
Hydroxylated Coumarin-Based Thiosemicarbazones as Dual Antityrosinase and Antioxidant Agents
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
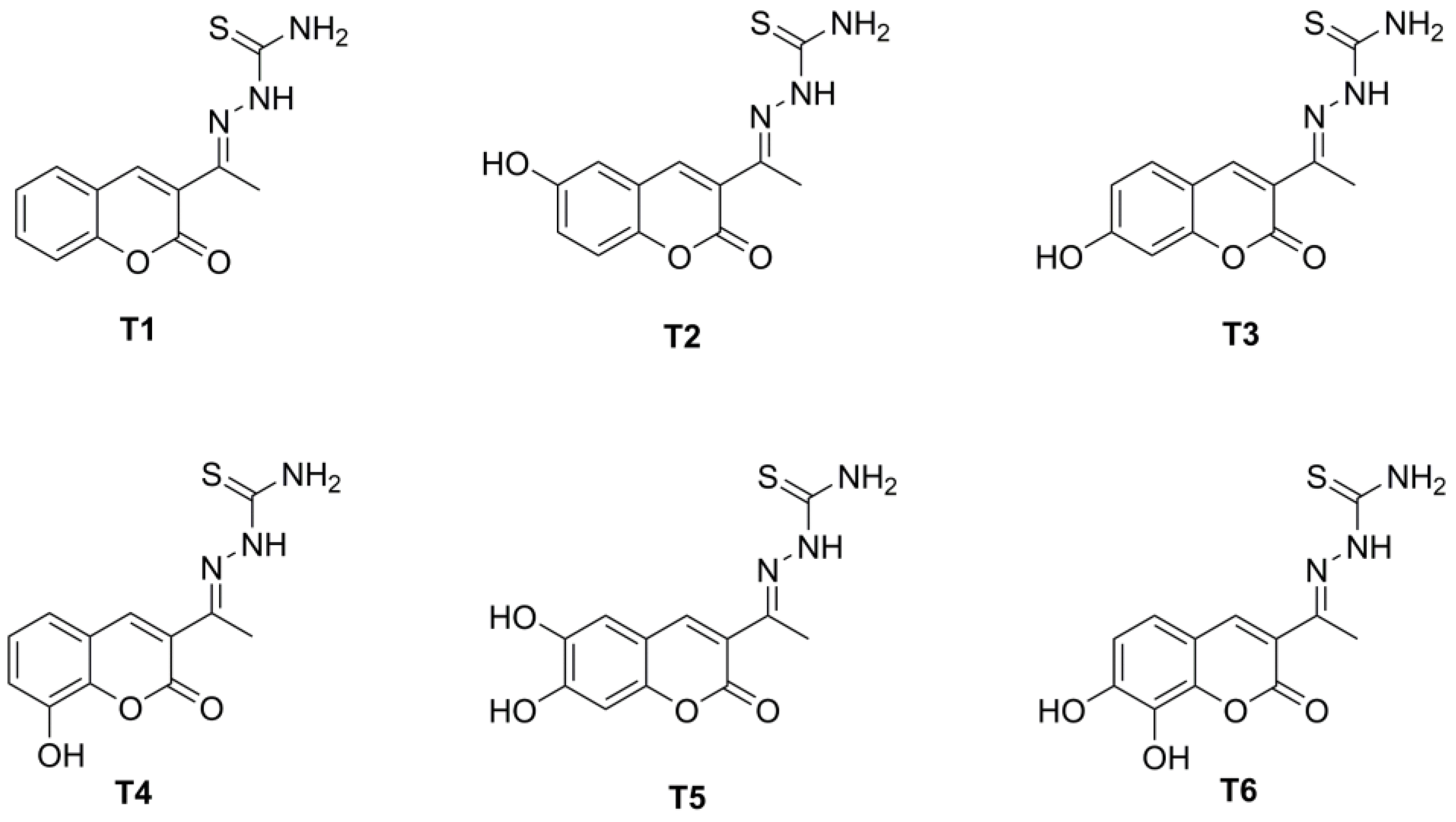
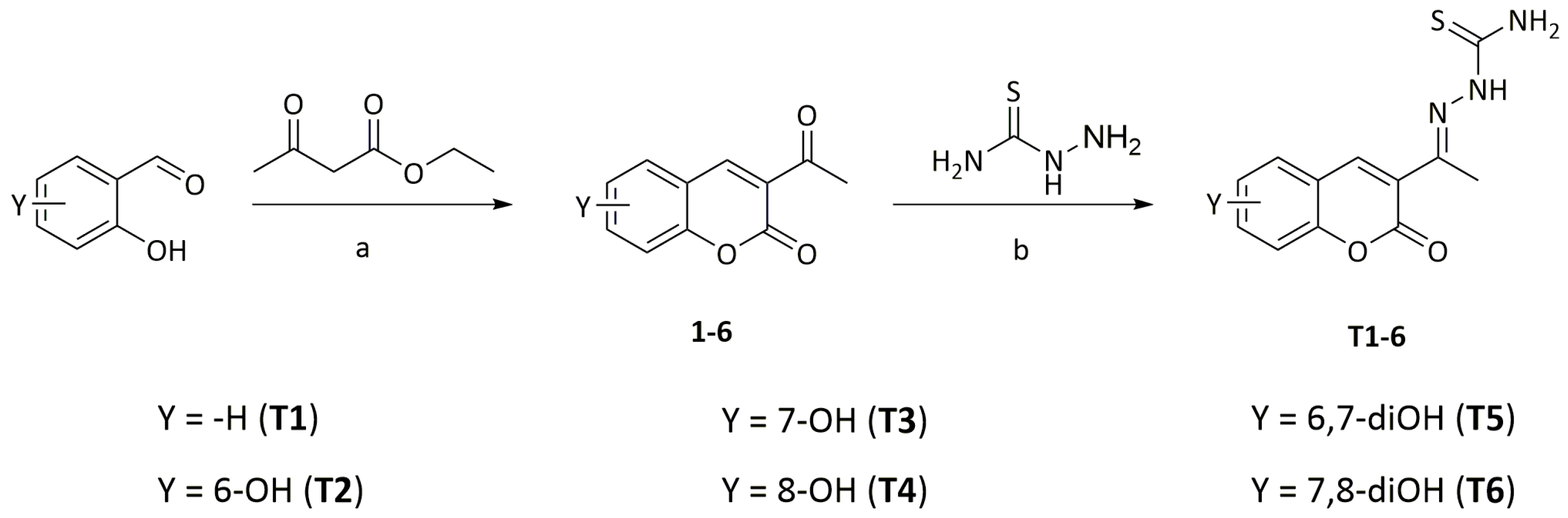
2.1. Synthesis and Chemical Characterization
2.2. Antioxidant Assay
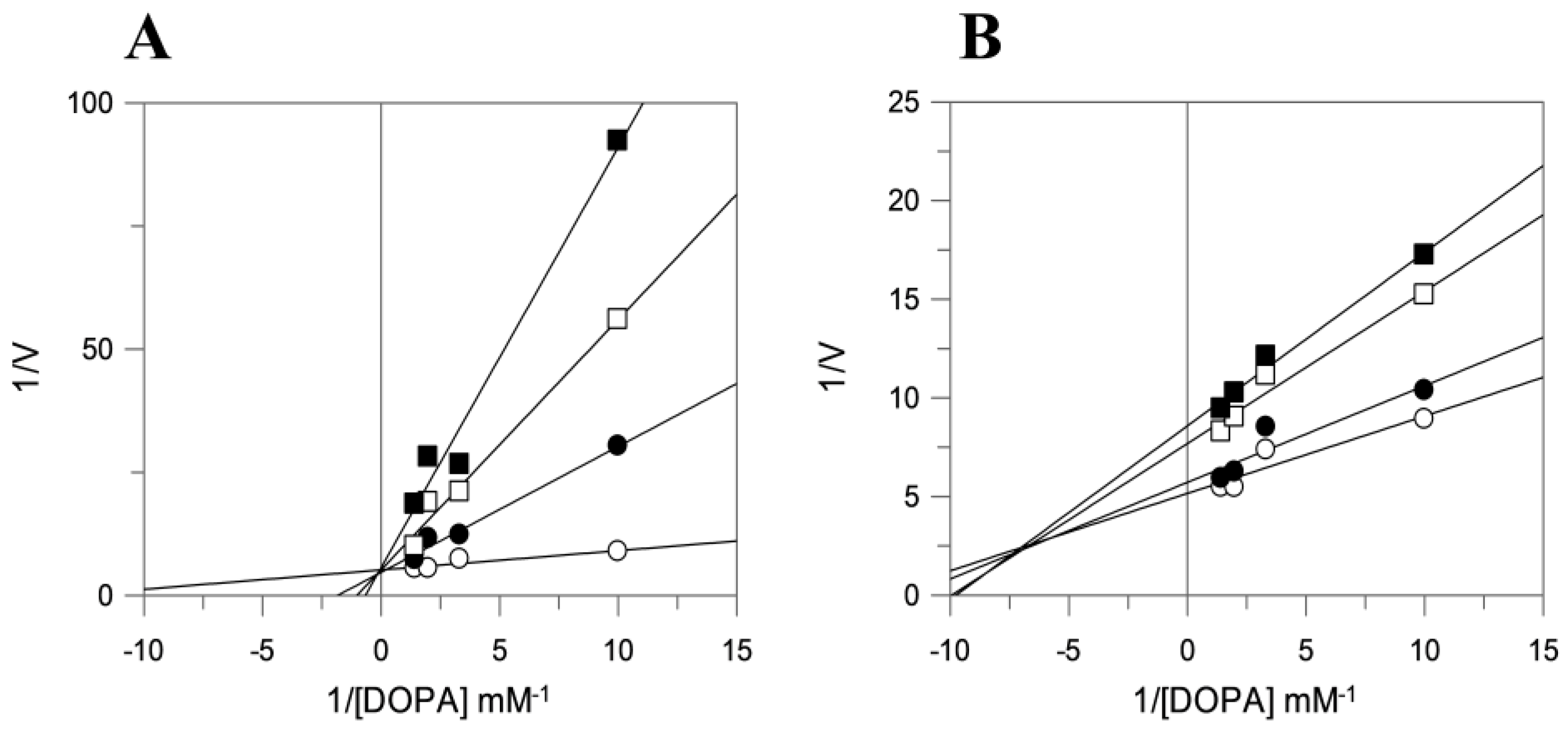
2.3. Tyrosinase Inhibition Assay
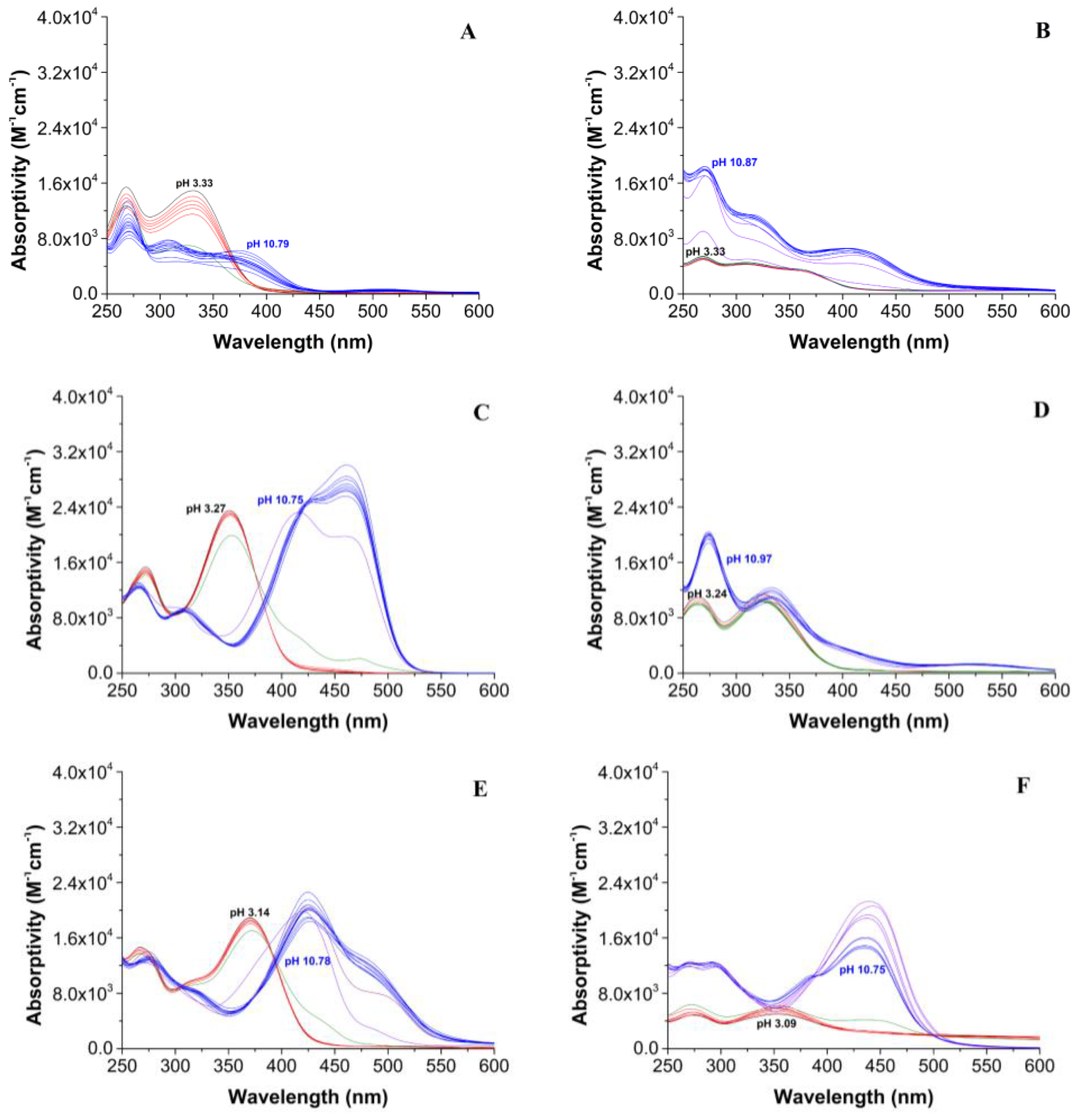
2.4. Drug-Likeness and Biospeciation Studies
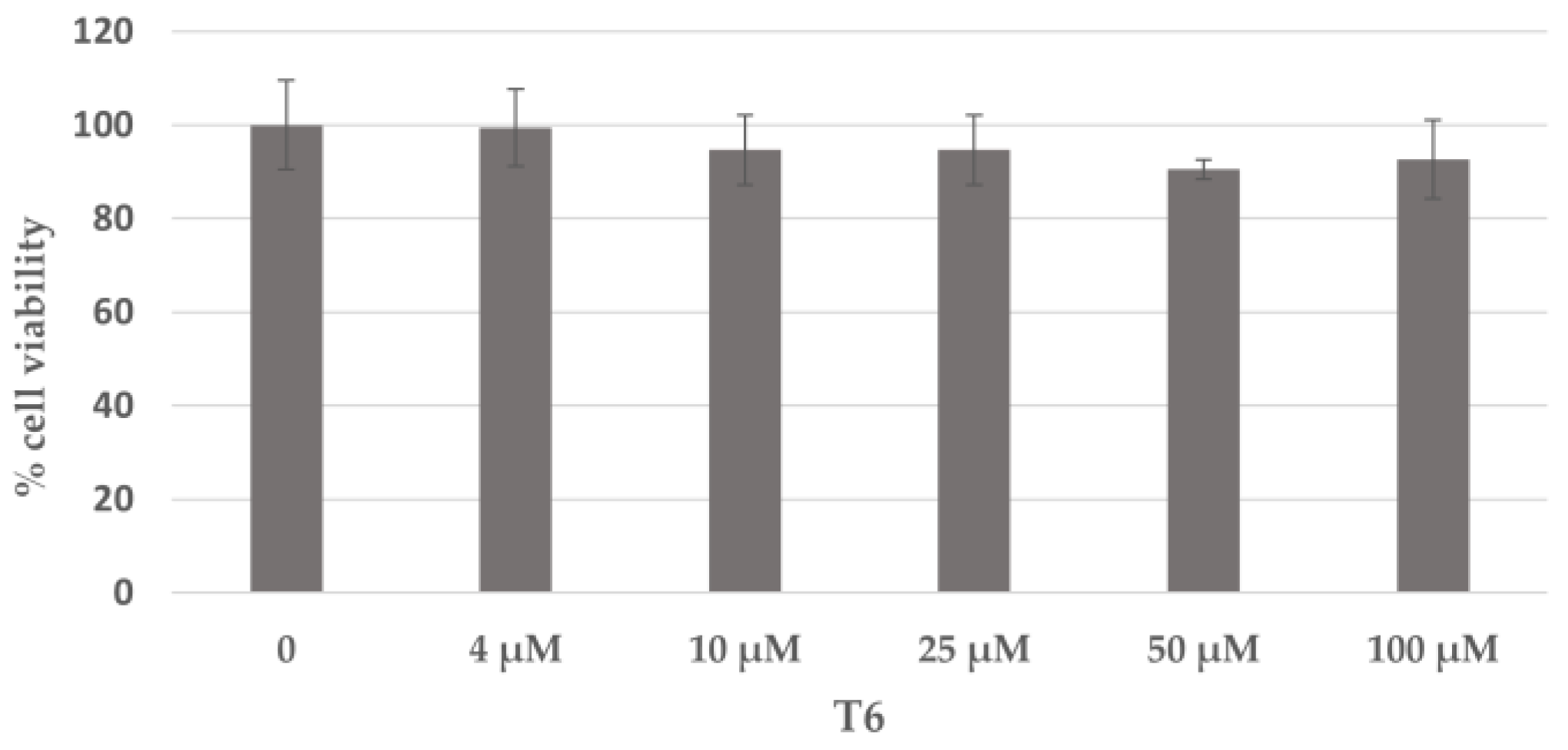
2.5. Cell Viability
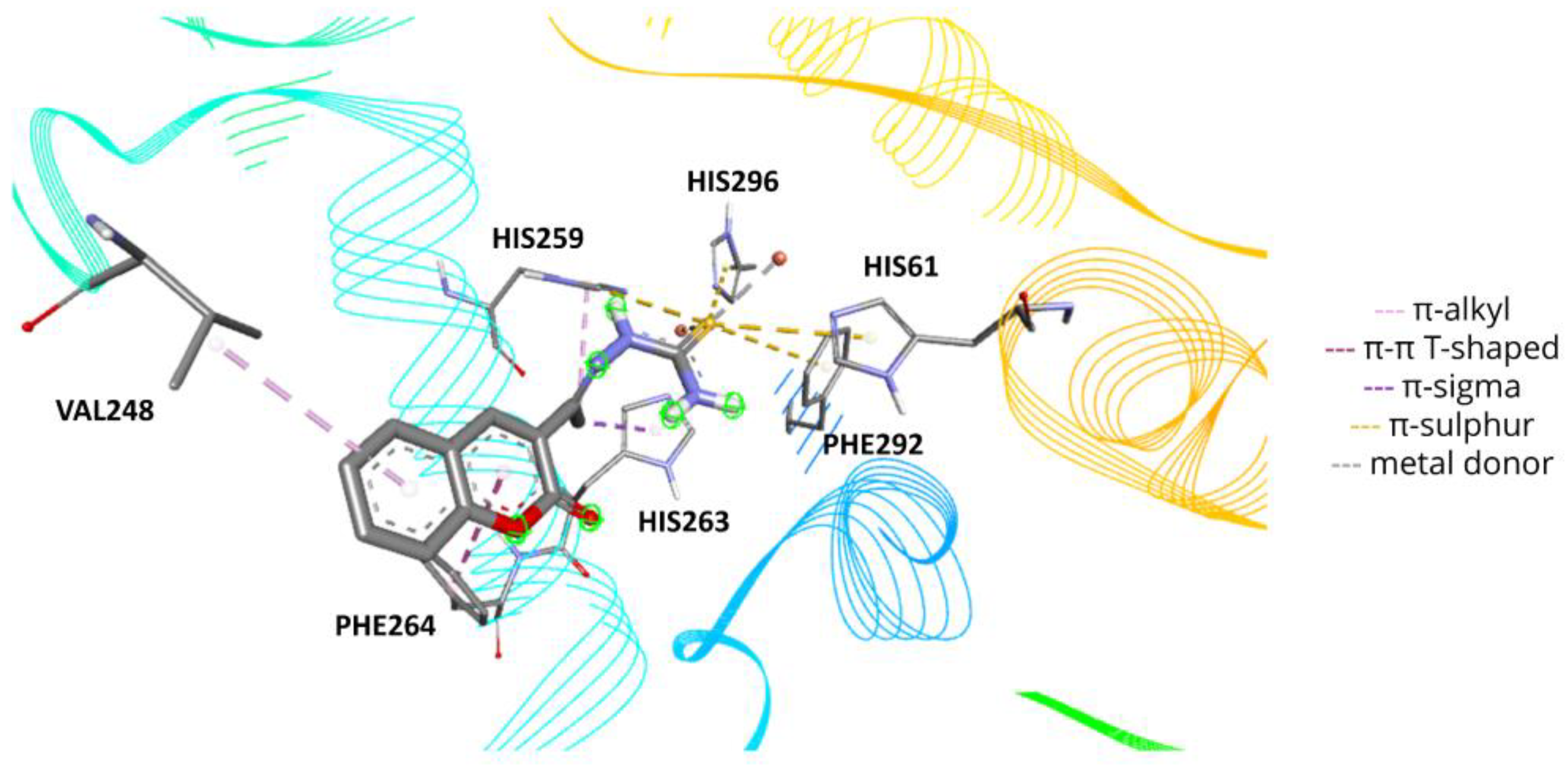
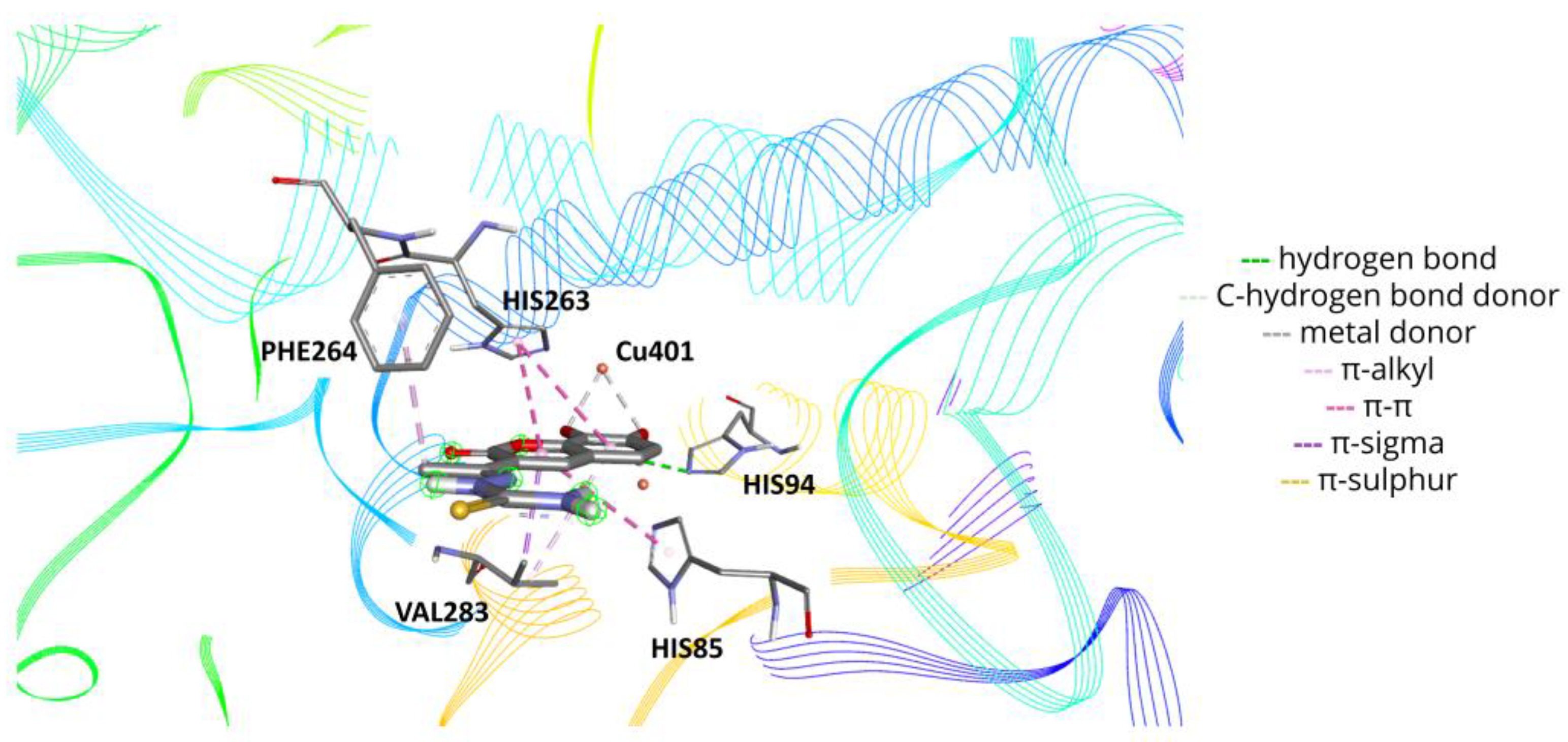
2.6. Molecular Docking
3. Materials and Methods
3.1. Synthesis and Characterization Techniques
3.2. General Procedure for the Synthesis of Substituted 3-Acetyl-2H-chromen-2-one Derivatives
3.3. General Procedure for the Synthesis of Substituted (1E)-2-(1-(2-oxo-2H-chromen-3-yl)ethylidene)hydrazine-1-carbothioamide Compounds [13]
3.4. Potentiometric and Spectrophotometric Titrations
3.5. DFT Calculations
3.6. Drug-Likeness Descriptors
3.7. Antioxidant Assays
3.8. Tyrosinase Inhibition Assay
3.9. Cell Viability Assay
3.10. Molecular Docking
3.11. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, J.; Feng, L.; Liu, L.; Wang, F.; Ouyang, L.; Zhang, L.; Hu, X.; Wang, G. Recent Advances in the Design and Discovery of Synthetic Tyrosinase Inhibitors. Eur. J. Med. Chem. 2021, 224, 113744. [Google Scholar] [CrossRef] [PubMed]
- Solano, F.; Briganti, S.; Picardo, M.; Ghanem, G. Hypopigmenting Agents: An Updated Review on Biological, Chemical and Clinical Aspects. Pigment Cell Res. 2006, 19, 550–571. [Google Scholar] [CrossRef] [PubMed]
