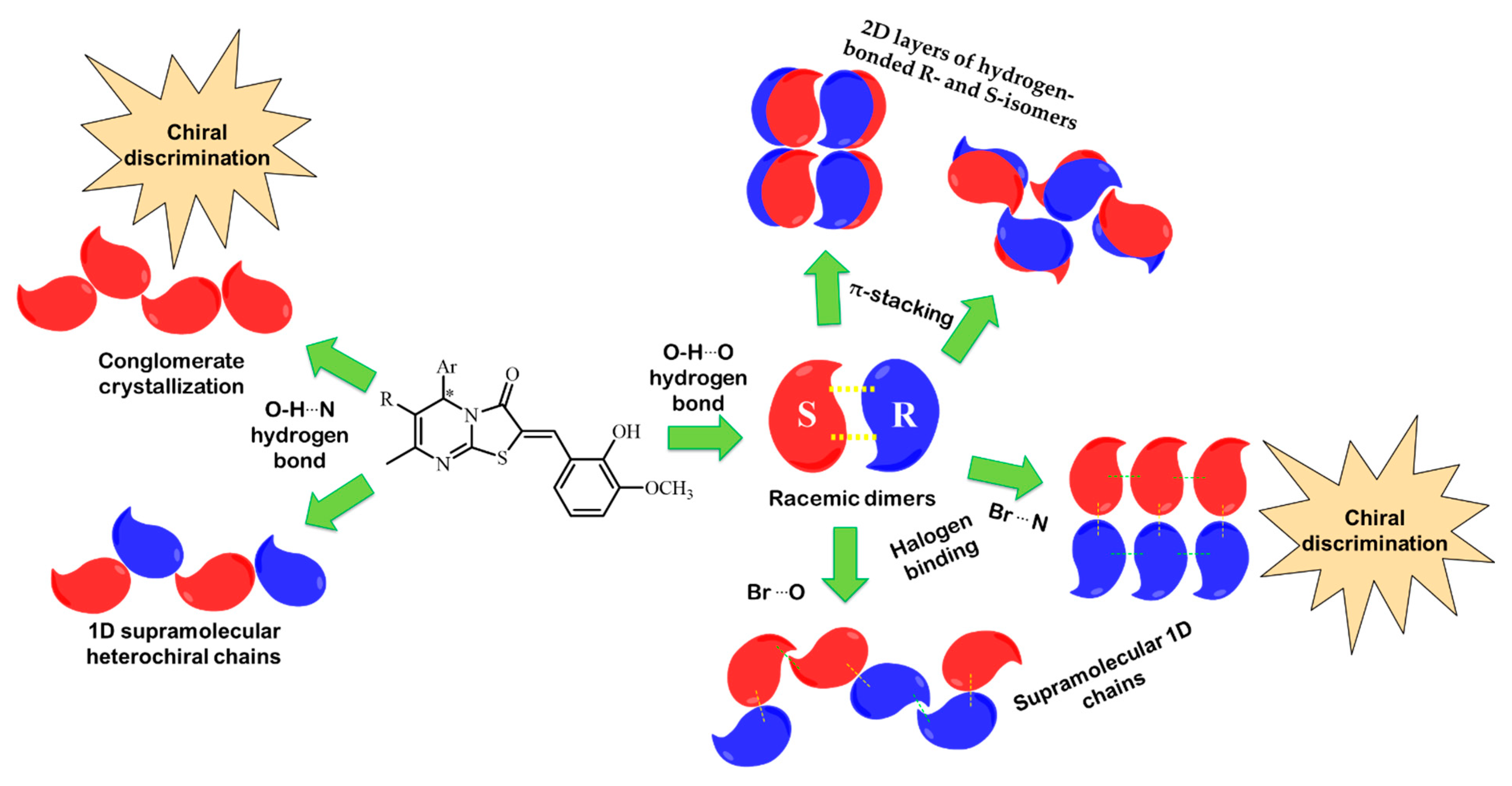
2. Results and Discussion
3. Materials and Methods
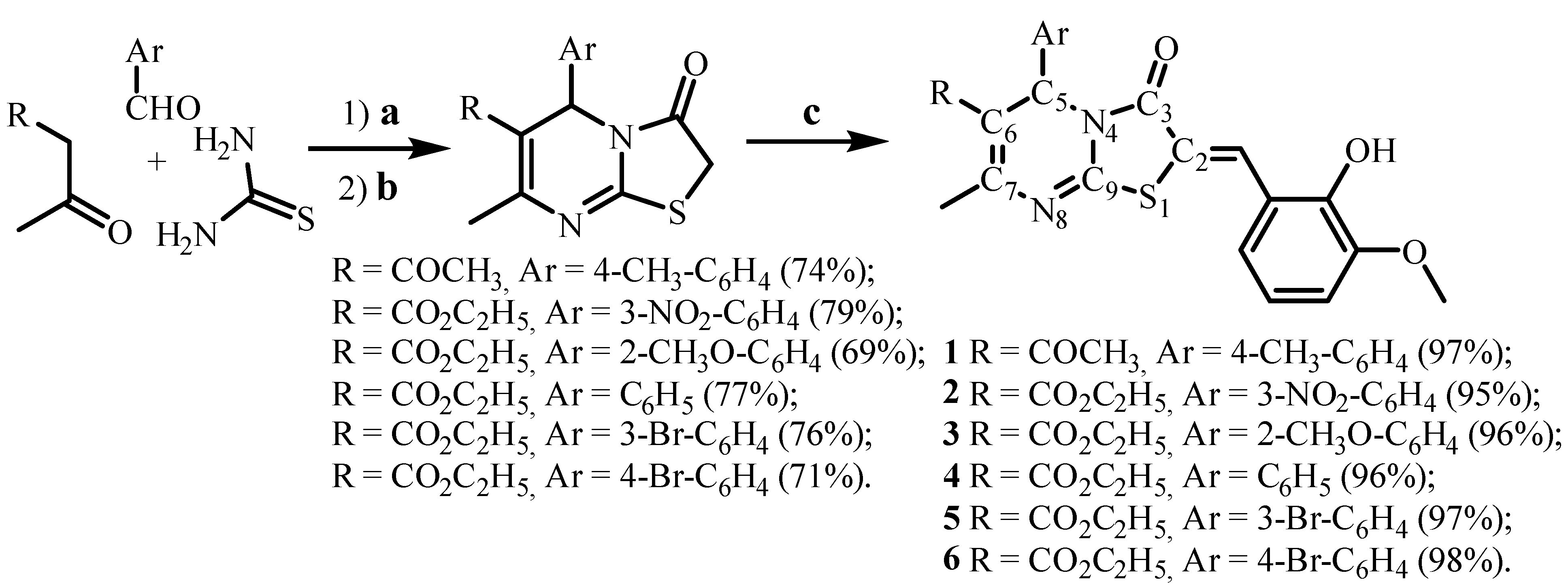
3.1. Synthesis and Characterisation
3.1.1. General Method for Compounds 1–6 Preparation
3.1.2. Crystallization Conditions
3.2. Hirshfeld Surface Analysis
3.3. Biological Study
3.3.1. Cells and Materials
3.3.2. MTT Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Volgraf, M.; Sellers, B.D.; Jiang, Y.; Wu, G.; Ly, C.Q.; Villemure, E.; Pastor, R.M.; Yuen, P.; Lu, A.; Luo, X.; et al. Discovery of GluN2A-Selective NMDA Receptor Positive Allosteric Modulators (PAMs): Tuning Deactivation Kinetics via Structure-Based Design. J. Med. Chem. 2016, 59, 2760–2779. [Google Scholar] [CrossRef] [PubMed]
- Basiri, A.; Xiao, M.; McCarthy, A.; Dutta, D.; Byrareddy, S.N.; Conda-Sheridan, M. Design and synthesis of new piperidone grafted acetylcholinesterase inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Shahid Nadeem, M.; Azam Khan, J.; Kazmi, I.; Rashid, U. Design, Synthesis, and Bioevaluation of Indole Core Containing 2-Arylidine Derivatives of Thiazolopyrimidine as Multitarget Inhibitors of Cholinesterases and Monoamine Oxidase A/B for the Treatment of Alzheimer Disease. ACS Omega 2022, 7, 9369–9379. [Google Scholar] [CrossRef] [PubMed]
- Istanbullu, H.; Bayraktar, G.; Akbaba, H.; Cavus, I.; Coban, G.; Debelec Butuner, B.; Kilimcioglu, A.A.; Ozbilgin, A.; Alptuzun, V.; Erciyas, E. Design, synthesis, and in vitro biological evaluation of novel thiazolopyrimidine derivatives as antileishmanial compounds. Arch. Pharm. 2020, 353, 1900325. [Google Scholar] [CrossRef] [PubMed]
- Sukanya, S.H.; Venkatesh, T.; Rao, S.A.; Joy, M.N. Efficient L-Proline catalyzed synthesis of some new (4-substituted-phenyl)-1,5-dihydro-2H-pyrimido[4,5-d][1,3]thiazolo[3,2-a]pyrimidine-2,4(3H)-diones bearing thiazolopyrimidine derivatives and evaluation of their pharmacological activities. J. Mol. Struct. 2022, 1247, 131324. [Google Scholar] [CrossRef]
