
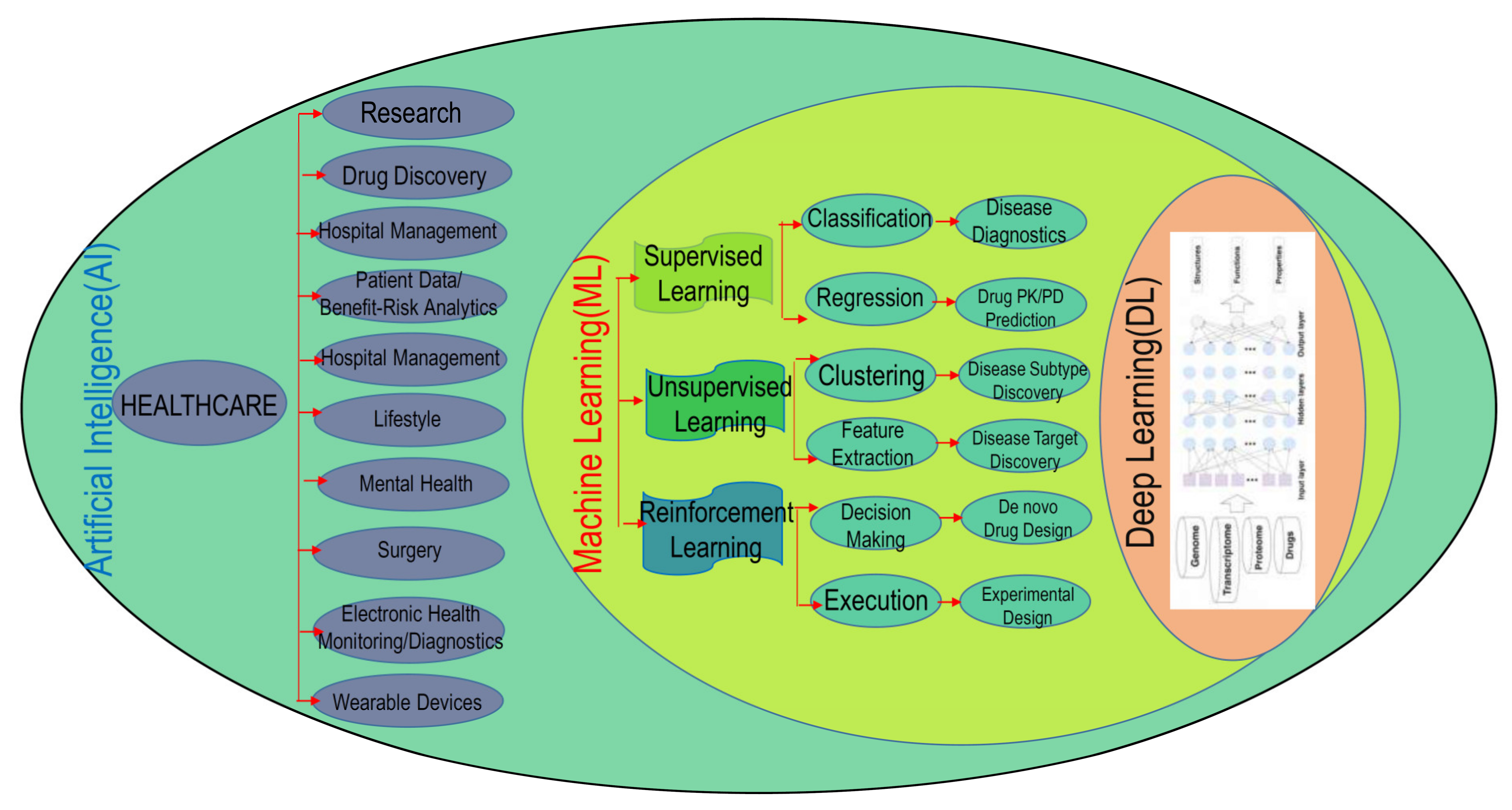
Artificial Intelligence and Machine Learning Technology Driven Modern Drug Discovery and Development

 , ,
, ,
Abstract
:1. Introduction
1.1. Artificial Intelligence: Facts to Ponder
1.2. AI: Networks and Tools
2. Futuristic Applications of AI in Drug Design
2.1. The Structure and Function of Proteins
2.1.1. Prognostication of Protein Folding from Sequence (Predicting the 3D Structure of a Target Protein)
2.1.2. Prognostication of Protein–Protein Interactions
- text mining of scientific documents,
- interactions estimated from genomic attributes, and
- interactions conveyed from model organisms, depending upon orthology.
2.1.3. Prognosticating Drug–Protein Interactions
2.1.4. De Novo Drug Design
2.2. Hit Discovery
2.2.1. Drug Repurposing
2.2.2. Virtual Screening (VS)
2.2.3. Activity Scoring
2.3. Hit-to-Lead Optimization
2.3.1. Quantitative Structure-Activity Relationship (QSAR)/Quantitative Structure-Property Relationship (QSPR) and Structure-Aided Modelling with AI
2.3.2. Generative Schemes for De Novo Drug Design with AI
2.3.3. Automated Chemical Synthesis Planning with AI
Foretelling the Retrosynthesis Roadmap
Prediction of Yield of Reaction and Understanding of Reaction Scheme
Synthesis Methods Digitized and Standardized
AI-Enabled Mechanized Reaction Space Sampling
2.4. In Silico Assessment of ADME/T Attributes
2.4.1. Physico-Chemical Characteristics
2.4.2. Pharmacokinetic Parameters (Absorption, Distribution, Biotransformation and Excretion)
2.4.3. Toxicity and the ADME/T Multi-Task Neural Network
3. Machine Learning Schemes and Usable Algorithms for Drug Design Scenarios
3.1. Approaches for Molecular Depiction
3.2. Transfer Learning Engagement for Low Data
3.3. The Process of Cross-Validation
3.4. What It Takes to Train the Deep Neural Networks
3.5. The Accessible Drug Design AI Source Code
4. Contribution of AI in the Lifecycle of Pharmaceutical Items
4.1. AI in Promoting Pharmaceutical Product Advancement
4.2. Contribution of AI towards Manufacturing of Pharmaceutical Products
4.3. Role of AI in Managing and Ensuring Quality
4.4. Role of AI Algorithms in Determining Clinical Trial Blueprints
4.5. Role of AI in Pharmaceutical Product Management
4.5.1. Role of AI in Market Positioning
4.5.2. Role of AI in Market Forecasting and Scrutiny
4.5.3. Role of AI in Product Cost
4.6. A Snapshot of AI-Based Advanced Implementations
4.6.1. Drug Delivery Technologies Engaging AI-Grounded Nanorobots
4.6.2. Role of AI in Concerted Drug Delivery and Augury of Synergism/Antagonism
4.6.3. The Materialization of AI in Nanomedicine
5. The Market Potential of AI Applications for Drug Discovery and Development
6. Continuing Bottlenecks in Accepting AI: Hints on Methods to Conquer
7. Conclusions and Future Promise
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, J.R. New Drug Development; Springer: New York, NY, USA, 2010. [Google Scholar]
- Hassan Baig, M.; Ahmad, K.; Roy, S.; Mohammad Ashraf, J.; Adil, M.; Haris Siddiqui, M.; Khan, S.; Amjad Kamal, M.; Provazník, I.; Choi, I. Computer aided drug design: Success and limitations. Curr. Pharm. Des. 2015, 22, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.S. Introduction to the volume and overview of computer assisted drug design in the drug discovery process. In Comprehensive Medicinal Chemistry II; Taylor, J.B., Triggle, D.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 1–11. [Google Scholar]
- Swaminathan, K.; Meller, J. Artificial Intelligence Approaches for Rational Drug Design and Discovery. Curr. Pharm. Des. 2007, 13, 1497–1508. [Google Scholar] [CrossRef] [Green Version]
- Lavecchia, A.; Di Giovanni, C. Virtual screening strategies in drug discovery: A critical review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef] [PubMed]
- Melville, J.L.; Burke, E.K.; Hirst, J.D. Machine Learning in Virtual Screening. Comb. Chem. High Throughput Screen. 2009, 12, 332–343. [Google Scholar] [CrossRef]
- Ma, J.; Sheridan, R.P.; Liaw, A.; Dahl, G.E.; Svetnik, V. Deep neural nets as a method for quantitative structure-activity relationships. J. Chem. Inf. Model. 2015, 55, 263–274. [Google Scholar] [CrossRef]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Schmidhuber, J. Deep Learning in Neural Networks: An Overview. Neural Netw. 2015, 61, 85–117. [Google Scholar] [CrossRef] [Green Version]
- Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; The MIT Press: Cambridge, UK, 2016. [Google Scholar]
- Ramesh, A.; Kambhampati, C.; Monson, J.R.; Drew, P.J. Artificial intelligence in medicine. Ann. R. Coll. Surg. Engl. 2004, 86, 334–338. [Google Scholar] [CrossRef]
- Miles, J.; Walker, A. The potential application of artificial intelligence in transport. IEEE Proc. Intell. Transp. Syst. 2006, 153, 183–198. [Google Scholar] [CrossRef]
- Yang, Y.; Siau, K. A Qualitative Research on Marketing and Sales in the Artificial Intelligence Age. In Proceedings of the Midwest United States Association for Information Systems(MWAIS), St. Louis, MO, USA, 17–18 May 2018. [Google Scholar]
- Wirtz, B.W.; Weyerer, J.C.; Geyer, C. Artificial Intelligence and the Public Sector—Applications and Challenges. Int. J. Public Adm. 2018, 42, 596–615. [Google Scholar] [CrossRef]
- Smith, R.G.; Farquhar, A. The road ahead for knowledge management: An AI perspective. AI Mag. 2000, 21, 17. [Google Scholar]
- Lamberti, M.J.; Wilkinson, M.; Donzanti, B.A.; Wohlhieter, G.E.; Parikh, S.; Wilkins, R.G.; Getz, K. A Study on the Application and Use of Artificial Intelligence to Support Drug Development. Clin. Ther. 2019, 41, 1414–1426. [Google Scholar] [CrossRef] [Green Version]
- Beneke, F.; Mackenrodt, M.-O. Artificial intelligence and collusion. IIC Int. Rev. Intellect. Prop. Compet. Law 2019, 50, 109–134. [Google Scholar] [CrossRef] [Green Version]
- Steels, L.; Brooks, R. The Artificial Life Route to Artificial Intelligence: Building Embodied, Situated Agents; Routledge: London, UK, 2018. [Google Scholar]
- Bielecki, A.; Bielecki, A. Foundations of artificial neural networks. In Models of Neurons and Perceptrons: Selected Problems and Challenges; Janusz, K., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 15–28. [Google Scholar]
- Kalyane, D.; Sanap, G.; Paul, D.; Shenoy, S.; Anup, N.; Polaka, S.; Tambe, V.; Tekade, R.K. Artificial intelligence in the pharmaceutical sector: Current scene and future prospect. In The Future of Pharmaceutical Product Development and Research; Academic Press: Cambridge, MA, USA, 2020; pp. 73–107. [Google Scholar] [CrossRef]
- da Silva, I.N.; Spatti, D.H.; Flauzino, R.A.; Liboni, L.H.B.; Alves, S.F.D.R. Artificial Neural Network Architectures and Training Processes. In Artificial Neural Networks; Springer: Cham, Switzerland, 2016; pp. 21–28. [Google Scholar] [CrossRef]
- Medsker, L.; Jain, L.C. Recurrent Neural Networks: Design and Applications; CRC Press: Boca Raton, FL, USA, 1999. [Google Scholar]
- Hanggi, M.; Moschytz, G.S. Cellular Neural Networks: Analysis, Design and Optimization; Springer Science & Business Media: Berlin, Germany, 2000. [Google Scholar]
- Rouse, M. IBM Watson Supercomputer. 2017. Available online: https://searchenterpriseai.techtarget.com/definition/IBM-Watson-supercomputer (accessed on 13 October 2020).
- Vyas, M.; Thakur, S.; Riyaz, B.; Bansal, K.K.; Tomar, B.; Mishra, V. Artificial intelligence: The beginning of a new era in pharmacy profession. Asian J. Pharm. 2018, 12, 72–76. [Google Scholar]