- Loizzo, M.R.; Tundis, R.; Menichini, F. Natural and Synthetic Tyrosinase Inhibitors as Antibrowning Agents: An Update. Compr. Rev. Food Sci. Food Saf. 2012, 11, 378–398. [Google Scholar] [CrossRef]
- Masamoto, Y.; Murata, Y.; Baba, K.; Shimoishi, Y.; Tada, M.; Takahata, K. Inhibitory Effects of Esculetin on Melanin Biosynthesis. Biol. Pharm. Bull. 2004, 27, 422–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pivetta, T.; Masuri, S.; Cabiddu, M.G.; Caltagirone, C.; Pintus, A.; Massa, M.; Isaia, F.; Cadoni, E. A Novel Ratiometric and Turn-on Fluorescent Coumarin-Based Probe for Fe(Iii). New J. Chem. 2019, 43, 12032–12041. [Google Scholar] [CrossRef]
- Zolghadri, S.; Bahrami, A.; Hassan Khan, M.T.; Munoz-Munoz, J.; Garcia-Molina, F.; Garcia-Canovas, F.; Saboury, A.A. A Comprehensive Review on Tyrosinase Inhibitors. J. Enzyme Inhib. Med. Chem. 2019, 34, 279–309. [Google Scholar] [CrossRef] [Green Version]
- Matos, M.J.; Santana, L.; Uriarte, E.; Delogu, G.; Corda, M.; Fadda, M.B.; Era, B.; Fais, A. New Halogenated Phenylcoumarins as Tyrosinase Inhibitors. Bioorganic Med. Chem. Lett. 2011, 21, 3342–3345. [Google Scholar] [CrossRef]
- Matos, M.J.; Varela, C.; Vilar, S.; Hripcsak, G.; Borges, F.; Santana, L.; Uriarte, E.; Fais, A.; Di Petrillo, A.; Pintus, F.; et al. Design and Discovery of Tyrosinase Inhibitors Based on a Coumarin Scaffold. RSC Adv. 2015, 5, 94227–94235. [Google Scholar] [CrossRef]
