
Learning in Transcriptional Network Models: Computational Discovery of Pathway-Level Memory and Effective Interventions

Abstract
:1. Introduction
2. Results
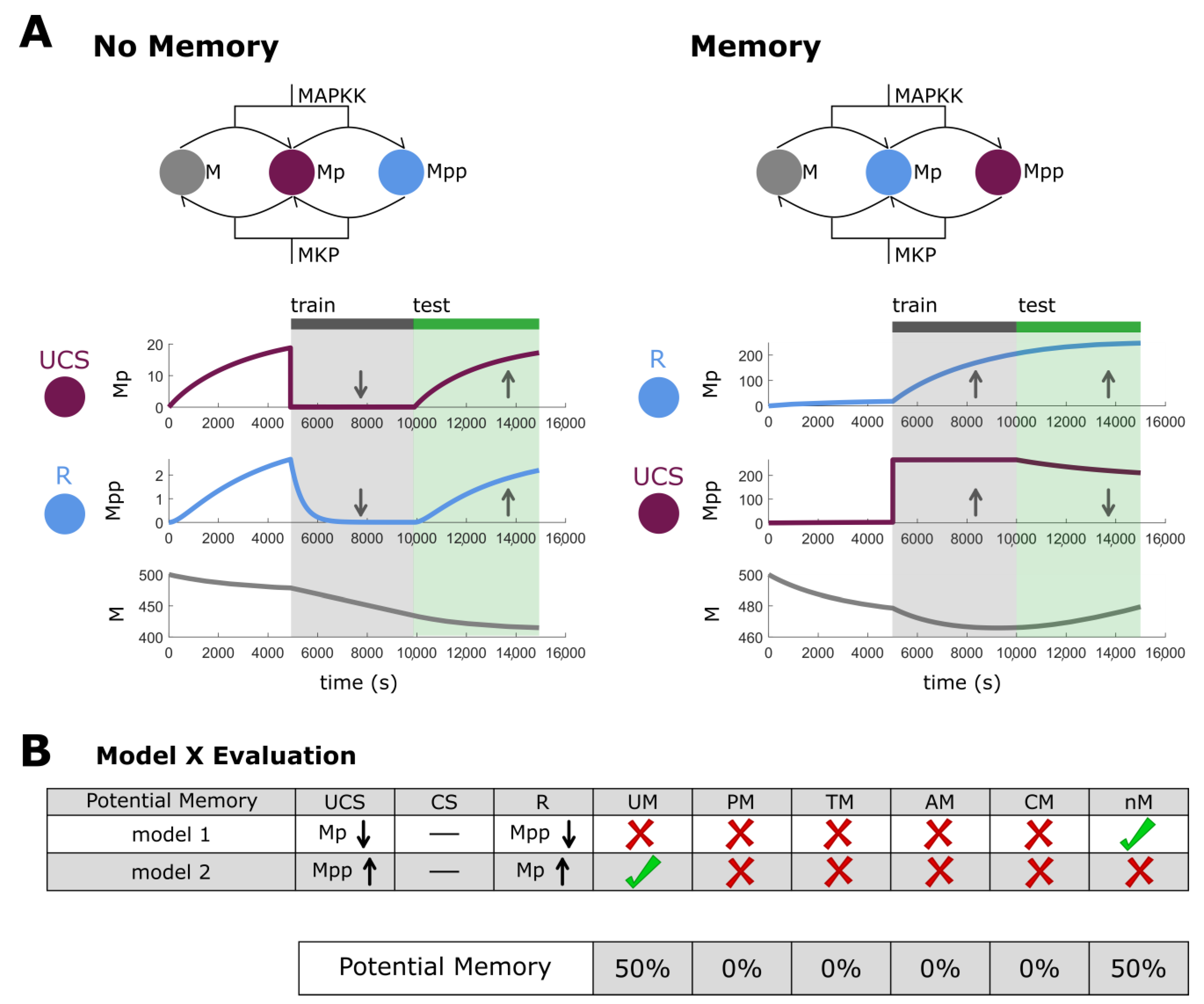
2.1. Biological Networks May Possess Multiple Types of Memory
2.2. Memory Effects Are Robust to Different Strengths of Stimulus
2.3. Memory Effects Are Robust to Noise
2.4. Memory Effects Are Stable over Long Periods
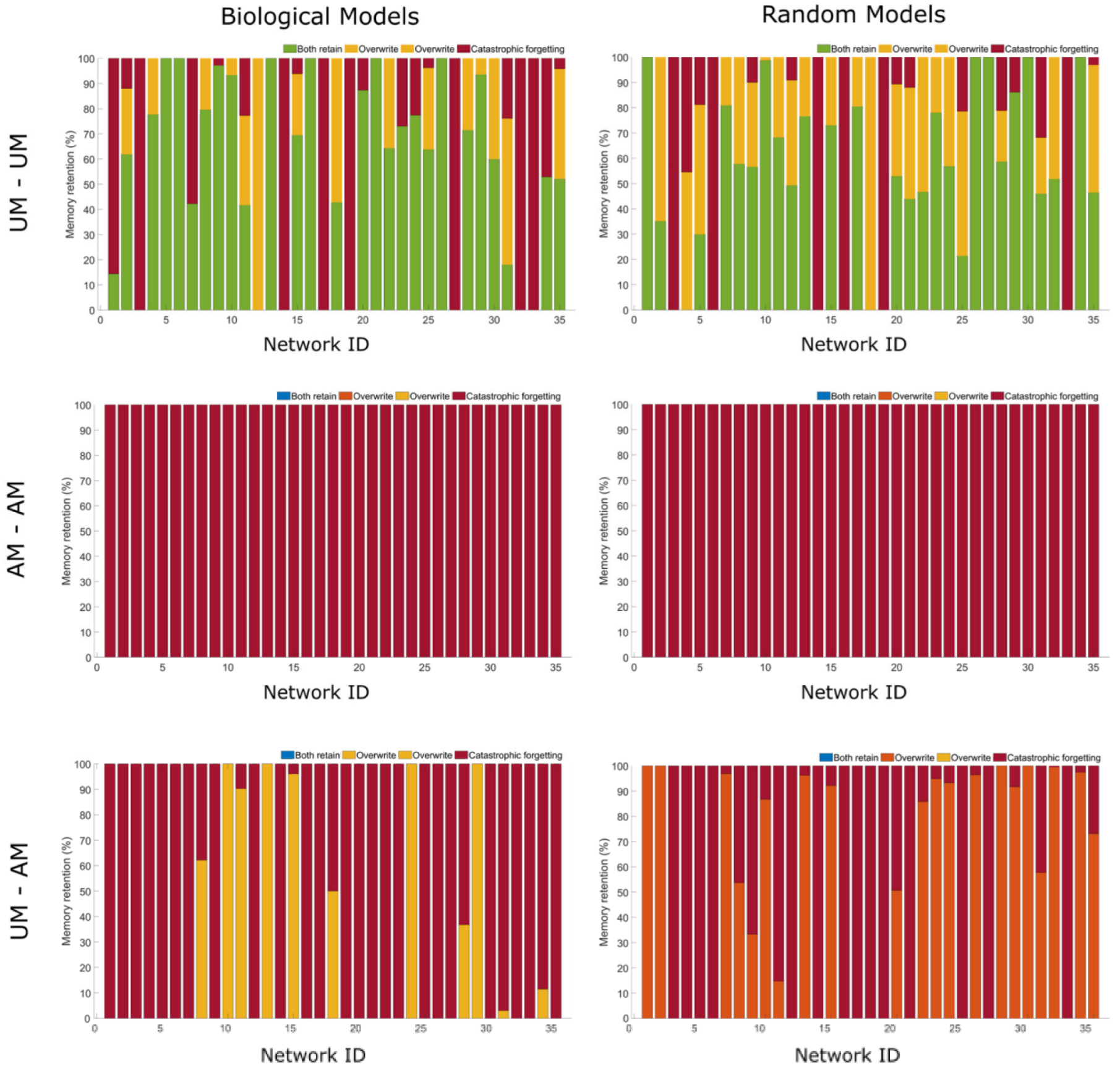
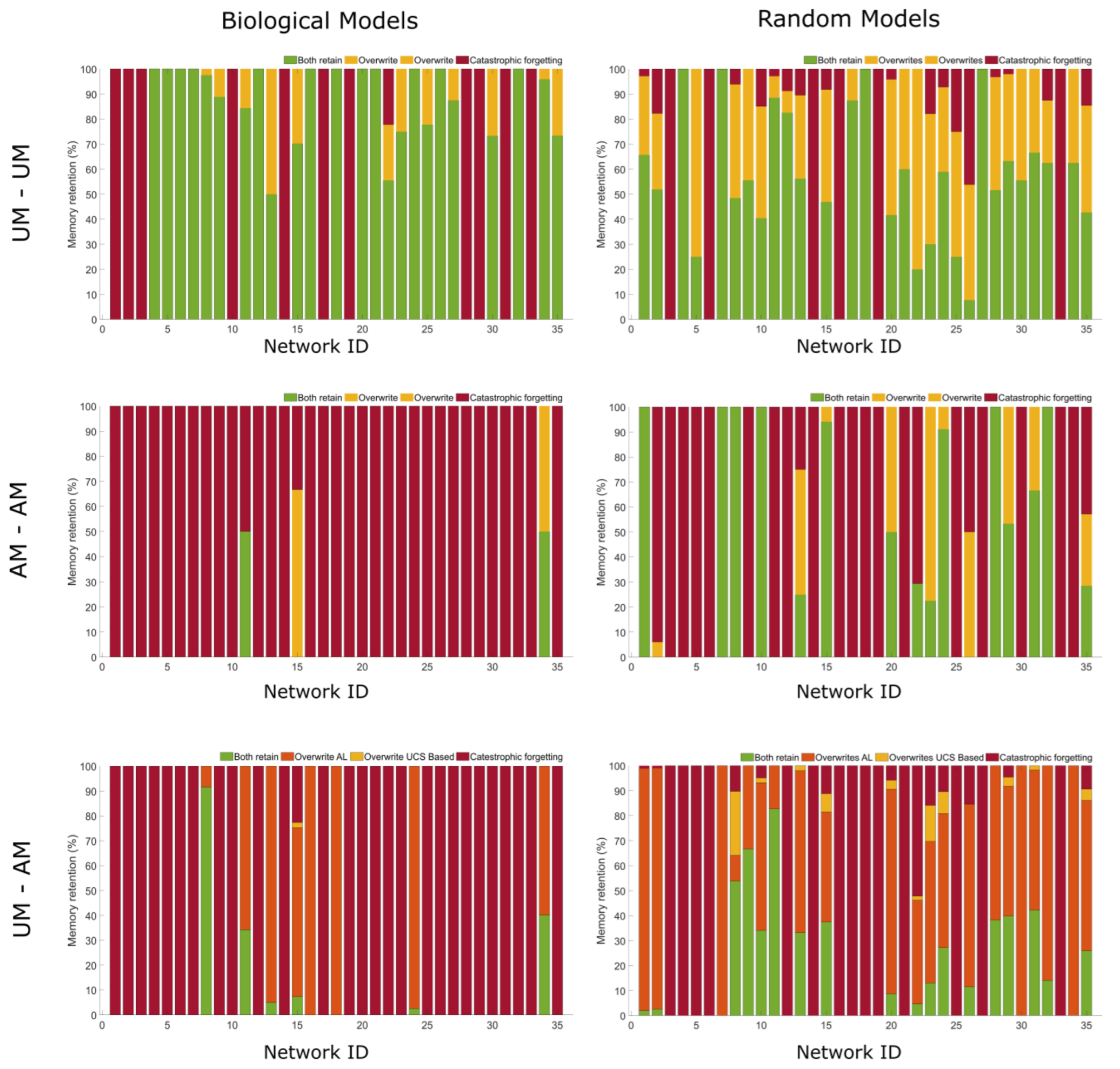
2.5. Can Networks Be Trained to Keep More Than One Memory?
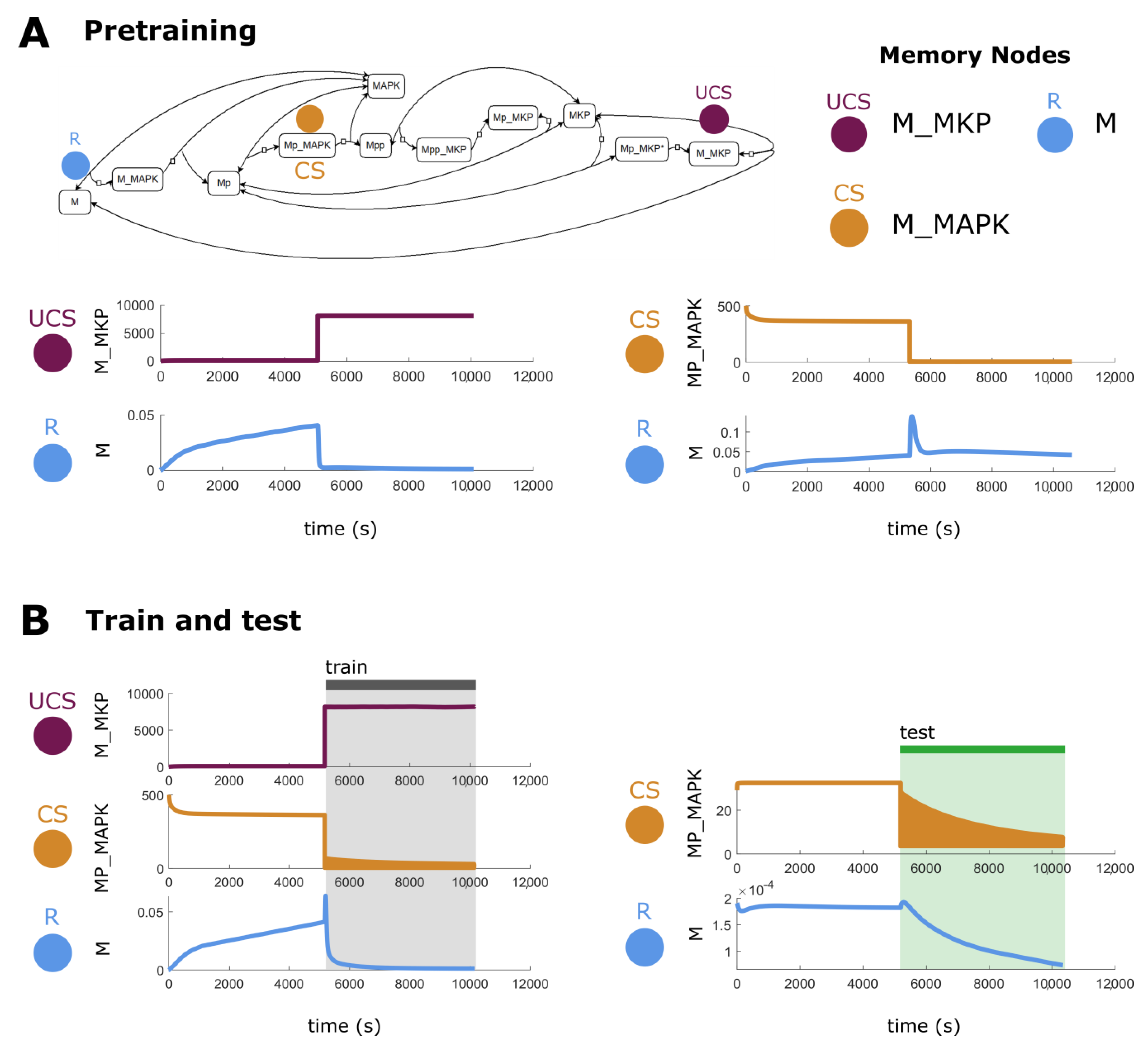