- Spencer, M.; Eickholt, J.; Cheng, J. A Deep Learning Network Approach to ab initio Protein Secondary Structure Prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 12, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Hou, J.; Adhikari, B.; Lyu, Q.; Cheng, J. Deep learning methods for protein torsion angle prediction. BMC Bioinform. 2017, 18, 417. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Sun, S.; Li, Z.; Zhang, R.; Xu, J. Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model. PLoS Comput. Biol. 2017, 13, e1005324. [Google Scholar] [CrossRef] [Green Version]
- Schaarschmidt, J.; Monastyrskyy, B.; Kryshtafovych, A.; Bonvin, A.M. Assessment of contact predictions in CASP12: Co-evolution and deep learning coming of age. Proteins Struct. Funct. Bioinform. 2017, 86, 51–66. [Google Scholar] [CrossRef]
- Falchi, F.; Caporuscio, F.; Recanatini, M. Structure-based design of small-molecule protein–protein interaction modulators: The story so far. Futur. Med. Chem. 2014, 6, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein-protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-Protein Interaction Networks, Integrated Over the Tree of Life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Cukuroglu, E.; Engin, H.B.; Gursoy, A.; Keskin, O. Hot spots in protein–protein interfaces: Towards drug discovery. Prog. Biophys. Mol. Biol. 2014, 116, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Higueruelo, A.P.; Jubb, H.; Blundell, T.L. Protein–protein interactions as druggable targets: Recent technological advances. Curr. Opin. Pharmacol. 2013, 13, 791–796. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef]
- Labbé, C.M.; Kuenemann, M.A.; Zarzycka, B.; Vriend, G.; Nicolaes, G.A.F.; Lagorce, D.; Miteva, M.A.; Villoutreix, B.O.; Sperandio, O. iPPI-DB: An online database of modulators of protein-protein interactions. Nucleic Acids Res. 2016, 44, D542–D547. [Google Scholar] [CrossRef]
- Shin, W.-H.; Christoffer, C.W.; Kihara, D. In silico structure-based approaches to discover protein-protein interaction-targeting drugs. Methods 2017, 131, 22–32. [Google Scholar] [CrossRef]
- Valkov, E.; Sharpe, T.; Marsh, M.; Greive, S.; Hyvonen, M. Targeting protein-proteininteractions and fragment-based drug discovery. Top. Curr. Chem. 2012, 317, 145–179. [Google Scholar]
- Wang, J.; Luo, C.; Shan, C.; You, Q.; Lu, J.; Elf, S.; Zhou, Y.; Wen, Y.; Vinkenborg, J.L.; Fan, J.; et al. Inhibition of human copper trafficking by a small molecule significantly attenuates cancer cell proliferation. Nat. Chem. 2015, 7, 968–979. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.C.; Dobbs, D.; Bonvin, A.M.; Honavar, V. Computational prediction of protein interfaces: A review of data driven methods. FEBS Lett. 2015, 589, 3516–3526. [Google Scholar] [CrossRef]
- Zhang, Q.C.; Petrey, D.; Norel, R.; Honig, B.H. Protein interface conservation across structure space. Proc. Natl. Acad. Sci. USA 2010, 107, 10896–10901. [Google Scholar] [CrossRef] [Green Version]
- Maheshwari, S.; Brylinski, M. Template-based identification of protein–protein interfaces using eFindSitePPI. Methods 2016, 93, 64–71. [Google Scholar] [CrossRef]
- Chen, R.; Li, L.; Weng, Z. ZDOCK: An initial-stage protein-docking algorithm. Proteins Struct. Funct. Bioinform. 2003, 52, 80–87. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock andSymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Vakser, I.A. Protein-Protein Docking: From Interaction to Interactome. Biophys. J. 2014, 107, 1785–1793. [Google Scholar] [CrossRef] [Green Version]
- Du, T.; Liao, L.; Wu, C.H.; Sun, B. Prediction of residue-residue contact matrix forprotein-protein interaction with Fisher score features and deep learning. Methods 2016, 110, 97–105. [Google Scholar] [CrossRef]
- Bai, F.; Morcos, F.; Cheng, R.R.; Jiang, H.; Onuchic, J.N. Elucidating the druggable interface of protein−protein interactions using fragment docking and coevolutionary analysis. Proc. Natl. Acad. Sci. USA 2016, 113, E8051–E8058. [Google Scholar] [CrossRef] [Green Version]
- Wan, F.; Zeng, J. Deep learning with feature embedding for compound– protein interaction prediction. bioRxiv 2016, 086033. [Google Scholar]
- AlQuraishi, M. End-to-End Differentiable Learning of Protein Structure. Cell Syst. 2019, 8, 292–301.e3. [Google Scholar] [CrossRef]
- Hutson, M. AI protein-folding algorithms solve structures faster than ever. Nature 2019. [Google Scholar] [CrossRef] [PubMed]
- Avdagic, Z.; Purisevic, E.; Omanovic, S.; Coralic, Z. Artificial Intelligence in Prediction of Secondary Protein Structure Using CB513 Database. Summit Transl. Bioinform. 2009, 2009, 1–5. [Google Scholar] [PubMed]
- Tian, K.; Shao, M.; Wang, Y.; Guan, J.; Zhou, S. Boosting compound-protein interaction prediction by deep learning. Methods 2016, 110, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, D.; Wang, H.; Luo, C.; Zheng, M.; Liu, H.; Zhu, W.; Luo, X.; Zhang, J.; Jiang, H. Computational Screening for Active Compounds Targeting Protein Sequences: Methodology and Experimental Validation. J. Chem. Inf. Model. 2011, 51, 2821–2828. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chen, J.; Xu, X.; Li, Y.; Zhao, H.; Fang, Y.; Li, X.; Zhou, W.; Wang, W.; Wang, Y. A systematic prediction of multiple drug–target interactions from chemical, genomic, and pharmacological data. PLoS ONE 2012, 7, e37608. [Google Scholar] [CrossRef]
- Xiao, X.; Min, J.L.; Lin, W.Z.; Liu, Z.; Cheng, X.; Chou, K.C. iDrug-Target: Predicting the interactions between drug compounds and target proteins in cellular networking via benchmark dataset optimization approach. J. Biomol. Struct. Dyn. 2015, 33, 2221–2233. [Google Scholar] [CrossRef]
- Mak, K.-K.; Pichika, M.R. Artificial intelligence in drug development: Present status and future prospects. Drug Discov. Today 2018, 24, 773–780. [Google Scholar] [CrossRef]
- Persidis, A. The benefits of drug repositioning. Drug Discov. World 2011, 12, 9–12. [Google Scholar]
- Koromina, M.; Pandi, M.T.; Patrinos, G.P. Rethinking drug repositioning anddevelopment with artificial intelligence, machine learning, and omics. Omics 2019, 23, 539–548. [Google Scholar] [CrossRef]
- Park, K. A review of computational drug repurposing. Transl. Clin. Pharmacol. 2019, 27, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Zhu, S.; Lu, W.; Liu, Z.; Huang, J.; Zhou, Y.; Fang, J.; Huang, Y.; Guo, H.; Li, L.; et al. Target identification among known drugs by deep learning from heterogeneous networks. Chem. Sci. 2020, 11, 1775–1797. [Google Scholar] [CrossRef] [Green Version]
- Achenbach, J.; Tiikkainen, P.; Franke, L.; Proschak, E. Computational tools for polypharmacology and repurposing. Futur. Med. Chem. 2011, 3, 961–968. [Google Scholar] [CrossRef]
- Ke, Y.-Y.; Peng, T.-T.; Yeh, T.-K.; Huang, W.-Z.; Chang, S.-E.; Wu, S.-H.; Hung, H.-C.; Hsu, T.-A.; Lee, S.-J.; Song, J.-S.; et al. Artificial intelligence approach fighting COVID-19 with repurposing drugs. Biomed. J. 2020, 43, 355–362. [Google Scholar] [CrossRef]
- Li, X.; Xu, Y.; Cui, H.; Huang, T.; Wang, D.; Lian, B.; Li, W.; Qin, G.; Chen, L.; Xie, L. Prediction of synergistic anti-cancer drug combinations based on drug target network and drug induced gene expression profiles. Artif. Intell. Med. 2017, 83, 35–43. [Google Scholar] [CrossRef]