- Pintus, F.; Matos, M.J.; Vilar, S.; Hripcsak, G.; Varela, C.; Uriarte, E.; Santana, L.; Borges, F.; Medda, R.; Di Petrillo, A.; et al. New Insights into Highly Potent Tyrosinase Inhibitors Based on 3-Heteroarylcoumarins: Anti-Melanogenesis and Antioxidant Activities, and Computational Molecular Modeling Studies. Bioorganic Med. Chem. 2017, 25, 1687–1695. [Google Scholar] [CrossRef]
- Denat, L.; Kadekaro, A.L.; Marrot, L.; Leachman, S.A.; Abdel-Malek, Z.A. Melanocytes as Instigators and Victims of Oxidative Stress. J. Investig. Dermatol. 2014, 134, 1512–1518. [Google Scholar] [CrossRef]
- Lu, T.M.; Ko, H.H.; Ng, L.T.; Hsieh, Y.P. Free-Radical-Scavenging, Antityrosinase, and Cellular Melanogenesis Inhibitory Activities of Synthetic Isoflavones. Chem. Biodivers. 2015, 12, 963–979. [Google Scholar] [CrossRef] [PubMed]
- Hałdys, K.; Latajka, R. Thiosemicarbazones with Tyrosinase Inhibitory Activity. Medchemcomm 2019, 10, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, F.; Chen, L.; Zhao, L.; Zhao, Z.; Wang, M.; Lei, S. Biological Evaluation of Coumarin Derivatives as Mushroom Tyrosinase Inhibitors. Food Chem. 2012, 135, 2872–2878. [Google Scholar] [CrossRef] [PubMed]
- Floris, S.; Fais, A.; Rosa, A.; Piras, A.; Marzouki, H.; Medda, R.; González-Paramás, A.M.; Kumar, A.; Santos-Buelga, C.; Era, B. Phytochemical Composition and the Cholinesterase and Xanthine Oxidase Inhibitory Properties of Seed Extracts from the Washingtonia Filifera Palm Fruit. RSC Adv. 2019, 9, 21278–21287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuri, S.; Cadoni, E.; Cabiddu, M.G.; Isaia, F.; Demuru, M.G.; Morán, L.; Morán, L.; Bucek, D.; Vanhara, P.; Vanhara, P.; et