- Mohamed, M.M.; Khalil, A.K.; Abbass, E.M.; El-Naggar, A.M. Design, synthesis of new pyrimidine derivatives as anticancer and antimicrobial agents. Synth. Commun. 2017, 47, 1441–1457. [Google Scholar] [CrossRef]
- Cai, D.; Zhang, Z.H.; Chen, Y.; Yan, X.J.; Zhang, S.T.; Zou, L.J.; Meng, L.-H.; Li, F.; Fu, B.J. Synthesis of some new thiazolo[3,2-a]pyrimidine derivatives and screening of their in vitro antibacterial and antitubercular activities. Med. Chem. Res. 2016, 25, 292–302. [Google Scholar] [CrossRef]
- Batool, I.; Saeed, A.; Qureshi, I.Z.; Kalsoom, S.; Razzaq, A. Synthesis, molecular docking and biological evaluation of new thiazolopyrimidine carboxylates as potential antidiabetic and antibacterial agents. Res. Chem. Intermed. 2016, 42, 1139–1163. [Google Scholar] [CrossRef]
- Banoth, S.; Boda, S.; Perugu, S.; Balabadra, S.; Manga, V. Design, synthesis, biological evaluation and in silico molecular docking studies of novel benzochromeno[2,3-d]thiazolopyrimidine derivatives. Res. Chem. Intermed. 2018, 44, 1833–1846. [Google Scholar] [CrossRef]
- Basiony, E.A.; Hassan, A.A.; Al-Amshany, Z.M.; Abd-Rabou, A.A.; Abdel-Rahman, A.A.-H.; Hassan, N.A.; El-Sayed, W.A. Synthesis and Cytotoxic Activity of New Thiazolopyrimidine Sugar Hydrazones and Their Derived Acyclic Nucleoside Analogues. Molecules 2020, 25, 399. [Google Scholar] [CrossRef]
- Agarkov, A.S.; Gabitova, E.R.; Galieva, F.B.; Ovsyannikov, A.S.; Voloshina, A.D.; Shiryaev, A.K.; Litvinov, I.A.; Solovieva, S.E.; Antipin, I.S. Structure and Biological Properties of 2-Phenylhydrazone Derivatives of Thiazolopyrimidines. Dokl. Chem. 2022, 503, 45–50. [Google Scholar] [CrossRef]
- Agarkov, A.S.; Nefedova, A.A.; Gabitova, E.R.; Ovsyannikov, A.S.; Amerhanova, S.K.; Lyubina, A.P.; Voloshina, A.D.; Dorovatovskii, P.V.; Litvinov, I.A.; Solovieva, S.E.; et al. Synthesis, Self-Assembly in Crystalline Phase and Anti-Tumor Activity of 2-(2-/4-Hydroxybenzylidene)thiazolo[3,2-a]pyrimidines. Molecules 2022, 27, 7747. [Google Scholar] [CrossRef]