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug discovery for the future. Expert Rev. Clin. Pharmacol. 2013, 6, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, X.; Liu, X.; Fu, Z.; Xiong, Z.; Wu, X.; Tan, X.; Zhao, J.; Zhong, F.; Wan, X.; et al. KinomeX: A web application for predicting kinome-wide polypharmacology effect of small molecules. Bioinformatics 2019, 35, 5354–5356. [Google Scholar] [CrossRef]
- Cyclica Launches Ligand ExpressTM, a Disruptive Cloud–Based Platform to Revolutionize Drug Discovery. Business Wire, 30 November 2017.
- Hessler, G.; Baringhaus, K.-H. Artificial Intelligence in Drug Design. Molecules 2018, 23, 2520. [Google Scholar] [CrossRef] [Green Version]
- Corey, E.J.; Wipke, W.T. Computer-Assisted Design of Complex Organic Syntheses. Science 1969, 166, 178–192. [Google Scholar] [CrossRef]
- Grzybowski, B.A.; Szymkuć, S.; Gajewska, E.P.; Molga, K.; Dittwald, P.; Wołos, A.; Klucznik, T. Chematica: A Story of Computer Code That Started to Think like a Chemist. Chem 2018, 4, 390–398. [Google Scholar] [CrossRef]
- Klucznik, T.; Mikulak-Klucznik, B.; McCormack, M.P.; Lima, H.; Szymkuć, S.; Bhowmick, M.; Molga, K.; Zhou, Y.; Rickershauser, L.; Gajewska, E.P.; et al. Efficient Syntheses of Diverse, Medicinally Relevant Targets Planned by Computer and Executed in the Laboratory. Chem 2018, 4, 522–532. [Google Scholar] [CrossRef] [Green Version]
- Segler, M.H.S.; Preuss, M.; Waller, M.P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018, 555, 604–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, H.C.S.; Shan, H.; Dahoun, T.; Vogel, H.; Yuan, S. Advancing Drug Discovery via Artificial Intelligence. Trends Pharmacol. Sci. 2019, 40, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Putin, E.; Asadulaev, A.; Ivanenkov, Y.; Aladinskiy, V.; Sanchez-Lengeling, B.; Aspuru-Guzik, A.; Zhavoronkov, A. Reinforced adversarial neural computer for de novo molecular design. J. Chem. Inform. Modeling 2018, 58, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Segler, M.H.S.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent. Sci. 2017, 4, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef] [Green Version]
- Merk, D.; Friedrich, L.; Grisoni, F.; Schneider, G. De Novo Design of Bioactive Small Molecules by Artificial Intelligence. Mol. Inform. 2018, 37, 1700153. [Google Scholar] [CrossRef] [Green Version]
- Schneider, G.; Clark, D.E. Automated de novo drug design: Are we nearly there yet? Angew. Chem. 2019, 131, 10906–10917. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Shahreza, M.L.; Ghadiri, N.; Mousavi, S.R.; Varshosaz, J.; Green, J. A review of network-based approaches to drug repositioning. Briefings Bioinform. 2017, 19, 878–892. [Google Scholar] [CrossRef]
- Klaeger, S.; Heinzlmeir, S.; Wilhelm, M.; Polzer, H.; Vick, B.; Koenig, P.-A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358, eaan4368. [Google Scholar] [CrossRef] [Green Version]
- Cabreiro, F.; Au, C.; Leung, K.-Y.; Vergara-Irigaray, N.; Cochemé, H.M.; Noori, T.; Weinkove, D.; Schuster, E.; Greene, N.D.; Gems, D. Metformin Retards Aging in C. elegans by Altering Microbial Folate and Methionine Metabolism. Cell 2013, 153, 228–239. [Google Scholar] [CrossRef] [Green Version]
- De Haes, W.; Frooninckx, L.; Van Assche, R.; Smolders, A.; Depuydt, G.; Billen, J.; Braeckman, B.P.; Schoofs, L.; Temmerman, L. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc. Natl. Acad. Sci. USA 2014, 111, E2501–E2509. [Google Scholar] [CrossRef] [Green Version]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.-J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Araki, M.; Gutteridge, A.; Honda, W.; Kanehisa, M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, i232–i240. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Zhao, X.; Zhou, J.; Yang, J.; Zhang, Y.; Kuang, W.; Peng, J.; Chen, L.; Zeng, J. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 2017, 8, 573. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Kim, N.D.; Seong, B.L. Pharmacophore-based virtual screening: A review of recent applications. Expert Opin. Drug Discov. 2010, 5, 205–222. [Google Scholar] [CrossRef]
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef] [Green Version]
- Leelananda, S.P.; Lindert, S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef]
- Talele, T.; Khedkar, S.; Rigby, A. Successful applications of computer aided drug discovery: Moving drugs from concept to the clinic. Curr. Top. Med. Chem. 2010, 10, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X. Inclusion of solvation and entropy in the knowledge-based scoringfunction for protein-ligand interactions. J. Chem. Inf. Model. 2010, 50, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. The dynamics of drug-target interactions: Drug-target residence time and its impact on efficacy and safety. Expert Opin. Drug Discov. 2010, 5, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Lu, W.; Liu, R.; Wang, Y.; Xie, Y.; Zhang, H.; Shi, Z.; Jiang, H.; Liu, Y.-C.; Chen, K.; et al. Machine-Learning-Assisted Approach for Discovering Novel Inhibitors Targeting Bromodomain-Containing Protein 4. J. Chem. Inf. Model. 2017, 57, 1677–1690. [Google Scholar] [CrossRef] [PubMed]
- Liew, C.Y.; Ma, X.H.; Liu, X.; Yap, C.W. SVM Model for Virtual Screening of Lck Inhibitors. J. Chem. Inf. Model. 2009, 49, 877–885. [Google Scholar] [CrossRef]
- Ma, X.; Jia, J.; Zhu, F.; Xue, Y.; Li, Z.; Chen, Y. Comparative analysis of machine learning methods in ligand-based virtual screening of large compound libraries. Comb. Chem. High Throughput Screen. 2009, 12, 344–357. [Google Scholar] [CrossRef]
- Unterthiner, T.; Mayr, A.; Klambauer, G.; Steijaert, M.; Ceulemans, H.; Wegner, J.K.; Hochreiter, S. Deep learning as an opportunity in virtual screening. In Proceedings of the The Workshop on Deep Learning & Representation Learning, Montreal, QC, Canada, 12 December 2014. [Google Scholar]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Kadurin, A.; Aliper, A.; Kazennov, A.; Mamoshina, P.; Vanhaelen, Q.; Khrabrov, K.; Zhavoronkov, A. The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 2016, 8, 10883–10890. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.-Y.; Grinter, S.Z.; Zou, X. Scoring functions and their evaluation methods for protein–ligand docking: Recent advances and future directions. Phys. Chem. Chem. Phys. 2010, 12, 12899–12908. [Google Scholar] [CrossRef]
- Khamis, M.A.; Gomaa, W.; Ahmed, W.F. Machine learning in computational docking. Artif. Intell. Med. 2015, 63, 135–152. [Google Scholar] [CrossRef]
- Ain, Q.U.; Aleksandrova, A.; Roessler, F.D.; Ballester, P.J. Machine-learning scoringfunctions to improve structure-based binding affinity prediction and virtual screening. WIREs Comput. Mol. Sci. 2015, 5, 405–424. [Google Scholar] [CrossRef]
- Kinnings, S.L.; Liu, N.; Tonge, P.J.; Jackson, R.M.; Xie, L.; Bourne, P.E. A Machine Learning-Based Method To Improve Docking Scoring Functions and Its Application to Drug Repurposing. J. Chem. Inf. Model. 2011, 51, 408–419. [Google Scholar] [CrossRef] [Green Version]
- Zsoldos, Z.; Reid, D.; Simon, A.; Sadjad, S.B.; Johnson, A.P. eHiTS: A new fast, exhaustive flexible ligand docking system. J. Mol. Graph. Model. 2007, 26, 198–212. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, Y. Improving scoring-docking-screening powers of protein-ligand scoring functions using random forest. J. Comput. Chem. 2016, 38, 169–177. [Google Scholar] [CrossRef]