al. The First Copper(Ii) Complex with 1, 10-Phenanthroline and Salubrinal with Interesting Biochemical Properties. Metallomics 2020, 12, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Masuri, S.; Cabiddu, M.G.; Cadoni, E.; Pivetta, T. Hydroxylated 3-(Pyridin-2-Yl) Coumarins as Radical Scavengers with Potent Lipoxygenase Inhibitor Activity. New J. Chem. 2021, 45, 10749–10760. [Google Scholar] [CrossRef]
- Nurchi, V.M.; Crisponi, G.; Lachowicz, J.I.; Murgia, S.; Pivetta, T.; Remelli, M.; Rescigno, A.; Niclós-Gutíerrez, J.; González-Pérez, J.M.; Domínguez-Martín, A.; et al. Iron(III) and Aluminum(III) Complexes with Hydroxypyrone Ligands Aimed to Design Kojic Acid Derivatives with New Perspectives. J. Inorg. Biochem. 2010, 104, 560–569. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal Chemical Properties of Successful Central Nervous System Drugs. NeuroRX 2005, 2, 541–553. [Google Scholar] [CrossRef]
- Zhou, R.; Moench, P.; Heran, C.; Lu, X.; Mathias, N.; Faria, T.N.; Wall, D.A.; Hussain, M.A.; Smith, R.L.; Sun, D. PH-Dependent Dissolution in Vitro and Absorption in Vivo of Weakly Basic Drugs: Development of a Canine Model. Pharm. Res. 2005, 22, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Dressman, J.B.; Amidon, G.L.; Reppas, C.; Shah, V.P. Dissolution Testing as a Prognostic Tool for Oral Drug. Pharm. Res. 1998, 15, 11–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranade, D.S.; Bapat, A.M.; Ramteke, S.N.; Joshi, B.N.; Roussel, P.; Tomas, A.; Deschamps, P.; Kulkarni, P.P. Thiosemicarbazone Modification of 3-Acetyl Coumarin Inhibits Aβ Peptide Aggregation and Protect against Aβ-Induced Cytotoxicity. Eur. J. Med. Chem. 2016, 121, 803–809. [Google Scholar] [CrossRef]