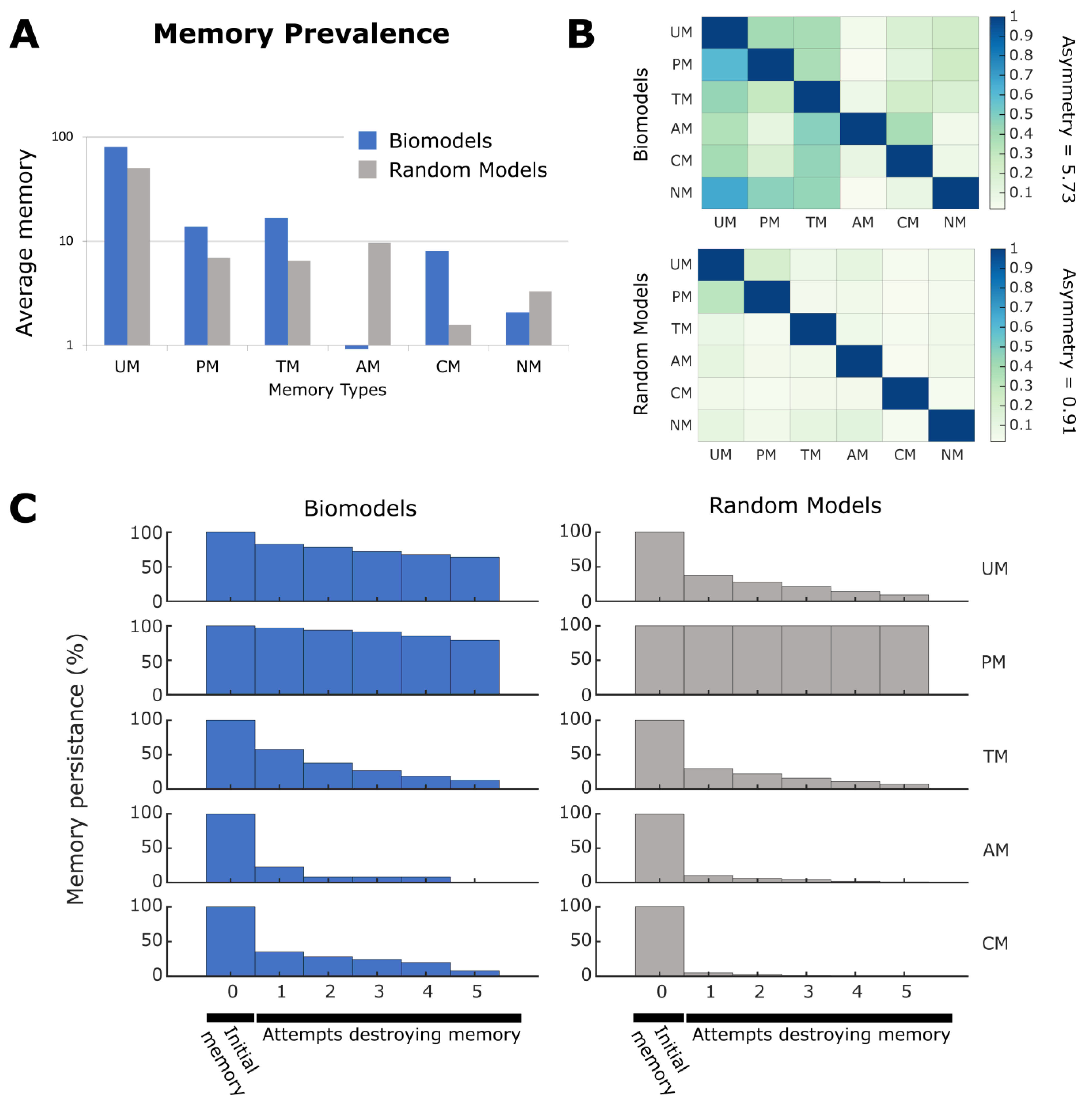
2.6. Biological Networks Have Unique Memory Profiles
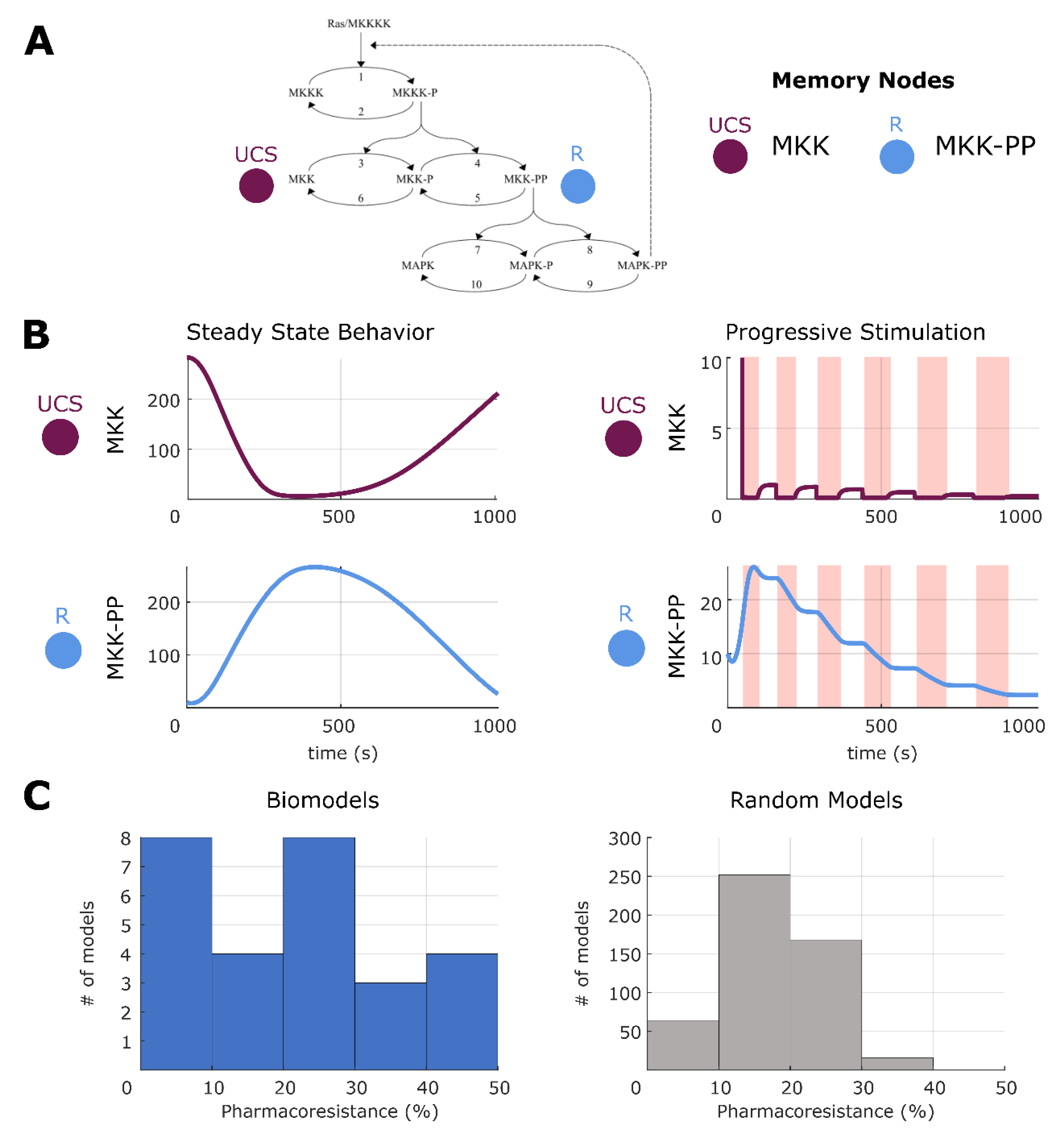
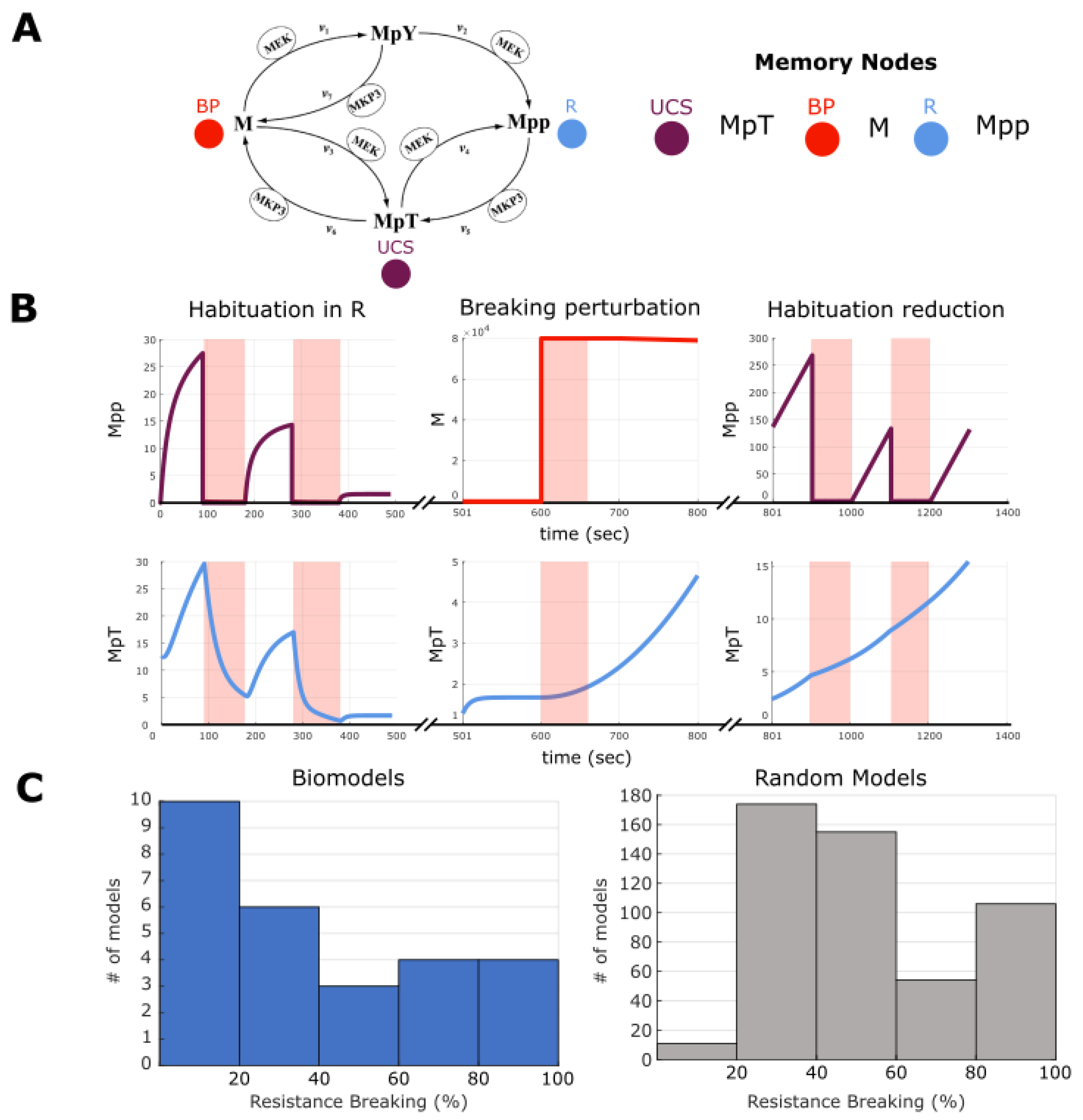
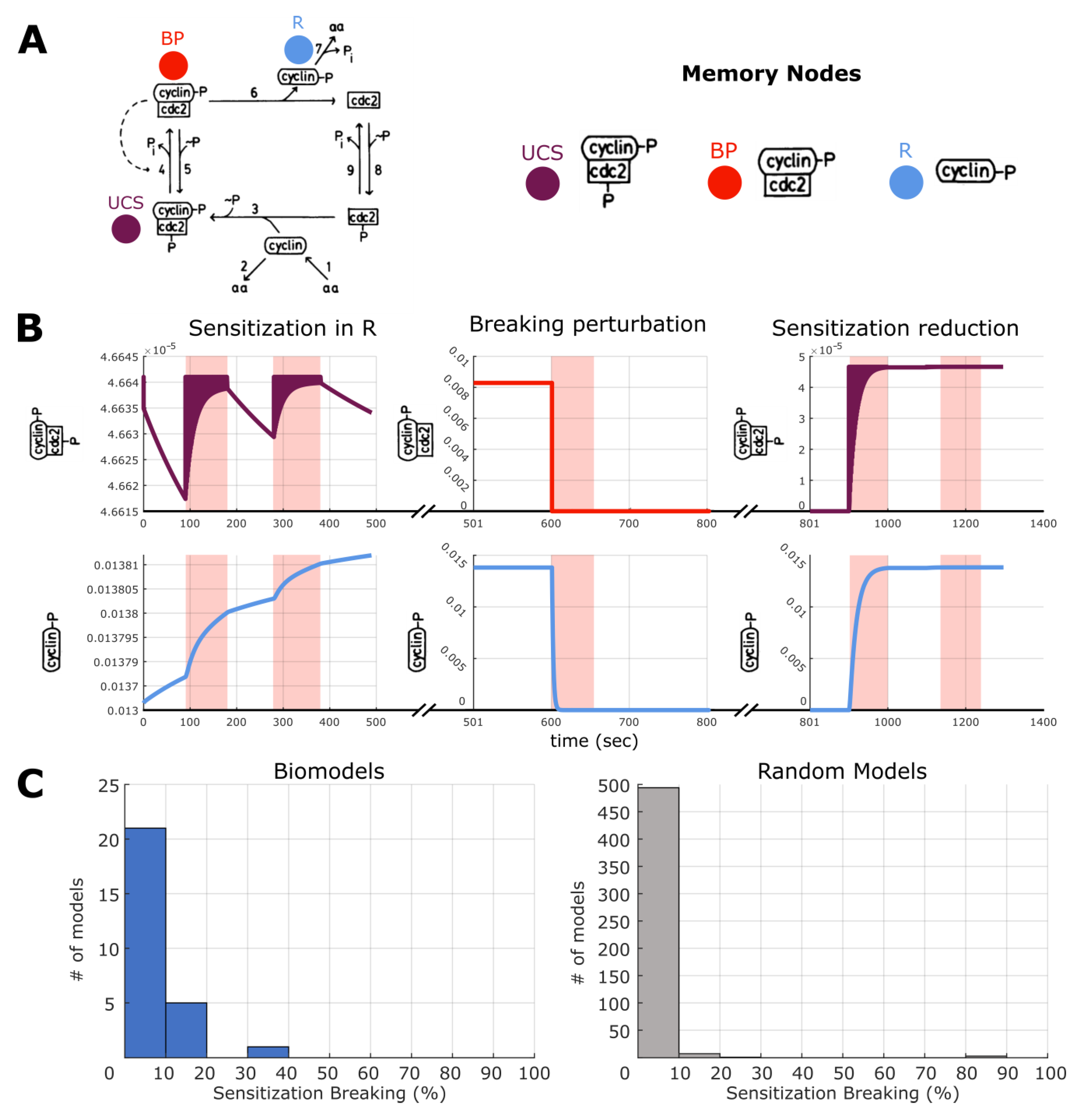
2.7. Making and Breaking Pharmacoresistance
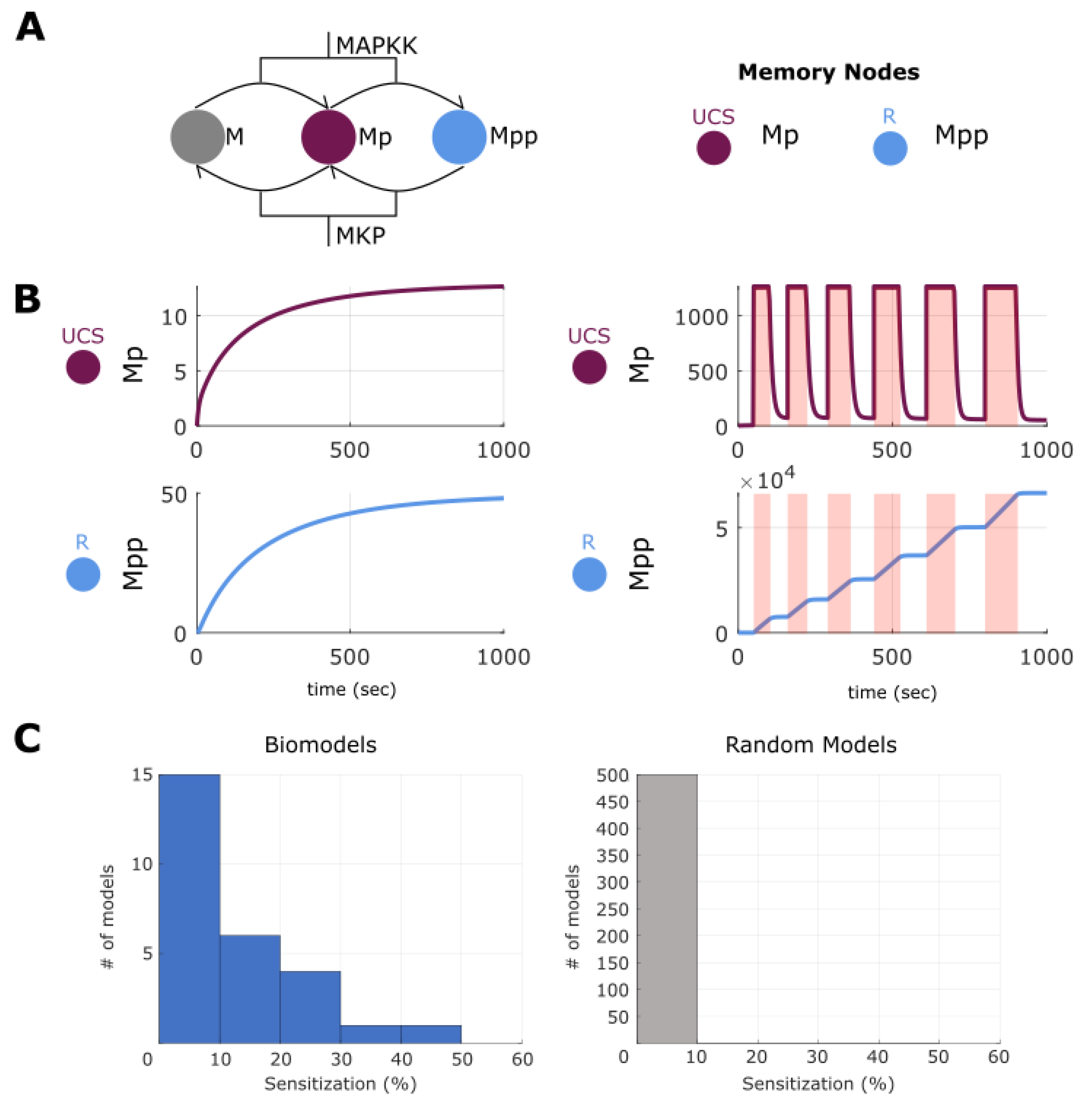
2.8. Making and Breaking Sensitization
3. Discussion
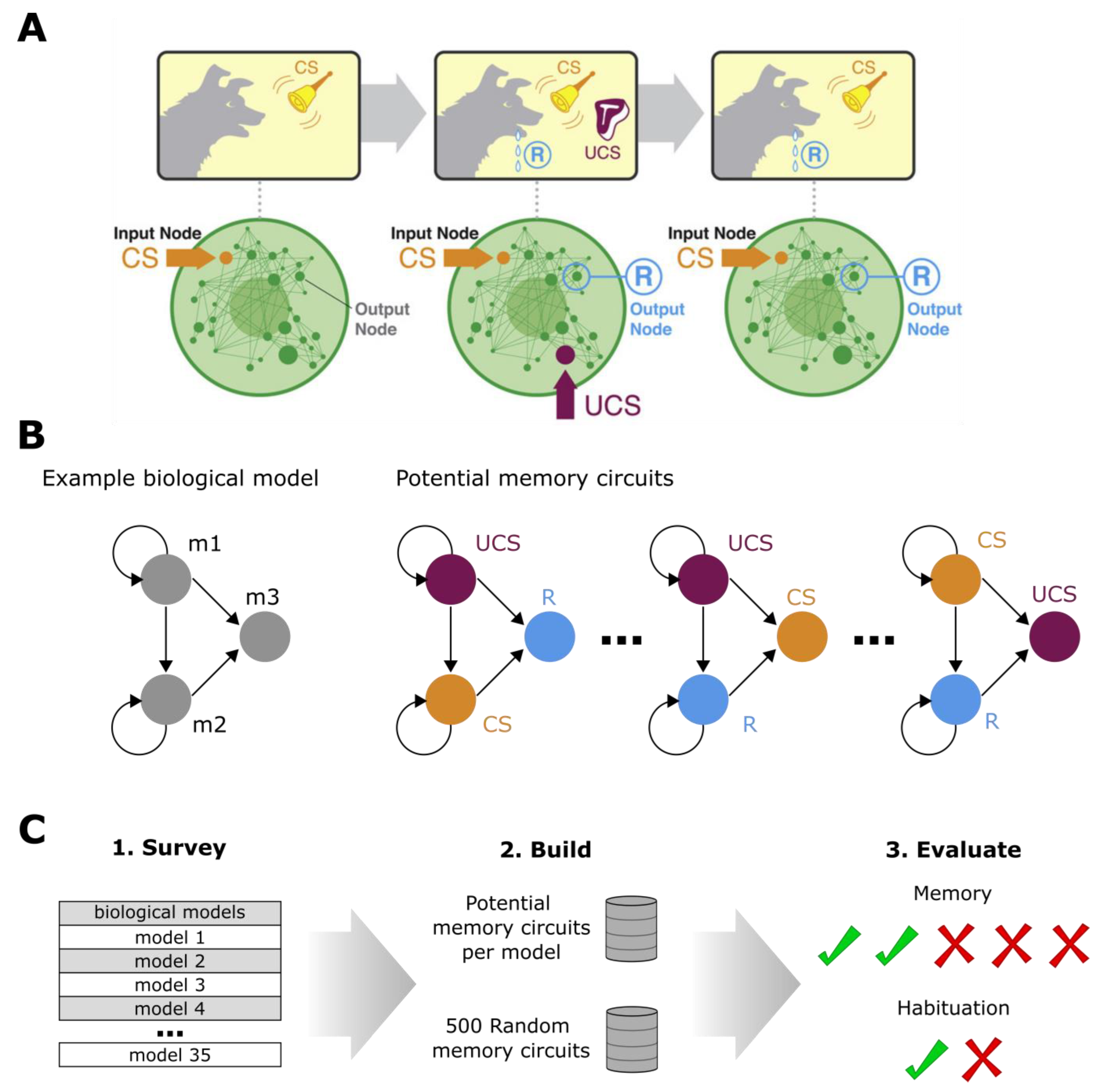
4. Materials and Methods
4.1. Biological Protein/Genetic Models
4.2. Random Synthetic Biology Models for Comparison
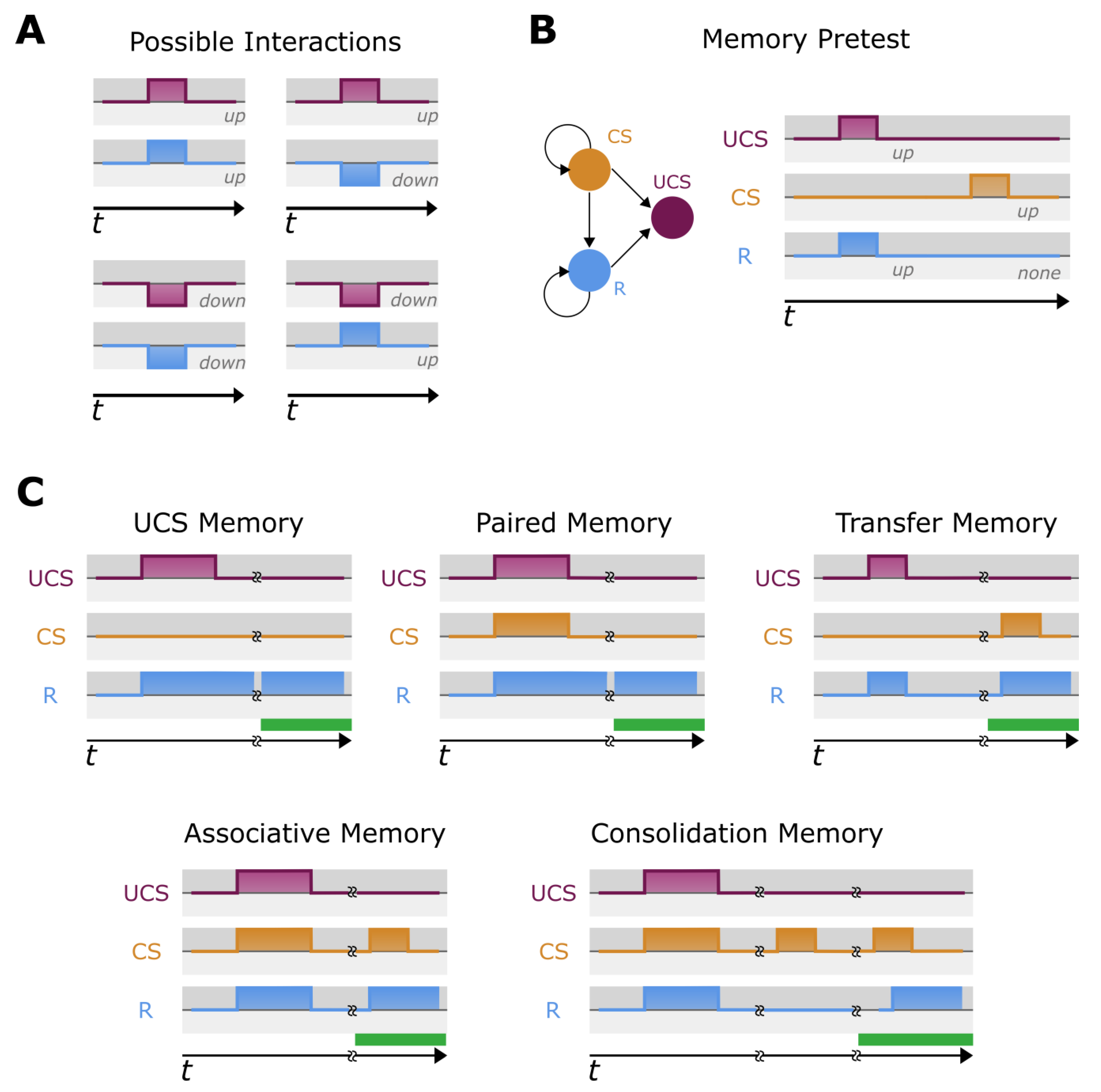
4.3. Memory Evaluation