- Repasky, M.P.; Shelley, M.; Friesner, R.A. Flexible Ligand Docking with Glide; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007. [Google Scholar]
- Jimenez, J.; Skalic, M.; Martinez-Rosell, G.; De Fabritiis, G. KDEEP: Protein-ligand absolute binding affinity prediction via 3D convolutional neural networks. J. Chem. Inf. Model. 2018, 58, 287–296. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM?A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Pereira, J.C.; Caffarena, E.R.; Dos Santos, C.N. Boosting docking-based virtualscreening with deep learning. J. Chem. Inf. Model. 2016, 56, 2495. [Google Scholar] [CrossRef] [Green Version]
- Esposito, E.X.; Hopfinger, A.J.; Madura, J.D. Methods for Applying the Quantitative Structure-Activity Relationship Paradigm. Artif. Intell. Med. 2004, 275, 131–213. [Google Scholar] [CrossRef]
- Myint, K.Z.; Xie, X.-Q. Recent Advances in Fragment-Based QSAR and Multi-Dimensional QSAR Methods. Int. J. Mol. Sci. 2010, 11, 3846–3866. [Google Scholar] [CrossRef]
- Hansch, C.; Fujita, T. Additions and corrections -ρ-σ-π analysis. A method for the correlation of biological activity and chemical structure. J. Am. Chem. Soc. 1964, 86, 5710. [Google Scholar] [CrossRef]
- Free, S.M.; Wilson, J.W. A Mathematical Contribution to Structure-Activity Studies. J. Med. Chem. 1964, 7, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Dobchev, D.A.; Pillai, G.; Karelson, M. In Silico Machine Learning Methods in Drug Development. Curr. Top. Med. Chem. 2014, 14, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Arodz, T.; Galvez, J. Computational Methods in Developing Quantitative Structure-Activity Relationships (QSAR): A Review. Comb. Chem. High Throughput Screen. 2006, 9, 213–228. [Google Scholar] [CrossRef]
- Ning, X.; Karypis, G. In silico structure-activity-relationship (SAR) models from machine learning: A review. Drug Dev. Res. 2010, 72, 138–146. [Google Scholar] [CrossRef]
- Dahl, G.E.; Jaitly, N.; Salakhutdinov, R. Multi-task neural networks for QSAR predictions. arXiv 2014, arXiv:1406.1231v1. [Google Scholar]
- Ramsundar, B.; Liu, B.; Wu, Z.; Verras, A.; Tudor, M.; Sheridan, R.P.; Pande, V. Is multitask deep learning practical for pharma? J. Chem. Inf. Model. 2017, 57, 2068–2076. [Google Scholar] [CrossRef]
- Subramanian, G.; Ramsundar, B.; Pande, V.; Denny, R.A. Computational Modeling of β-Secretase 1 (BACE-1) Inhibitors Using Ligand Based Approaches. J. Chem. Inf. Model. 2016, 56, 1936–1949. [Google Scholar] [CrossRef]
- Hartenfeller, M.; Schneider, G. De novo drug design. Methods Mol. Biol. 2011, 672, 299–323. [Google Scholar]
- Schneider, G.; Funatsu, K.; Okuno, Y.; Winkler, D. De novo Drug Design—Ye olde Scoring Problem Revisited. Mol. Inform. 2017, 36, 1681031. [Google Scholar] [CrossRef] [Green Version]
- Mullard, A. The drug-maker’s guide to the galaxy. Nature 2017, 549, 445–447. [Google Scholar] [CrossRef] [Green Version]
- Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. Molecular de-novo designthrough deep reinforcement learning. J. Cheminform. 2017, 9, 48. [Google Scholar] [CrossRef] [Green Version]
- Mnih, V.; Kavukcuoglu, K.; Silver, D.; Rusu, A.A.; Veness, J.; Bellemare, M.G.; Graves, A.; Riedmiller, M.; Fidjeland, A.K.; Ostrovski, G.; et al. Human-level control through deep reinforcement learning. Nature 2015, 518, 529–533. [Google Scholar] [CrossRef]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.K.; Hernandez-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Pu, Y.; Wang, W.; Henao, R.; Chen, L.; Gan, Z.; Li, C.; Carin, L. Adversarial symmetric variational autoencoder. arXiv 2017, arXiv:1711.04915v2. [Google Scholar]
- Kadurin, A.; Nikolenko, S.; Khrabrov, K.; Aliper, A.; Zhavoronkov, A. druGAN: An advanced generative adversarial autoencoder model for de novo generation of new moleculeswith desired molecular properties in silico. Mol. Pharm. 2017, 14, 3098–3104. [Google Scholar] [CrossRef]
- Coley, C.W.; Barzilay, R.; Green, W.H.; Jaakkola, T.S.; Jensen, K.F. Convolutional Embedding of Attributed Molecular Graphs for Physical Property Prediction. J. Chem. Inf. Model. 2017, 57, 1757–1772. [Google Scholar] [CrossRef]
- Andras, P. High-Dimensional Function Approximation With Neural Networks for Large Volumes of Data. IEEE Trans. Neural Netw. Learn. Syst. 2017, 29, 500–508. [Google Scholar] [CrossRef]
- Coley, C.W.; Green, W.H.; Jensen, K.F. Machine Learning in Computer-Aided Synthesis Planning. Accounts Chem. Res. 2018, 51, 1281–1289. [Google Scholar] [CrossRef]
- Maryasin, B.; Marquetand, P.; Maulide, N. Machine learning for organic synthesis: Are robots replacing chemists ? Angew. Chem. Int. Ed. 2018, 57, 6978–6980. [Google Scholar] [CrossRef]
- Santos, C.B.R.; Lobato, C.C.; Braga, F.S.; Morais, S.S.S.; Santos, C.F.; Fernandes, C.P.; Brasil, D.S.B.; Hage-Melim, L.I.S.; Macêdo, W.J.C.; Carvalho, J.C.T. Application of Hartree-Fock Method for Modeling of Bioactive Molecules Using SAR and QSPR. Comput. Mol. Biosci. 2014, 4, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Collins, K.D.; Glorius, F. A robustness screen for the rapid assessment of chemical reactions. Nat. Chem. 2013, 5, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Browne, C.B.; Powley, E.; Whitehouse, D.; Lucas, S.M.; Cowling, P.I.; Rohlfshagen, P.; Tavener, S.; Perez, D.; Samothrakis, S.; Colton, S. A Survey of Monte Carlo Tree Search Methods. IEEE Trans. Comput. Intell. AI Games 2012, 4, 1–43. [Google Scholar] [CrossRef] [Green Version]
- Kayala, M.A.; Azencott, C.A.; Chen, J.H.; Baldi, P. Learning to predict chemicalreactions. J. Chem. Inf. Model. 2011, 51, 2209–2222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, A.; Johnson, A.P.; Law, J.; Mirzazadeh, M.; Ravitz, O.; Simon, A. Computer-aided synthesis design: 40 years on. WIREs Comput. Mol. Sci. 2011, 2, 79–107. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Waller, M.P. Neural-Symbolic Machine Learning for Retrosynthesis and Reaction Prediction. Chem. A Eur. J. 2017, 23, 5966–5971. [Google Scholar] [CrossRef]
- Silver, D.; Huang, A.; Maddison, C.J.; Guez, A.; Sifre, L.; van den Driessche, G.; Schrittwieser, J.; Antonoglou, I.; Panneershelvam, V.; Lanctot, M.; et al. Mastering the game of Go with deep neural networks and tree search. Nature 2016, 529, 484–489. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, X.; Zare, R.N. Optimizing Chemical Reactions with Deep Reinforcement Learning. ACS Cent. Sci. 2017, 3, 1337–1344. [Google Scholar] [CrossRef] [Green Version]
- Coley, C.W.; Barzilay, R.; Jaakkola, T.S.; Green, W.H.; Jensen, K.F. Prediction of Organic Reaction Outcomes Using Machine Learning. ACS Cent. Sci. 2017, 3, 434–443. [Google Scholar] [CrossRef] [Green Version]
- Monemian, S.A.; Shahsavan, H.; Bolouri, O.; Taranejoo, S.; Goodarzi, V.; Torabi-Angaji, M. A stacked neural network approach for yield prediction of propylene polymerization. J. Appl. Polym. Sci. 2010, 116, 1237–1246. [Google Scholar] [CrossRef]
- Rahman, M.B.A.; Chaibakhsh, N.; Basri, M.; Salleh, A.B.; Rahman, R.N.Z.R.A. Application of Artificial Neural Network for Yield Prediction of Lipase-Catalyzed Synthesis of Dioctyl Adipate. Appl. Biochem. Biotechnol. 2009, 158, 722–735. [Google Scholar] [CrossRef]