- Geist, J.G.; Lauw, S.; Illarionova, V.; Illarionov, B.; Fischer, M.; Gräwert, T.; Rohdich, F.; Eisenreich, W.; Kaiser, J.; Groll, M.; et al. Thiazolopyrimidine inhibitors of 2-methylerythritol 2, 4-cyclodiphosphate synthase (IspF) from Mycobacterium tuberculosis and Plasmodium falciparum. ChemMedChem 2010, 5, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Waldeck, B. Biological significance of the enantiomeric purity of drugs. Chirality 1993, 5, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Agarkov, A.S.; Litvinov, I.A.; Gabitova, E.R.; Ovsyannikov, A.S.; Dorovatovskii, P.V.; Shiryaev, A.K.; Solovieva, S.E.; Antipin, I.S. Crystalline State Hydrogen Bonding of 2-(2-Hydroxybenzylidene)Thiazolo[3,2-a]Pyrimidines: A Way to Non-Centrosymmetric Crystals. Crystals 2022, 12, 494. [Google Scholar] [CrossRef]
- Shiryaev, A.K.; Baranovskaya, N.S.; Eremin, M.S. Synthesis of 5H-thiazolo [3, 2-a] pyrimidines. Chem. Heterocycl. Compd. 2013, 48, 1550–1554. [Google Scholar] [CrossRef]
- Karpfen, A.; Legon, A.C.; Pennington, W.T.; Hanks, T.W.; Arman, H.D.; Metrangolo, P.; Resnati, G.; Pilati, T.; Biella, S.; Rosokha, S.V.; et al. Halogen Bonding: Fundamentals and Applications; Metrangolo, P., Resnati, G., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 1–221. [Google Scholar]
- Cavallo, G.; Metrangolo, P.; Pilati, T.; Resnati, G.; Sansotera, M.; Terraneo, G. Halogen bonding: A general route in anion recognition and coordination. Chem. Soc. Rev. 2010, 39, 3772–3783. [Google Scholar] [CrossRef]
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen Bonds in Crystal Engineering: Like Hydrogen Bonds yet Different. Acc. Chem. Res. 2014, 47, 2514–2524. [Google Scholar] [CrossRef]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.G.; Murray, J.S. An overview of halogen bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other sigma-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef]
- Bertani, R.; Sgarbossa, P.; Venzo, A.; Lelj, F.; Amati, M.; Resnati, G.; Pilati, T.; Metrangolo, P.; Terraneo, G. Halogen bonding in metal-organic-supramolecular networks. Coord. Chem. Rev. 2010, 254, 677–695. [Google Scholar] [CrossRef]