- Hassani, S.; Haghbeen, K.; Fazli, M. Non-Specific Binding Sites Help to Explain Mixed Inhibition in Mushroom Tyrosinase Activities. Eur. J. Med. Chem. 2016, 122, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Martínez, F.J.; Razo-Hernández, R.S.; Peraza-Campos, A.L.; Villanueva-García, M.; Sumaya-Martínez, M.T.; Cano, D.J.; Gómez-Sandoval, Z. Synthesis and in Vitro Antioxidant Activity Evaluation of 3-Carboxycoumarin Derivatives and Qsar Study of Their Dpph Radical Scavenging Activity. Molecules 2012, 17, 14882–14898. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, C.; Navarrete-Encina, P.; Squella, J.A. Electrochemistry and Reactivity Against Superoxide Anion Radicals of Hydroxycoumarins and Its Derivatives. J. Electrochem. Soc. 2020, 167, 165502. [Google Scholar] [CrossRef]
- Starčević, Š.; Brožič, P.; Turk, S.; Cesar, J.; Lanišnik Rižner, T.; Gobec, S. Synthesis and Biological Evaluation of (6-and 7-Phenyl) Coumarin Derivatives as Selective Nonsteroidal Inhibitors of 17β-Hydroxysteroid Dehydrogenase Type 1. J. Med. Chem. 2011, 54, 248–261. [Google Scholar] [CrossRef]
- Liu, X.H.; Fan, J.C.; Liu, Y.; Shang, Z.C. L-Proline as an Efficient and Reusable Promoter for the Synthesis of Coumarins in Ionic Liquid. J. Zhejiang Univ. Sci. B 2008, 9, 990–995. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Xia, Y.L.; Zou, L.W.; Qian, X.K.; Dou, T.Y.; Jin, Q.; Li, S.Y.; Yu, Y.; Wang, D.D.; Luo, Q.; et al. An Optimized Two-Photon Fluorescent Probe for Biological Sensing and Imaging of Catechol-O-Methyltransferase. Chem. A Eur. J. 2017, 23, 10800–10807. [Google Scholar] [CrossRef]