- Ahneman, D.T.; Estrada, J.G.; Lin, S.; Dreher, S.D.; Doyle, A.G. Predicting reaction performance in C–N cross-coupling using machine learning. Science 2018, 360, 186–190. [Google Scholar] [CrossRef] [Green Version]
- Merrifield, R.B. Automated Synthesis of Peptides. Science 1965, 150, 178–185. [Google Scholar] [CrossRef]
- Alvarado-Urbina, G.; Sathe, G.M.; Liu, W.-C.; Gillen, M.F.; Duck, P.D.; Bender, R.; Ogilvie, K.K. Automated Synthesis of Gene Fragments. Science 1981, 214, 270–274. [Google Scholar] [CrossRef]
- Karp, P.D. Pathway Databases: A Case Study in Computational Symbolic Theories. Science 2001, 293, 2040–2044. [Google Scholar] [CrossRef] [Green Version]
- Steiner, S.; Wolf, J.; Glatzel, S.; Andreou, A.; Granda, J.M.; Keenan, G.; Hinkley, T.; Aragon-Camarasa, G.; Kitson, P.J.; Angelone, D.; et al. Organic synthesis in a modular robotic system driven by a chemical programming language. Science 2019, 363, eaav2211. [Google Scholar] [CrossRef] [Green Version]
- Fuhrman, J.A.; Schwalbach, M.S.; Stingl, U. Proteorhodopsins: An array of physiological roles? Nat. Rev. Microbiol. 2008, 6, 488–494. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- Zhu, H. Big Data and Artificial Intelligence Modeling for Drug Discovery. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 573–589. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, F.; Mehridehnavi, A.; Fassihi, A.; Pérez-Sánchez, H. Deep neural network in biological activity prediction using deep belief network. Appl. Soft Comput. 2018, 62, 251–258. [Google Scholar] [CrossRef]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. DeepTox: Toxicity Prediction using Deep Learning. Front. Environ. Sci. 2016, 3, 80. [Google Scholar] [CrossRef] [Green Version]
- Stork, C.; Chen, Y.; Šícho, M.; Kirchmair, J. Hit Dexter 2.0: Machine-Learning Models for the Prediction of Frequent Hitters. J. Chem. Inf. Model. 2019, 59, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- Urban, G.; Subrahmanya, N.; Baldi, P. Inner and Outer Recursive Neural Networks for Chemoinformatics Applications. J. Chem. Inf. Model. 2018, 58, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Barzilay, R.; Jaakkola, T. Junction tree variational autoencoder for molecular graph generation. In Proceedings of the 35th International Conference on Machine Learning, PMLR, Stockholmsmässan, Stockholm, Sweden, 10–15 July 2018; pp. 2323–2332. [Google Scholar]
- Duvenaud, D.K.; Maclaurin, D.; Iparraguirre, J.; Gomez-Bombarelli, R.; Hirzel, T.; Aspuru-Guzik, A.; Adams, R.P. Convolutional networks on graphs for learning molecular fingerprints. In Advances in Neural Information Processing Systems; Cortes, C., Lawrence, N., Lee, D., Sugiyama, M., Garnett, R., Eds.; Curran Associates, Inc.: Red Hook, NY, USA, 2015; pp. 1–9. [Google Scholar]
- Durrant, J.D.; McCammon, J.A. NNScore 2.0: A Neural-Network Receptor–Ligand Scoring Function. J. Chem. Inf. Model. 2011, 51, 2897–2903. [Google Scholar] [CrossRef] [PubMed]
- Wójcikowski, M.; Zielenkiewicz, P.; Siedlecki, P. Open Drug Discovery Toolkit (ODDT): A new open-source player in the drug discovery field. J. Cheminform. 2015, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lengeling, B.; Outeiral, C.; Guimaraes, G.L.; Aspuru-Guzik, A. Optimizing distributions over molecular space. An objective-reinforced generative adversarial network for inverse-design chemistry (ORGANIC). ChemRxiv 2017, 1–18. [Google Scholar]
- Feinberg, E.N.; Sur, D.; Wu, Z.; Husic, B.E.; Mai, H.; Li, Y.; Sun, S.; Yang, J.; Ramsundar, B.; Pande, V.S. PotentialNet for molecular property prediction. ACS Cent. Sci. 2018, 4, 1520–1530. [Google Scholar] [CrossRef]
- Awale, M.; Reymond, J.-L. Polypharmacology browser PPB2: Target prediction combining nearest neighbors with machine learning. J. Chem. Inf. Model. 2019, 59, 10–17. [Google Scholar] [CrossRef]
- Cho, A. No room for error. Science 2020, 369, 130–133. [Google Scholar] [CrossRef]
- Blaschke, T.; Arús-Pous, J.; Chen, H.; Margreitter, C.; Tyrchan, C.; Engkvist, O.; Papadopoulos, K.; Patronov, A. REINVENT 2.0: An AI Tool for De Novo Drug Design. J. Chem. Inf. Model. 2020, 60, 5918–5922. [Google Scholar] [CrossRef]
- Coley, C.W.; Rogers, L.; Green, W.H.; Jensen, K.F. SCScore: Synthetic Complexity Learned from a Reaction Corpus. J. Chem. Inf. Model. 2018, 58, 252–261. [Google Scholar] [CrossRef]
- Yasuo, N.; Sekijima, M. Improved Method of Structure-Based Virtual Screening via Interaction-Energy-Based Learning. J. Chem. Inf. Model. 2019, 59, 1050–1061. [Google Scholar] [CrossRef] [Green Version]
- Caramelli, D.; Salley, D.; Henson, A.; Camarasa, G.A.; Sharabi, S.; Keenan, G.; Cronin, L. Networking chemical robots for reaction multitasking. Nat. Commun. 2018, 9, 3406. [Google Scholar] [CrossRef]
- Coomans, D.; Jonckheer, M.; Massart, D.; Broeckaert, I.; Blockx, P. The application of linear discriminant analysis in the diagnosis of thyroid diseases. Anal. Chim. Acta 1978, 103, 409–415. [Google Scholar] [CrossRef]
- Granda, J.M.; Donina, L.; Dragone, V.; Long, D.-L.; Cronin, L. Controlling an organic synthesis robot with machine learning to search for new reactivity. Nature 2018, 559, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Perera, D.; Tucker, J.W.; Brahmbhatt, S.; Helal, C.J.; Chong, A.; Farrell, W.; Richardson, P.; Sach, N.W. A platform for automated nanomole-scale reaction screening and micromole-scale synthesis in flow. Science 2018, 359, 429–434. [Google Scholar] [CrossRef] [Green Version]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Lusci, A.; Pollastri, G.; Baldi, P. Deep Architectures and Deep Learning in Chemoinformatics: The Prediction of Aqueous Solubility for Drug-Like Molecules. J. Chem. Inf. Model. 2013, 53, 1563–1575. [Google Scholar] [CrossRef]
- Artursson, P.; Karlsson, J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem. Biophys. Res. Commun. 1991, 175, 880–885. [Google Scholar] [CrossRef]
- Hubatsch, I.; Ragnarsson, E.G.E.; Artursson, P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2007, 2, 2111–2119. [Google Scholar] [CrossRef]
- Selvaraj, C.; Chandra, I.; Singh, S.K. Artificial intelligence and machine learning approaches for drug design: Challenges and opportunities for the pharmaceutical industries. Mol. Divers. 2021, 26, 1893–1913. [Google Scholar] [CrossRef] [PubMed]
- OECD. Guidance document on the validation of (quantitative) structure-activity relationships [(Q) SAR] models. In OECD Series on Testing and Assessment No. 69; ENV/JM/MONO; OECD: Paris, France, 2007; Volume 2, p. 154. [Google Scholar]
- Tian, S.; Li, Y.; Wang, J.; Zhang, J.; Hou, T. ADME Evaluation in Drug Discovery. 9. Prediction of Oral Bioavailability in Humans Based on Molecular Properties and Structural Fingerprints. Mol. Pharm. 2011, 8, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Sim, D.S.M. Drug Distribution; Springer International Publishing: Cham, Switzerland, 2015. [Google Scholar]
- Lombardo, F.; Jing, Y. In Silico Prediction of Volume of Distribution in Humans. Extensive Data Set and the Exploration of Linear and Nonlinear Methods Coupled with Molecular Interaction Fields Descriptors. J. Chem. Inf. Model. 2016, 56, 2042–2052. [Google Scholar] [CrossRef] [PubMed]