- Persch, E.; Dumele, O.; Diederich, F. Molecular Recognition in Chemical and Biological Systems. Angew. Chem.-Int. Edit. 2015, 54, 3290–3327. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.S. Biomolecular Halogen Bonds; Springer: Berlin/Heidelberg, Germany, 2015; pp. 241–276. [Google Scholar]
- Bulfield, D.; Huber, S.M. Halogen Bonding in Organic Synthesis and Organocatalysis. Chem.-Eur. J. 2016, 22, 14434–14450. [Google Scholar] [CrossRef] [PubMed]
- Landenberger, K.B.; Bolton, O.; Matzger, A.J. Energetic-Energetic Cocrystals of Diacetone Diperoxide (DADP): Dramatic and Divergent Sensitivity Modifications via Cocrystallization. J. Am. Chem. Soc. 2015, 137, 5074–5079. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hu, R.X.; Pang, X.; Gao, H.Y.; Jin, W.J. The phosphorescent co-crystals of 1,4-diiodotetrafluorobenzene and bent 3-ring-N-heterocyclic hydrocarbons by C–I•••N and C–I•••π halogen bonds. CrystEngComm 2014, 16, 7942–7948. [Google Scholar] [CrossRef]
- Sivchik, V.V.; Solomatina, A.I.; Chen, Y.-T.; Karttunen, A.J.; Tunik, S.P.; Chou, P.-T.; Koshevoy, I.O. Halogen Bonding to Amplify Luminescence: A Case Study Using a Platinum Cyclometalated Complex. Angew. Chem.-Int. Ed. 2015, 54, 14057–14060. [Google Scholar] [CrossRef]
- Lashmanova, E.A.; Rybakov, V.B.; Shiryaev, A.K. Synthesis of adamantylated pyrimidines using the Biginelli reaction. Synthesis 2016, 48, 3965–3970. [Google Scholar]
- Lashmanova, E.A.; Agarkov, A.S.; Rybakov, V.B.; Shiryaev, A.K. Rearrangement of thiazolo[3,2-a]pyrimidines into triazolo[4,3-a]pyrimidines induced by C=N bond reduction. Chem. Heterocycl. Compd. 2019, 55, 1217–1221. [Google Scholar] [CrossRef]
- Lebedyeva, I.O.; Povstyanoy, M.V.; Ryabitskii, A.B.; Povstyanoy, V.M. Ternary condensation of Biginelli thiones, chloroacetic acid, and aldehydes as an effective approach towards thiazolo[3,2-a]pyrimidines and 5-arylidenethiazolidine-2,4-diones. J. Heterocycl. Chem. 2010, 47, 368–372. [Google Scholar] [CrossRef]
- Agarkov, A.S.; Kozhikhov, A.A.; Nefedova, A.A.; Ovsyannikov, A.S.; Islamov, D.R.; Solovieva, S.E.; Antipin, I.S. New Method for the Preparation of 2,3-Disubstituted 2,3-Dihydrothiazolo[3,2-a]pyrimidines. Dokl. Chem. 2022, 505, 177–183. [Google Scholar]