- (1)
- UCS Based memory (UM) [65]—This type of memory is associated with the stimulation of UCS alone. During the memory evaluation, we stimulate UCS and check if R is regulated. After a delay, we halt the stimulation and observe if the R retains its regulated state. Conceptually, we are evaluating if stimulation of UCS causes a long-term (as compared to the stimulation) change in the behavior of R.
- (2)
- Pairing memory (PM)—This type of memory is associated with the stimulation of paired UCS and NS. Here, we stimulate the paired [UCS, NS] and examine if R is regulated. If so, we relax the model and observe if R still continues retaining regulated state. Conceptually, we are evaluating whether the paired stimulation of UCS causes a long-term (as compared to the stimulation) change in the behavior of R.
- (3)
- Transfer memory (TM) [74]: This memory does not demonstrate retention of the effect of stimuli on response R as in pairing, but it is based on change of behavior of the response by the stimulation of other stimuli. Here, initially we confirm UCS regulates R and NS does not regulate R. After, we stimulate UCS alone and after a short period, test to see if the NS begins regulating R. In other words, we check to see if NS becomes CS. Conceptually, we evaluate if the network shifts after the UCS stimulation, allowing R to be regulated by NS/CS.
- (4)
- Associative memory (AM): Like TM, we are interested in transforming NS to CS. The concept of AM resembles classical conditioning [160,161]. Here, we check if conditions like UCS regulates R and NS does not regulate R is. Next, we train the model by stimulating UCS and NS simultaneously. Finally, we test if NS now started regulating R. Conceptually, the regulation of R has been associated with NS/CS through the simultaneous stimulation with UCS.
- (5)
- Consolidation memory (CM): This memory is similar to AM, but with a temporal delay, as in classical consolidation. Here, we perform the same sequence as in AM detection, but crucially, the NS is not transformed into CS. However, after a relaxation period, we test again and see if NS has converted into CS (i.e., begins regulating R). Conceptually, the regulation of R has been associated with NS/CS through the simultaneous stimulation with UCS, but after a period of consolidation.