- Matlock, M.; Hughes, T.B.; Swamidass, S.J. XenoSite server: A web-available site of metabolism prediction tool. Bioinformatics 2014, 31, 1136–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaretzki, J.; Matlock, M.; Swamidass, S.J. XenoSite: Accurately Predicting CYP-Mediated Sites of Metabolism with Neural Networks. J. Chem. Inf. Model. 2013, 53, 3373–3383. [Google Scholar] [CrossRef] [PubMed]
- Le Dang, N.; Hughes, T.B.; Krishnamurthy, V.; Swamidass, S.J. A simple model predicts UGT-mediated metabolism. Bioinformatics 2016, 32, 3183–3189. [Google Scholar] [CrossRef] [Green Version]
- Sim, D.S.M. Drug elimination. In Pharmacological Basis of Acute Care; Chan, Y., Ng, K., Sim, D., Eds.; Springer: Cham, Switzerland, 2015; pp. 37–47. [Google Scholar]
- Lombardo, F.; Obach, R.S.; Varma, M.V.; Stringer, R.; Berellini, G. Clearance Mechanism Assignment and Total Clearance Prediction in Human Based upon in Silico Models. J. Med. Chem. 2014, 57, 4397–4405. [Google Scholar] [CrossRef]
- Guengerich, F.P. Mechanisms of Drug Toxicity and Relevance to Pharmaceutical Development. Drug Metab. Pharmacokinet. 2011, 26, 3–14. [Google Scholar] [CrossRef]
- Xu, Y.; Pei, J.; Lai, L. Deep Learning Based Regression and Multiclass Models for Acute Oral Toxicity Prediction with Automatic Chemical Feature Extraction. J. Chem. Inf. Model. 2017, 57, 2672–2685. [Google Scholar] [CrossRef]
- Sushko, I.; Salmina, E.; Potemkin, V.A.; Poda, G.; Tetko, I.V. ToxAlerts: A Web Server of Structural Alerts for Toxic Chemicals and Compounds with Potential Adverse Reactions. J. Chem. Inf. Model. 2012, 52, 2310–2316. [Google Scholar] [CrossRef]
- Kearnes, S.; Goldman, B.; Pande, V. Modeling industrial ADMET data with multitask networks. arXiv 2016, arXiv:1606.08793v3. [Google Scholar]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL Keys for Use in Drug Discovery. ChemInform 2003, 34. [Google Scholar] [CrossRef] [Green Version]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Bender, A.; Mussa, A.H.Y.; Glen, R.C.; Reiling, S. Similarity Searching of Chemical Databases Using Atom Environment Descriptors (MOLPRINT 2D): Evaluation of Performance. J. Chem. Inf. Comput. Sci. 2004, 44, 1708–1718. [Google Scholar] [CrossRef]
- Wallach, I.; Dzamba, M.; Heifets, A. AtomNet: A deep convolutional neural network for bioactivity prediction in structure based drug discovery. Math. Z. 2015, 47, 34–46. [Google Scholar]
- Kearnes, S.; McCloskey, K.; Berndl, M.; Pande, V.; Riley, P. Molecular graph convolutions: Moving beyond fingerprints. J. Comput. Mol. Des. 2016, 30, 595–608. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 2017, 9, 513–530. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.G.; Wiswesser, W.J. The Wiswesser Line-Formula Chemical Notation; McGraw-Hill: New York, NY, USA, 1975. [Google Scholar]
- Ash, S.; Cline, M.A.; Homer, R.W.; Hurst, T.; Smith, G.B. ChemInform Abstract: SYBYL Line Notation (SLN): A Versatile Language for Chemical Structure Representation. ChemInform 2010, 28. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical Language and Information System 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar]
- Heller, S.R.; McNaught, A.; Pletnev, I.V.; Stein, S.; Tchekhovskoi, D. InChI, the IUPAC International Chemical Identifier. J. Cheminform. 2015, 7, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Goh, G.B.; Hodas, N.O.; Siegel, C.; Vishnu, A. SMILES2Vec: An Interpretable General-Purpose Deep Neural Network for Predicting Chemical Properties. arXiv 2017, arXiv:1712.02034v2. [Google Scholar]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Sahoo, S.; Adhikari, C.; Kuanar, M.; Mishra, B. A Short Review of the Generation of Molecular Descriptors and Their Applications in Quantitative Structure Property/Activity Relationships. Curr. Comput. Aided-Drug Des. 2016, 12, 181–205. [Google Scholar] [CrossRef] [PubMed]
- Danishuddin; Khan, A.U. Descriptors and their selection methods in QSAR analysis: Paradigm for drug design. Drug Discov. Today 2016, 21, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Mauri, A.; Consonni, V.; Pavan, M.; Todeschini, R. DRAGON software: An easy approach to molecular descriptor calculations. Match Commun. Math Comput. Chem. 2006, 56, 237–248. [Google Scholar]
- Cao, D.-S.; Xu, Q.-S.; Hu, Q.-N.; Liang, Y.-Z. ChemoPy: Freely available python package for computational biology and chemoinformatics. Bioinformatics 2013, 29, 1092–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Hutchison, G.R. Cinfony—Combining Open Source cheminformatics toolkits behind a common interface. Chem. Central J. 2008, 2, 24. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.J.; Yang, Q. A Survey on Transfer Learning. IEEE Trans. Knowl. Data Eng. 2010, 22, 1345–1359. [Google Scholar] [CrossRef]
- Vinyals, O.; Blundell, C.; Lillicrap, T.; Kavukcuoglu, K.; Wierstra, D. Matching networks for one shot learning. In Proceedings of the Neural Information Processing Systems Conference, Barcelona, Spain 5–10 December 2016. [Google Scholar]
- Altae-Tran, H.; Ramsundar, B.; Pappu, A.S.; Pande, V. Low Data Drug Discovery with One-Shot Learning. ACS Cent. Sci. 2017, 3, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Cortes, C.; Kuznetsov, V.; Mohri, M. Ensemble methods for structured prediction. In Proceedings of the 31st International Conference on Machine Learning, Beijing, China, 21–26 June 2014; pp. 1134–1142. [Google Scholar]
- Chen, B.; Sheridan, R.P.; Hornak, V.; Voigt, J.H. Comparison of Random Forest and Pipeline Pilot Naïve Bayes in Prospective QSAR Predictions. J. Chem. Inf. Model. 2012, 52, 792–803. [Google Scholar] [CrossRef]
- Sheridan, R.P. Time-Split Cross-Validation as a Method for Estimating the Goodness of Prospective Prediction. J. Chem. Inf. Model. 2013, 53, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.I.; Peng, Y.; Augsburger, L.L. Generalization of a prototype intelligent hybrid system for hard gelatin capsule formulation development. AAPS PharmSciTech 2005, 6, E449–E457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, C.H.; Narayan, R.; Nayak, U.Y. Computational modeling for formulation design. Drug Discov. Today 2018, 24, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Jain, A.; Hailemariam, L.; Suresh, P.; Akkisetty, P.; Joglekar, G.; Venkatasubramanian, V.; Reklaitis, G.V.; Morris, K.; Basu, P. Toward intelligent decision support for pharmaceutical product development. J. Pharm. Innov. 2006, 1, 23–35. [Google Scholar] [CrossRef]
- Rantanen, J.; Khinast, J. The Future of Pharmaceutical Manufacturing Sciences. J. Pharm. Sci. 2015, 104, 3612–3638. [Google Scholar] [CrossRef] [Green Version]
- Ketterhagen, W.R.; Am Ende, M.T.; Hancock, B.C. Process Modeling in the Pharmaceutical Industry using the Discrete Element Method. J. Pharm. Sci. 2009, 98, 442–470. [Google Scholar] [CrossRef]
- Chen, W.; Desai, D.; Good, D.; Crison, J.; Timmins, P.; Paruchuri, S.; Wang, J.; Ha, K. Mathematical Model-Based Accelerated Development of Extended-release Metformin Hydrochloride Tablet Formulation. AAPS PharmSciTech 2015, 17, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Meziane, F.; Vadera, S.; Kobbacy, K.; Proudlove, N. Intelligent systems in manufacturing: Current developments and future prospects. Integr. Manuf. Syst. 2000, 11, 218–238. [Google Scholar] [CrossRef]
- Sahu, A.; Mishra, J.; Kushwaha, N. Artificial Intelligence (AI) in Drugs and Pharmaceuticals. Comb. Chem. High Throughput Screen. 2022, 25, 1818–1837. [Google Scholar] [CrossRef]