- Shiryaev, A.K.; Kolesnikova, N.G.; Kuznetsova, N.M.; Lashmanova, E.A. Alkylation of tetrahydropyrimidine-2-thions with ethyl chloroacetate. Chem. Heterocycl. Compd. 2013, 49, 1812–1817. [Google Scholar]
- Hu, J.; Wang, Y.; Wei, X.; Wu, X.; Chen, G.; Cao, G.; Shen, X.; Zhang, X.; Tang, Q.; Liang, G.; et al. Synthesis and biological evaluation of novel thiazolidinone derivatives as potential anti-inflammatory agents. Eur. J. Med. Chem. 2013, 64, 292–301. [Google Scholar] [CrossRef]
- Lazarenko, V.A.; Dorovatovskii, P.V.; Zubavichus, Y.V.; Burlov, A.S.; Koshchienko, Y.V.; Vlasenko, V.G.; Khrustalev, V.N. High-throughput small-molecule crystallography at the ‘Belok’beamline of the Kurchatov synchrotron radiation source: Transition metal complexes with azomethine ligands as a case study. Crystals 2017, 7, 325. [Google Scholar] [CrossRef] [Green Version]
- Svetogorov, R.D.; Dorovatovskii, P.V.; Lazarenko, V.A. Belok/XSA diffraction beamline for studying crystalline samples at Kurchatov Synchrotron Radiation Source. Cryst. Res. Technol. 2020, 55, 1900184. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Streek, J.V.D. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; The University of Western Australia: Perth, Australia, 2017. [Google Scholar]
- Mironov, V.F.; Nemtarev, A.V.; Tsepaeva, O.V.; Dimukhametov, M.N.; Litvinov, I.A.; Voloshina, A.D.; Pashirova, T.N.; Titov, E.A.; Lyubina, A.P.; Amerhanova, S.K.; et al. Rational design 2-hydroxypropylphosphonium salts as cancer cell mitochondria-targeted vectors: Synthesis, structure, and biological properties. Molecules 2021, 26, 6350. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Compounds | IC50 (µM) | ||||
|---|---|---|---|---|---|
| Cancer Cell Lines | Normal Cell Lines | ||||
| M-HeLa a | MCF−7 b | PC3 c | HuTu 80 d | Chang Liver (HeLa) e | |
| 1 | 88.0 ± 7.0 | >100 | 57.4 ± 4.5 | 88.0 ± 6.9 | >100 |
| 2 | 16.2 ± 1.3 | 72.0 ± 5.8 | >100 | 90.0 ± 8.0 | >100 |
| 3 | 98.1 ± 7.7 | >100 | 54.1 ± 4.3 | >100 | 96.0 ± 7.5 |