4.4. Detection of Simultaneous Memories
4.5. Methodologies for Making and Breaking of Pharmacoresistance in ODE MODELS
4.6. Methodologies for Making and Breaking of Sensitization in ODE Models
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Descartes, R.; Haldane, E.S.; Ross, G.R.T. The Philosophical Works of Descartes; University Press: Cambridge, UK, 1931; p. 2. [Google Scholar]
- Gould, S.J. Ontogeny and Phylogeny; Belknap Press of Harvard University Press: Cambridge, MA, USA, 1977; 501p. [Google Scholar]
- Lyon, P. The biogenic approach to cognition. Cogn. Process 2006, 7, 11–29. [Google Scholar] [CrossRef]
- Baluška, F.; Levin, M. On Having No Head: Cognition throughout Biological Systems. Front. Psychol. 2016, 7, 902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barandiaran, X.; Moreno, A. On what makes certain dynamical systems cognitive: A minimally cognitive organization program. Adapt. Behav. 2006, 14, 171–185. [Google Scholar] [CrossRef]
- Di Primio, F.; Muller, B.S.; Lengeler, J.W. Minimal Cognition in Unicellular Organisms. In Proceedings of the SAB2000 Sixth International Conference on Simulation of Adaptive Behavior: From Animals to Animats, Paris, France, 14 August 2000. [Google Scholar]
- McGivern, P. Active materials: Minimal models of cognition? Adapt. Behav. 2019, 28, 441–451. [Google Scholar] [CrossRef]
- Bongard, J.; Levin, M. Living Things Are Not (20th Century) Machines: Updating Mechanism Metaphors in Light of the Modern Science of Machine Behavior. Front. Ecol. Evol. 2021, 9, 650726. [Google Scholar] [CrossRef]
- Clawson, W.P.; Levin, M. Endless Forms Most Beautiful: Teleonomy and the bioengineering of chimeric and synthetic organisms. Biol. J. Linn. Soc. 2022; in press. [Google Scholar]
- Morgan, C.L. Other minds than ours. In An Introduction to Comparative Psychology; Scott, W., Ed.; Routledge: Milton Park, UK, 1903; p. 59. [Google Scholar]
- Levin, M. TAME: Technological Approach to Mind Everywhere. PsyArXiv 2022, arXiv:10.31234/osf.io/t6e8p. [Google Scholar]
- Mathews, J.; Levin, M. The body electric 2.0: Recent advances in developmental bioelectricity for regenerative and synthetic bioengineering. Curr. Opin. Biotechnol. 2018, 52, 134–144. [Google Scholar] [CrossRef]
- Pezzulo, G.; Levin, M. Re-membering the body: Applications of computational neuroscience to the top-down control of regeneration of limbs and other complex organs. Integr. Biol. 2015, 7, 1487–1517. [Google Scholar] [CrossRef] [Green Version]
- Pezzulo, G.; Levin, M. Top-down models in biology: Explanation and control of complex living systems above the molecular level. J. R. Soc. Interface 2016, 13, 555. [Google Scholar] [CrossRef] [Green Version]
- Fields, C.; Levin, M. Competency in Navigating Arbitrary Spaces: Intelligence as an Invariant for Analyzing Cognition in Diverse Embodiments. Entropy 2022. in review. [Google Scholar]
- Lobo, D.; Solano, M.; Bubenik, G.A.; Levin, M. A linear-encoding model explains the variability of the target morphology in regeneration. J. R. Soc. Interface 2014, 11, 20130918. [Google Scholar] [CrossRef]
- Davies, J.A.; Glykofrydis, F. Engineering pattern formation and morphogenesis. Biochem. Soc. Trans. 2020, 48, 1177–1185. [Google Scholar] [CrossRef]
- Davies, J.A.; Cachat, E. Synthetic biology meets tissue engineering. Biochem. Soc. Trans. 2016, 44, 696–701. [Google Scholar] [CrossRef]
- Davies, J.A. Mechanisms of Morphogenesis; Academic Press: Cambridge, MA, USA, 2013; Available online: https://shop.elsevier.com/books/mechanisms-of-morphogenesis/davies/978-0-12-391062-2#full-description (accessed on 30 September 2022).
- Doursat, R.; Sayama, H.; Michel, O. A review of morphogenetic engineering. Nat. Comput. 2013, 12, 517–535. [Google Scholar] [CrossRef] [Green Version]
- Solé, R.; Amor, D.R.; Duran-Nebreda, S.; Conde-Pueyo, N.; Carbonell-Ballestero, M.; Montanez, R. Synthetic collective intelligence. Biosystems 2016, 148, 47–61. [Google Scholar] [CrossRef]
- Abramson, C.I.; Levin, M. Behaviorist approaches to investigating memory and learning: A primer for synthetic biology and bioengineering. Commun. Integr. Biol. 2021, 14, 230–247. [Google Scholar] [CrossRef]
- Sanz-Ezquerro, J.J.; Münsterberg, A.E.; Stricker, S. Editorial: Signaling Pathways in Embryonic Development. Front. Cell Dev. Biol. 2017, 5, 76. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Buylla, E.R.; Balleza, E.; Benitez, M.; Espinosa-Soto, C.; Padilla-Longoria, P. Gene regulatory network models: A dynamic and integrative approach to development. SEB Exp. Biol. Ser. 2008, 61, 113–139. [Google Scholar]
- Huang, S.; Eichler, G.; Bar-Yam, Y.; Ingber, D.E. Cell fates as high-dimensional attractor states of a complex gene regulatory network. Phys. Rev. Lett. 2005, 94, 128701. [Google Scholar] [CrossRef] [Green Version]
- Peter, I.S.; Davidson, E.H. Evolution of gene regulatory networks controlling body plan development. Cell 2011, 144, 970–985. [Google Scholar] [CrossRef] [Green Version]
- Davidson, E.H. Emerging properties of animal gene regulatory networks. Nature 2010, 468, 911–920. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, M. Beyond Casual Resemblance: Rigorous Frameworks for Comparing Regeneration Across Species. Annu. Rev. Cell Dev. Biol. 2021, 37, 415–440. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Sayama, H. How Criticality of Gene Regulatory Networks Affects the Resulting Morphogenesis under Genetic Perturbations. Artif. Life 2018, 24, 85–105. [Google Scholar] [CrossRef] [PubMed]
- Ten Tusscher, K.H.; Hogeweg, P. Evolution of networks for body plan patterning; interplay of modularity, robustness and evolvability. PLoS Comput. Biol. 2011, 7, e1002208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.J.; Ramsey, S.A.; Filtz, T.M.; Kioussi, C. Differential gene regulatory networks in development and disease. Cell Mol. Life Sci. 2018, 75, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Qin, G.; Yang, L.; Ma, Y.; Liu, J.; Huo, Q. The exploration of disease-specific gene regulatory networks in esophageal carcinoma and stomach adenocarcinoma. BMC Bioinform. 2019, 20, 717. [Google Scholar] [CrossRef] [Green Version]
- Fazilaty, H.; Rago, L.; Kass Youssef, K.; Ocana, O.H.; Garcia-Asencio, F.; Arcas, A.; Galceran, J.; Nieto, M.A. A gene regulatory network to control EMT programs in development and disease. Nat. Commun. 2019, 10, 5115. [Google Scholar] [CrossRef] [Green Version]
- De Jong, H. Modeling and simulation of genetic regulatory systems: A literature review. J. Comput. Biol. 2002, 9, 67–103. [Google Scholar] [CrossRef] [Green Version]
- Delgado, F.M.; Gómez-Vela, F. Computational methods for Gene Regulatory Networks reconstruction and analysis: A review. Artif. Intell. Med. 2019, 95, 133–145. [Google Scholar] [CrossRef]
- Fetrow, J.S.; Babbitt, P.C. New computational approaches to understanding molecular protein function. PLoS Comput. Biol. 2018, 14, e1005756. [Google Scholar] [CrossRef] [Green Version]
- Schlitt, T.; Brazma, A. Current approaches to gene regulatory network modelling. BMC Bioinform. 2007, 8, S9. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Delgado, E.; Perez-Carrasco, R.; Briscoe, J.; Sollich, P. Memory functions reveal structural properties of gene regulatory networks. PLoS Comput. Biol. 2018, 14, e1006003. [Google Scholar] [CrossRef]