- Faure, A.; York, P.; Rowe, R. Process control and scale-up of pharmaceutical wet granulation processes: A review. Eur. J. Pharm. Biopharm. 2001, 52, 269–277. [Google Scholar] [CrossRef]
- Landin, M. Artificial intelligence tools for scaling up of high shear wet granulationprocess. J. Pharm. Sci. 2017, 106, 273–277. [Google Scholar] [CrossRef] [Green Version]
- Das, M.K.; Chakraborty, T. ANN in Pharmaceutical Product and Process Development. In Artificial Neural Network for Drug Design, Delivery and Disposition; Puri, M., Pathak, Y., Sutariya, V.K., Tipparaju, S., Moreno, W., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 277–293. [Google Scholar]
- Gams, M.; Horvat, M.; Ožek, M.; Luštrek, M.; Gradišek, A. Integrating Artificial and Human Intelligence into Tablet Production Process. AAPS PharmSciTech 2014, 15, 1447–1453. [Google Scholar] [CrossRef] [Green Version]
- Kraft, D.L. System and Methods for the Production of Personalized Drug Products. U.S. Patent 20120041778A1, 29 January 2019. [Google Scholar]
- Aksu, B.; Paradkar, A.; de Matas, M.; Özer, Ö.; Güneri, T.; York, P. A quality bydesign approach using artificial intelligence techniques to control the critical quality attributes of ramipril tablets manufactured by wet granulation. Pharm. Dev. Technol. 2013, 18, 236–245. [Google Scholar] [CrossRef]
- Goh, W.Y.; Lim, C.; Peh, K.; Subari, K. Application of a Recurrent Neural Network to Prediction of Drug Dissolution Profiles. Neural Comput. Appl. 2002, 10, 311–317. [Google Scholar] [CrossRef]
- Drăgoi, E.N.; Curteanu, S.; Fissore, D. On the Use of Artificial Neural Networks to Monitor a Pharmaceutical Freeze-Drying Process. Dry. Technol. 2013, 31, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Reklaitis, R. Towards Intelligent Decision Support for Pharmaceutical Product Development; PharmaHub: Olongapo, Philippines, 2008. [Google Scholar]
- Wang, X. Intelligent quality management using knowledge discovery in databases. In Proceedings of the 2009 International Conference on Computational Intelligence and Software Engineering, Wuhan, China, 11–13 December 2009; IEEE: Piscataway, NJ, USA, 2009; pp. 1–4. [Google Scholar]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef]
- Harrer, S.; Shah, P.; Antony, B.; Hu, J. Artificial Intelligence for Clinical Trial Design. Trends Pharmacol. Sci. 2019, 40, 577–591. [Google Scholar] [CrossRef] [Green Version]
- Fogel, D.B. Factors associated with clinical trials that fail and opportunities for improving the likelihood of success: A review. Contemp. Clin. Trials Commun. 2018, 11, 156–164. [Google Scholar] [CrossRef]
- Kalafatis, S.P.; Tsogas, M.H.; Blankson, C. Positioning strategies in business markets. J. Bus. Ind. Mark. 2000, 15, 416–437. [Google Scholar] [CrossRef]
- Jalkala, A.M.; Keränen, J. Brand positioning strategies for industrial firms providing customer solutions. J. Bus. Ind. Mark. 2014, 29, 253–264. [Google Scholar] [CrossRef]
- Ding, M.; Eliashberg, J.; Stremersch, S. Innovation and Marketing in the Pharmaceutical Industry; Springer: New York, NY, USA, 2016. [Google Scholar]
- Dou, W.; Lim, K.H.; Su, C.; Zhou, N.; Cui, N. Brand Positioning Strategy Using Search Engine Marketing. MIS Q. 2010, 34, 261. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.-Y.; Chen, Y.-F.; Kuo, I.-T.; Ku, H.C. An intelligent market segmentation system using k-means and particle swarm optimization. Expert Syst. Appl. 2009, 36, 4558–4565. [Google Scholar] [CrossRef]
- Toker, D.; Tozan, H.; Vayvai, O. A Decision Model for Pharmaceutical Marketing and a Case Study in Turkey. Econ. Res.-Ekon. 2013, 26, 101–114. [Google Scholar] [CrossRef]
- Singh, J.; Flaherty, K.; Sohi, R.S.; Deeter-Schmelz, D.; Habel, J.; Le Meunier-FitzHugh, K.; Malshe, A.; Mullins, R.; Onyemah, V. Sales profession and professionals in the age of digitization and artificial intelligence technologies: Concepts, priorities, and questions. J. Pers. Sell. Sales Manag. 2018, 39, 2–22. [Google Scholar] [CrossRef]
- Milgrom, P.R.; Tadelis, S. How Artificial Intelligence and Machine Learning Can Impact Market Design; National Bureau of Economic Research: Cambridge, MA, USA; University of Chicago Press: Chicago, IL, USA, 2019; pp. 567–586. [Google Scholar]
- Davenport, T.; Guha, A.; Grewal, D.; Bressgott, T. How artificial intelligence will change the future of marketing. J. Acad. Mark. Sci. 2020, 48, 24–42. [Google Scholar] [CrossRef] [Green Version]
- Syam, N.; Sharma, A. Waiting for a sales renaissance in the fourth industrial revolution: Machine learning and artificial intelligence in sales research and practice. Ind. Mark. Manag. 2018, 69, 135–146. [Google Scholar] [CrossRef]
- Duran, O.; Rodriguez, N.; Consalter, L.A. Neural networks for cost estimation of shelland tube heat exchangers. Expert Syst. Appl. 2009, 36, 7435–7440. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Goto, D.; Yang, K.; Downton, K.; LeComte, P.; Olson, M.; Mullins, C. A Literature Review of Factors Affecting Price and Competition in the Global Pharmaceutical Market. Value Health 2016, 19, A265. [Google Scholar] [CrossRef]
- de Jesus, A. AI for Pricing—Comparing 5 Current Applications. Emerj. Artif. Intell. Res. 2019. [Google Scholar]
- Hassanzadeh, P.; Atyabi, F.; Dinarvand, R. The significance of artificial intelligence in drug delivery system design. Adv. Drug Deliv. Rev. 2019, 151–152, 169–190. [Google Scholar] [CrossRef]
- Luo, M.; Feng, Y.; Wang, T.; Guan, J. Micro-/nanorobots at work in active drug delivery. Adv. Funct. Mater. 2018, 28, 1706100. [Google Scholar] [CrossRef]
- Fu, J.; Yan, H. Controlled drug release by a nanorobot. Nat. Biotechnol. 2012, 30, 407–408. [Google Scholar] [CrossRef]
- Calzolari, D.; Bruschi, S.; Coquin, L.; Schofield, J.; Feala, J.D.; Reed, J.C.; McCulloch, A.D.; Paternostro, G. Search Algorithms as a Framework for the Optimization of Drug Combinations. PLoS Comput. Biol. 2008, 4, e1000249. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.; Km, G. Artificial intelligence and related technologies enabled nanomedicine for advanced cancer treatment. Nanomedicine 2020, 15, 433–435. [Google Scholar] [CrossRef]
- Tsigelny, I.F. Artificial intelligence in drug combination therapy. Briefings Bioinform. 2019, 20, 1434–1448. [Google Scholar] [CrossRef]
- Mason, D.J.; Eastman, R.T.; Lewis, R.P.I.; Stott, I.P.; Guha, R.; Bender, A. Using Machine Learning to Predict Synergistic Antimalarial Compound Combinations With Novel Structures. Front. Pharmacol. 2018, 9, 1096. [Google Scholar] [CrossRef]
- Ho, D.; Wang, P.; Kee, T. Artificial intelligence in nanomedicine. Nanoscale Horiz. 2018, 4, 365–377. [Google Scholar] [CrossRef]
- Sacha, G.M.; Varona, P. Artificial intelligence in nanotechnology. Nanotechnology 2013, 24, 452002. [Google Scholar] [CrossRef] [Green Version]
- Pellat, G.; Anghelache, C. Governance in the EU Member States in the Era of Big Data; Editura EconomicĄ Distributie: Bucharest, Romania, 2019. [Google Scholar]
- van der Lee, M.; Swen, J.J. Artificial intelligence in pharmacology research and practice. Clin. Transl. Sci. 2022, 1–6. [Google Scholar] [CrossRef]
- Research and Markets Global Growth Insight—Role of AI in the Pharmaceutical Industry 2018–2022: Exploring Key Investment Trends, Companies-to Action, and Growth Opportunities for AI in the Pharmaceutical Industry; Research and Markets: Dublin, Ireland, 2019.