| 4 | >100 | >100 | >100 | >100 | >100 |
| 5 | >100 | >100 | >100 | 43.3 ± 3.4 | 51.4 ± 4.1 |
| 6 | 85±7.8 | >100 | 100±8.6 | 90.3±8.1 | 64.2±5.1 |
Sorafenib | 25.0 ± 1.8 | 27.5 ± 2.2 | 12.7 ± 1.1 | 13.0 ± 1.2 | 21.7 ± 1.7 |
| Compound | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Molecular formula | C24H22N2O4S | C24H21N3O7S | C24H22N2O6S | C24H22N2O5S | C24H21BrN2O5S | C24H21BrN2O5S |
| Sum formula | C24H22N2O4S | C24H21N3O7S | C24H22N2O6S | C24H22N2O5S | C24H21BrN2O5S | C24H21BrN2O5S |
| Formula weight | 434.50 | 495.50 | 480.52 | 450.50 | 529.39 | 529.39 |
| Crystal system | triclinic | monoclinic | monoclinic | monoclinic | triclinic | triclinic |
| Space group | P−1 (P1 bar) | P21/n | P21/c | P21 | P−1 (P1 bar) | P−1 (P1 bar) |
| Temp. of measurement, K | 110(2) | 105(2) | 100(2) | 100(2) | 105(2) | 100(2) |
| Cell parameters | a = 8.7755(3) Å, b = 10.8819(4) Å, c = 11.2091(4) Å; α = 96.579(1)° β = 92.727(1)° γ = 100.275(1)° | a = 8.1077(4) Å, b = 15.1003(7) Å, c = 18.1296(9) Å; α = 90° β = 96.517(2)° γ = 90° | a = 7.7100(15) Å, b = 23.790(5) Å, c = 11.830(2) Å; α = 90° β = 95.20(3)° γ = 90° | a = 7.8510(16) Å, b = 13.220(3) Å, c = 10.538(2) Å; α = 90° β = 108.42(3)° γ = 90° | a = 10.4542(6) Å, b = 10.8673(6) Å, c = 11.2907(6) Å; α = 110.578(2)° β = 90.151(2)° γ = 110.953(2)° | a = 9.6562(7) Å, b = 15.7076(12) Å, c = 16.0945(12) Å; α = 70.337(3)° β = 87.181(3)° γ = 77.925(3)° |
| V [Å3] | 1043.77(6) | 2205.24(19) | 2160.9(7) | 1037.7(4) | 1109.32(11) | 2247.3(3) |
| Z and Z′ | 2 and 1 | 4 and 1 | 4 and 1 | 2 and 1 | 2 and 1 | 4 and 2 |
| D(calc) [g/cm3] | 1.383 | 1.492 | 1.477 | 1.442 | 1.585 | 1.565 |
| λ (Å) | (MoKα) 0.71073 | (MoKα) 0.71073 | 0.74500 (synchrotron) | 0.74500 (synchrotron) | (MoKα) 0.71073 | (MoKα) 0.71073 |
| μ [/mm] | 0.190 | 0.201 | 0.220 | 0.220 | 1.987 | 1.962 |
| F(000) | 456 | 1032 | 1008 | 472 | 540 | 1080 |
| Theta Min-Max [Deg] | 1.9–30.0° | 2.6–28.4 | 2.0–31.0 | 2.9–31.0 | 1.9–29.2° | 2.2–28.9° |
| Reflections measured | 77,493 | 55,478 | 46,934 | 20,079 | 71,035 | 76,480 |
| Independent reflections | 6080 | 5516 | 5985 | 5672 | 5996 | 11163 |