- Kalaiarasi, G.; Mohamed Subarkhan, M.; Fathima Safwana, C.K.; Sruthi, S.; Sathiya Kamatchi, T.; Keerthana, B.; Ashok Kumar, S.L. New Organoruthenium(II) Complexes Containing N, X-Donor (X = O, S) Heterocyclic Chelators: Synthesis, Spectral Characterization, in Vitro Cytotoxicity and Apoptosis Investigation. Inorganica Chim. Acta 2022, 535, 120863. [Google Scholar] [CrossRef]
- Gans, P.; O’Sullivan, B. GLEE, a New Computer Program for Glass Electrode Calibration. Talanta 2000, 51, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of Equilibria in Solution. Determination of Equilibrium Constants with the HYPERQUAD Suite of Programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef] [PubMed]
- Alderighi, L.; Gans, P.; Ienco, A.; Peters, D.; Sabatini, A.; Vacca, A. Hyperquad Simulation and Speciation (HySS): A Utility Program for the Investigation of Equilibria Involving Soluble and Partially Soluble Species. Coord. Chem. Rev. 1999, 184, 311–318. [Google Scholar] [CrossRef]
- Neese, F. The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Bartmess, J.E. Thermodynamics of the Electron and the Proton. J. Phys. Chem. 1994, 98, 6420–6424. [Google Scholar] [CrossRef]
- Marković, Z.; Tošović, J.; Milenković, D.; Marković, S. Revisiting the Solvation Enthalpies and Free Energies of the Proton and Electron in Various Solvents. Comput. Theor. Chem. 2016, 1077, 11–17. [Google Scholar] [CrossRef]