- Dong, J.; Zhao, M.; Liu, Y.; Su, Y.; Zeng, X. Deep learning in retrosynthesis planning: Datasets, models and tools. Briefings Bioinform. 2022, 23, bbab391. [Google Scholar] [CrossRef]
- Jämsä-Jounela, S.-L. Future trends in process automation. Annu. Rev. Control 2007, 31, 211–220. [Google Scholar] [CrossRef]
- Davenport, T.H.; Ronanki, R. Artificial intelligence for the real world. Harv. Bus. Rev. 2018, 96, 108–116. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Tools | Feature(s) | Website(s) | Reference(s) |
|---|---|---|---|
| AlphaFold | Protein 3D (tertiary) structure presage employing DNN | https://deepmind.com/blog/alphafold (accessed on 28 November 2022) https://www.sciencemag.org/news/2018/12/google-s-deepmind-aces-protein-folding (accessed on 28 November 2022) | [151] |
| Chemputer | An exhaustive regulated schema for documenting a chemical synthesis method (Furnishes comprehensive compound synthesis recipe) | https://zenodo.org/record/1481731 (accessed on 28 November 2022) | [149] |
| Conv_qsar_fast | Foretells molecular attributes aided by CNN algorithm | https://github.com/connorcoley/conv_qsar_fast (accessed on 28 November 2022) | [130] |
| Chemical VAE | Mechanized chemical crafting employing variational autoencoder (VAE) | https://github.com/aspuru-guzik-group/chemical_vae (accessed on 28 November 2022) | [133] |
| DeepChem | A Python-aided AI technique for various drug discovery workflow predictions utilizing a DL algorithm for molecule recognition | https://github.com/deepchem/deepchem (accessed on 28 November 2022) | [152] |
| DeepNeuralNet-QSAR | Foretells molecular activity engaging multilevel DNN | https://github.com/Merck/DeepNeuralNet-QSAR (accessed on 28 November 2022) | [153] |
| DeepTox | Toxicity predictions of chemical agents utilizing a DL algorithm | www.bioinf.jku.at/research/DeepTox (accessed on 28 November 2022) | [154] |
| DeltaVina | Presages small molecule interaction affinity with drug employing an amalgamation of random forest (RF) as well as AutoDock scoring function) | https://github.com/chengwang88/deltavina (accessed on 28 November 2022) | [111] |
| Hit Dexter | ML schemes for the presage of compounds that could be sensitive to biochemical assays by engaging ML techniques | http://hitdexter2.zbh.uni-hamburg.de (accessed on 28 November 2022) | [155] |
| InnerOuterRNN | Foretells the chemical, physical, and biological attributes utilizing inner- and outer RNNs | https://github.com/Chemoinformatics/InnerOuterRNN (accessed on 28 November 2022) | [156] |
| JunctionTree VAE | De novo molecule origination utilizing junction tree variational autoencoder (VAE) | https://github.com/wengong-jin/icml18-jtnn (accessed on 28 November 2022) | [157] |
| Neural Graph Fingerprints | Attribute augury of novel molecules employing CNN algorithms | https://github.com/HIPS/neural-fingerprint (accessed on 28 November 2022) | [158] |
| NNScore | Foretells the affinity of protein–ligand binding utilizing neural network-aided scoring function | http://rocce-vm0.ucsd.edu/data/sw/hosted/nnscore/ (accessed on 28 November 2022) http://www.nbcr.net/software/nnscore (accessed on 28 November 2022) | [159] |
| Open Drug Discovery Toolkit (ODDT) | An exhaustive toolkit utilized for chemoinformatics and molecular modelling employing random forest score (RF)-Score as well as NNScore | https://github.com/oddt/oddt (accessed on 28 November 2022) | [160] |
| ORGANIC | A competent molecular generation tool to originate molecules with favourable attributes employing ML schemes | https://github.com/aspuru-guzik-group/ORGANIC (accessed on 28 November 2022) | [161] |
| PotentialNet | Foretells ligand-binding affinity engaging graph CNN | https://pubs.acs.org/doi/full/10.1021/acscentsci.8b00507 (accessed on 28 November 2022) | [162] |
| PPB2 | Poly-pharmacology prediction employing nearest neighbour as well as ML schemes | http://ppb2.gdb.tools/ (accessed on 28 November 2022) | [163] |
| QML | A Python toolkit for quantum ML (utilizing qubits leading to incremented computational speed, data storage capacity, and learning optimization) | https://www.qmlcode.org (accessed on 28 November 2022) https://github.com/qmlcode/qm (accessed on 28 November 2022) | [164] |
| REINVENT | De novo design of molecule employing RNN (recurrent neural network) as well as RL (reinforcement learning) | https://github.com/MarcusOlivecrona/REINVENT (accessed on 28 November 2022) | [165] |
| SCScore | A scoring scheme to figure out the synthesis complexity of a compound | https://github.com/connorcoley/scscore (accessed on 28 November 2022) | [166] |
| SIEVE-Score | An upgraded technique of structure-aided virtual screening through interaction-energy-based learning | https://github.com/sekijima-lab/SIEVE-Score (accessed on 28 November 2022) | [167] |
| Company/ Firm | Utilization of AI | Partnership with the Pharmaceutical Establishment | Platform Advanced/Lead Agents for Clinical Trials |
|---|---|---|---|
| Numerate San Francisco, CA 94107, USA | A scheme for AI-facilitated drug design addressing oncology and gastroenterology specialities | Takeda | Agent S48168 in Phase 1 of clinical testing for Ryanodine receptor 2 |
| Numerate San Francisco, CA 94107, USA | A scheme for AI-facilitated drug design addressing oncology and gastroenterology specialities | Servier | Drug advancement related to oncology, central nervous system, and gastroenterologic maladies |
| Atomwise San Francisco, CA 94103, USA | A scheme for AI-enabled structural modelling | Lilly | Agent BBT-401 in Phase 2 of clinical testing |
| Atomwise San Francisco, CA 94103, USA | A scheme for AI-enabled structural modelling | Bridge Biotherapeutics | Augmentation of Pellino Inhibitor Pipeline; Agent BBT-401 evaluated in Phase-2a of clinical testing |
| Benevolent AI London, UK | AI-facilitated Judgement Augmented Cognition System (JACS) for originating and advancing novel clinical lead agents effective in neurodegenerative ailments | Janssen | Fresh set of drug compounds to be advanced via such collaboration |
| Benevolent AI London, UK | AI aided schemes to advance novel clinical lead agents effective in chronic kidney ailments | AstraZeneca | Drug candidate evaluated in Phase 2b clinical testing as a lead agent effective in chronic kidney ailments |
| Exscientia Oxford, UK | A scheme for AI-enabled drug discovery and lead refinement | Sanofi | Drug Discovery Research in obsessive-compulsive disorder, Agent DSP-1181 in Phase I clinical testing. Advance Centaur Chemist™ scheme for AI-enabled drug discovery |
| IBM Watson Health Cambridge, MA 02142, USA | Furnishes a scheme for clinical and health-associated data evaluation | Pfizer | Accelerating drug discovery efforts in immuno-oncology |
| IBM Watson Health Cambridge, MA 02142, USA | Furnishes a scheme for clinical and health-associated data evaluation | Novartis | Real-time surveillance of patients to augment breast cancer patient intervention results |
| Microsoft Redmond, WA 98052, USA | A scheme for image processing as well as cell and gene-aided therapeutic interventions | Novartis | Engendering an AI Innovation lab to augment the drug discovery mechanism as well as its commercialization |
| Owkin Broadway, New York, NY, USA | Furnish a scheme for clinical testing aided by ML technique | Roche | Originated and advanced Owkin’s Studio platform utilizing AI technology |
| Sensyne health Headington, Oxfordshire, UK | A tool serving clinical AI schemes | Bayer | Originated and advanced Sensyne Health’s proprietary clinical AI technology package |
| XtalPi Shenzhen, Guangdong, China | A package enabling Target identification and validation incorporating QM as well as ML schemes | Pfizer | Presage and refinement of crystalline entities of drug candidates utilizable in early stages of drug screening |
| BioXcel therapeutics New Haven, CT, USA | A scheme facilitating drug discovery services incorporating AI mechanisms | Pfizer | Lead agent BXCL501-in assessment in Phase 3 clinical testing; Drug agent BXCL701-in assessment in Phase 2 clinical assessment |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarkar, C.; Das, B.; Rawat, V.S.; Wahlang, J.B.; Nongpiur, A.; Tiewsoh, I.; Lyngdoh, N.M.; Das, D.; Bidarolli, M.; Sony, H.T. Artificial Intelligence and Machine Learning Technology Driven Modern Drug Discovery and Development. Int. J. Mol. Sci. 2023, 24, 2026. https://doi.org/10.3390/ijms24032026
Sarkar C, Das B, Rawat VS, Wahlang JB, Nongpiur A, Tiewsoh I, Lyngdoh NM, Das D, Bidarolli M, Sony HT. Artificial Intelligence and Machine Learning Technology Driven Modern Drug Discovery and Development. International Journal of Molecular Sciences. 2023; 24(3):2026. https://doi.org/10.3390/ijms24032026
Chicago/Turabian StyleSarkar, Chayna, Biswadeep Das, Vikram Singh Rawat, Julie Birdie Wahlang, Arvind Nongpiur, Iadarilang Tiewsoh, Nari M. Lyngdoh, Debasmita Das, Manjunath Bidarolli, and Hannah Theresa Sony. 2023. "Artificial Intelligence and Machine Learning Technology Driven Modern Drug Discovery and Development" International Journal of Molecular Sciences 24, no. 3: 2026. https://doi.org/10.3390/ijms24032026