| Observed reflections [I > 2σ(I)] | 5099 | 4061 | 5277 | 5487 | 4527 | 7060 |
| Goodness of fit | 1.020 | 1.017 | 1.032 | 1.054 | 0.998 | 1.031 |
| R [I > 2σ(I)] | R1 = 0.0393, wR2 = 0.1073 | R1 = 0.0387, wR2 = 0.0930 | R1 = 0.0360, wR2 = 0.0882 | R1 = 0.0295, wR2 = 0.0733 | R1 = 0.0398, wR2 = 0.0909 | R1 = 0.0471, wR2 = 0.0962 |
| R (all reflections) | R1 = 0.0497, wR2 = 0.1135 | R1 = 0.0645, wR2 = 0.1000 | R1 = 0.0418, wR2 = 0.0918 | R1 = 0.0310, wR2 = 0.0741 | R1 = 0.0656, wR2 = 0.0990 | R1 = 0.1230, wR2 = 0.1078 |
| Max. and Min. Resd. Dens. [e/Å−3] | 0.80 and –0.49 | 0.34 and –0.32 | 0.31 and −0.31 | 0.28 and −0.32 | 0.54 and –0.59 | 0.55 and −0.84 |
| Depositor numbers in CCDC | 2,232,800 | 2,232,801 | 2,231,737 | 2,231,736 | 2,232,802 | 2,232,803 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agarkov, A.S.; Nefedova, A.A.; Gabitova, E.R.; Mingazhetdinova, D.O.; Ovsyannikov, A.S.; Islamov, D.R.; Amerhanova, S.K.; Lyubina, A.P.; Voloshina, A.D.; Litvinov, I.A.; et al. (2-Hydroxy-3-Methoxybenzylidene)thiazolo[3,2-a]pyrimidines: Synthesis, Self-Assembly in the Crystalline Phase and Cytotoxic Activity. Int. J. Mol. Sci. 2023, 24, 2084. https://doi.org/10.3390/ijms24032084
Agarkov AS, Nefedova AA, Gabitova ER, Mingazhetdinova DO, Ovsyannikov AS, Islamov DR, Amerhanova SK, Lyubina AP, Voloshina AD, Litvinov IA, et al. (2-Hydroxy-3-Methoxybenzylidene)thiazolo[3,2-a]pyrimidines: Synthesis, Self-Assembly in the Crystalline Phase and Cytotoxic Activity. International Journal of Molecular Sciences. 2023; 24(3):2084. https://doi.org/10.3390/ijms24032084
Chicago/Turabian StyleAgarkov, Artem S., Anna A. Nefedova, Elina R. Gabitova, Dilyara O. Mingazhetdinova, Alexander S. Ovsyannikov, Daut R. Islamov, Syumbelya K. Amerhanova, Anna P. Lyubina, Alexandra D. Voloshina, Igor A. Litvinov, and et al. 2023. "(2-Hydroxy-3-Methoxybenzylidene)thiazolo[3,2-a]pyrimidines: Synthesis, Self-Assembly in the Crystalline Phase and Cytotoxic Activity" International Journal of Molecular Sciences 24, no. 3: 2084. https://doi.org/10.3390/ijms24032084