- Era, B.; Floris, S.; Sogos, V.; Porcedda, C.; Piras, A.; Medda, R.; Fais, A.; Pintus, F. Anti-Aging Potential of Extracts from Washingtonia Filifera Seeds. Plants 2021, 10, 151. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Bio. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismaya, W.T.; Rozeboom, H.J.; Weijn, A.; Mes, J.J.; Fusetti, F.; Wichers, H.J.; Dijkstra, B.W. Crystal Structure of Agaricus Bisporus Mushroom Tyrosinase: Identity of the Tetramer Subunits and Interaction with Tropolone. Biochemistry 2011, 50, 5477–5486. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DPPH | ABTS | |
|---|---|---|
| Compound | EC50 (µM) | EC50 (µM) |
| T1 | >250 a | 72 ± 6 a |
| T2 | >250 a | 25.7 ± 0.4 b |
| T3 | >250 a | 36 ± 1 c |
| T4 | 28 ± 2 b | 21.8 ± 0.8 b |
| T5 | 7.1 ± 0.3 c | 9 ± 1 d |
| T6 | 17.9 ± 0.4 d | 8.8 ± 0.4 d |
| Ascorbic Acid | 18.6 ± 0.6 d | - |
| Trolox | - | 13 ± 1 d |
| Compound | IC50 (µM) |
|---|---|
| T1 | 4 ± 2 a |
| T2 | 14 ± 7 b |
| T3 | 5 ± 2 a,c |
| T4 | 5.7 ± 0.8 a |
| T5 | 6 ± 2 a |
| T6 | 4.1 ± 0.7 a |
| Kojic acid | 18 ± 1 b |
| T1 | T2 | T3 | T4 | T5 | T6 | |
|---|---|---|---|---|---|---|
| miLogP a | 1.80 | 1.30 | 1.30 | 1.53 | 0.81 | 1.04 |
| TPSA (Å2) b | 80.62 | 100.85 | 100.85 | 100.85 | 121.08 | 121.08 |
| n-atoms c | 18 | 19 | 19 | 19 | 20 | 20 |
| MW (Da) | 261.31 | 277.31 | 277.31 | 277.31 | 293.30 | 293.30 |
| n-ON d | 5 | 6 | 6 | 6 | 7 | 7 |
| n-OHNH e | 3 | 4 | 4 | 4 | 5 | 5 |
| n-violations f | 0 | 0 | 0 | 0 | 0 | 0 |
| n-rotb g | 3 | 3 | 3 | 3 | 3 | 3 |
| Volume (Å3) h | 219.96 | 227.98 | 227.98 | 227.98 | 236.00 | 236.00 |
| Compound | Equilibrium | Logβ | pK |
|---|---|---|---|
| T1 | 8.46 (9) | 8.5 | |
| 13.47 (9) | 5.0 | ||
| 17.45 (8) | 4.0 | ||
| T2 | 10.20 (8) | 10.2 | |
| 17.24 (6) | 6.8 | ||
| 22.4 (1) | 5.4 | ||
| 26.7 (1) | 4.3 | ||
| T3 | 10.5 (1) | 10.5 | |
| 17.6 (1) | 7.1 | ||
| 23.26 (7) | 5.7 | ||
| 26.7 (1) | 3.4 | ||
| T4 | 10.4 (1) | 10.4 | |
| 17.26 (1) | 6.9 | ||
| 22.94 (5) | 5.7 | ||
| 26.5 (2) | 3.6 | ||
| T5 | 10.7 (1) | 10.7 | |
| 18.46 (9) | 7.8 | ||
| 24.76 (7) | 6.3 | ||
| 29.50 (6) | 4.7 | ||
| 33.2 (1) | 3.7 | ||
| T6 | 10.5 (2) | 10.5 | |
| 19.3 (1) | 8.8 | ||
| 25.1 (1) | 5.8 | ||
| 29.5 (1) | 4.4 | ||
| 33.0 (1) | 3.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masuri, S.; Era, B.; Pintus, F.; Cadoni, E.; Cabiddu, M.G.; Fais, A.; Pivetta, T. Hydroxylated Coumarin-Based Thiosemicarbazones as Dual Antityrosinase and Antioxidant Agents. Int. J. Mol. Sci. 2023, 24, 1678. https://doi.org/10.3390/ijms24021678
Masuri S, Era B, Pintus F, Cadoni E, Cabiddu MG, Fais A, Pivetta T. Hydroxylated Coumarin-Based Thiosemicarbazones as Dual Antityrosinase and Antioxidant Agents. International Journal of Molecular Sciences. 2023; 24(2):1678. https://doi.org/10.3390/ijms24021678
Chicago/Turabian StyleMasuri, Sebastiano, Benedetta Era, Francesca Pintus, Enzo Cadoni, Maria Grazia Cabiddu, Antonella Fais, and Tiziana Pivetta. 2023. "Hydroxylated Coumarin-Based Thiosemicarbazones as Dual Antityrosinase and Antioxidant Agents" International Journal of Molecular Sciences 24, no. 2: 1678. https://doi.org/10.3390/ijms24021678