- Zagorski, M.; Tabata, Y.; Brandenberg, N.; Lutolf, M.P.; Tkacik, G.; Bollenbach, T.; Briscoe, J.; Kicheva, A. Decoding of position in the developing neural tube from antiparallel morphogen gradients. Science 2017, 356, 1379–1383. [Google Scholar] [CrossRef]
- Zanudo, J.G.T.; Yang, G.; Albert, R. Structure-based control of complex networks with nonlinear dynamics. Proc. Natl. Acad. Sci. USA 2017, 114, 7234–7239. [Google Scholar] [CrossRef] [Green Version]
- Choo, S.-M.; Ban, B.; Joo, J.I.; Cho, K.-H. The phenotype control kernel of a biomolecular regulatory network. BMC Syst. Biol. 2018, 12, 49. [Google Scholar] [CrossRef] [Green Version]
- Choo, S.-M.; Park, S.-M.; Cho, K.-H. Minimal intervening control of biomolecular networks leading to a desired cellular state. Sci. Rep. 2019, 9, 13124. [Google Scholar] [CrossRef] [Green Version]
- Csermely, P.; Kunsic, N.; Mendik, P.; Kerestély, M.; Faragó, T.; Veres, D.V.; Tompa, P. Learning of Signaling Networks: Molecular Mechanisms. Trends Biochem. Sci. 2020, 45, 284–294. [Google Scholar] [CrossRef] [Green Version]
- Zañudo, J.G.T.; Albert, R. Cell Fate Reprogramming by Control of Intracellular Network Dynamics. PLOS Comput. Biol. 2015, 11, e1004193. [Google Scholar] [CrossRef] [Green Version]
- Steinway, S.N.; Zañudo, J.G.T.; Michel, P.J.; Feith, D.J.; Loughran, T.P.; Albert, R. Combinatorial interventions inhibit TGFβ-driven epithelial-to-mesenchymal transition and support hybrid cellular phenotypes. NPJ Syst. Biol. Appl. 2015, 1, 15014. [Google Scholar] [CrossRef] [Green Version]
- Cifuentes Fontanals, L.; Tonello, E.; Siebert, H. Control Strategy Identification via Trap Spaces in Boolean Networks. In Computational Methods in Systems Biology. CMSB 2020; Abate, A., Petrov, T., Wolf, V., Eds.; Lecture Notes in Computer Science; Springer: Cham, Switzerland, 2020; Volume 12314. [Google Scholar]
- Murrugarra, D.; Veliz-Cuba, A.; Aguilar, B.; Laubenbacher, R. Identification of control targets in Boolean molecular network models via computational algebra. BMC Syst. Biol. 2016, 10, 94. [Google Scholar] [CrossRef]
- Manicka, S.; Levin, M. The Cognitive Lens: A primer on conceptual tools for analysing information processing in developmental and regenerative morphogenesis. Philos. Trans. R Soc. Lond. B Biol. Sci. 2019, 374, 20180369. [Google Scholar] [CrossRef] [Green Version]
- Szabó, Á.; Vattay, G.; Kondor, D. A cell signaling model as a trainable neural nanonetwork. Nano Commun. Netw. 2012, 3, 57–64. [Google Scholar] [CrossRef]
- Turner, C.H.; Robling, A.G.; Duncan, R.L.; Burr, D.B. Do bone cells behave like a neuronal network? Calcif. Tissue Int. 2002, 70, 435–442. [Google Scholar] [CrossRef]
- Goel, P.; Mehta, A. Learning theories reveal loss of pancreatic electrical connectivity in diabetes as an adaptive response. PLoS ONE 2013, 8, e70366. [Google Scholar] [CrossRef] [Green Version]
- Nashun, B.; Hill, P.W.; Hajkova, P. Reprogramming of cell fate: Epigenetic memory and the erasure of memories past. EMBO J. 2015, 34, 1296–1308. [Google Scholar] [CrossRef]
- Quintin, J.; Cheng, S.C.; van der Meer, J.W.; Netea, M.G. Innate immune memory: Towards a better understanding of host defense mechanisms. Curr. Opin. Immunol. 2014, 29, 1–7. [Google Scholar] [CrossRef]
- Corre, G.; Stockholm, D.; Arnaud, O.; Kaneko, G.; Vinuelas, J.; Yamagata, Y.; Neildez-Nguyen, T.M.; Kupiec, J.J.; Beslon, G.; Gandrillon, O.; et al. Stochastic fluctuations and distributed control of gene expression impact cellular memory. PLoS ONE 2014, 9, e115574. [Google Scholar] [CrossRef]
- Zediak, V.P.; Wherry, E.J.; Berger, S.L. The contribution of epigenetic memory to immunologic memory. Curr. Opin. Genet. Dev. 2011, 21, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.A.; Buckley, C.L.; Mills, R.; Davies, A. Associative memory in gene regulation networks. In Proceedings of the Artificial Life Conference XII, Odense, Denmark, 19–23 August 2010; pp. 194–201. [Google Scholar]
- Watson, R.A.; Mills, R.; Buckley, C.L. Global adaptation in networks of selfish components: Emergent associative memory at the system scale. Artif. Life 2011, 17, 147–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Association for the Advancement of Science. Maturing from Memory. Sci. Signal. 2003, 2003, tw462. [Google Scholar]
- Sible, J.C. Thanks for the memory. Nature 2003, 426, 392–393. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Ferrell, J.E. A positive-feedback-based bistable ‘memory module’that governs a cell fate decision. Nature 2003, 426, 460–465. [Google Scholar] [CrossRef]
- Levine, J.H.; Lin, Y.; Elowitz, M.B. Functional roles of pulsing in genetic circuits. Science 2013, 342, 1193–1200. [Google Scholar] [CrossRef] [Green Version]
- Urrios, A.; Macia, J.; Manzoni, R.; Conde, N.; Bonforti, A.; de Nadal, E.l.; Posas, F.; Solé, R. A synthetic multicellular memory device. ACS Synth. Biol. 2016, 5, 862–873. [Google Scholar] [CrossRef]
- Macia, J.; Vidiella, B.; Solé, R.V. Synthetic associative learning in engineered multicellular consortia. J. R. Soc. Interface 2017, 14, 20170158. [Google Scholar] [CrossRef] [Green Version]
- Kandel, E.R.; Dudai, Y.; Mayford, M.R. The molecular and systems biology of memory. Cell 2014, 157, 163–186. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.J.; Roy, D.S.; Pignatelli, M.; Arons, A.; Tonegawa, S. Engram cells retain memory under retrograde amnesia. Science 2015, 348, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Rozum, J.; Albert, R. Leveraging network structure in nonlinear control. NPJ Syst. Biol. Appl. 2022, 8, 36. [Google Scholar] [CrossRef]
- Biswas, S.; Manicka, S.; Hoel, E.; Levin, M. Gene regulatory networks exhibit several kinds of memory: Quantification of memory in biological and random transcriptional networks. Iscience 2021, 24, 102131. [Google Scholar] [CrossRef]
- Kauffman, S.A. At Home in the Universe: The Search for Laws of Self-Organization and Complexity; Oxford University Press: New York, NY, USA, 1995; p. viii, 321. [Google Scholar]
- Kauffman, S.A. The Origins of Order: Self Organization and Selection in Evolution; Oxford University Press: New York, NY, USA, 1993; p. 709. [Google Scholar]
- Mann, M.W.; Pons, G. Various pharmacogenetic aspects of antiepileptic drug therapy: A review. CNS Drugs 2007, 21, 143–164. [Google Scholar] [CrossRef]
- Beck, H. Plasticity of antiepileptic drug targets. Epilepsia 2007, 48 (Suppl. 1), 14–18. [Google Scholar] [CrossRef]
- Remy, S.; Beck, H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain 2006, 129, 18–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carosi, G. Pharmacoresistance: An emerging clinical problem. J. Biol. Regul. Homeost. Agents 1998, 12, 9–14. [Google Scholar] [PubMed]
- Rohwer, J.M.; Meadow, N.D.; Roseman, S.; Westerhoff, H.V.; Postma, P.W. Understanding glucose transport by the bacterial phosphoenolpyruvate: Glycose phosphotransferase system on the basis of kinetic measurements in vitro. J. Biol. Chem. 2000, 275, 34909–34921. [Google Scholar] [CrossRef]
- Markevich, N.I.; Hoek, J.B.; Kholodenko, B.N. Signaling switches and bistability arising from multisite phosphorylation in protein kinase cascades. J. Cell Biol. 2004, 164, 353–359. [Google Scholar] [CrossRef]
- Kholodenko, B.N. Negative feedback and ultrasensitivity can bring about oscillations in the mitogen-activated protein kinase cascades. Eur. J. Biochem. 2000, 267, 1583–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyson, J.J. Modeling the cell division cycle: cdc2 and cyclin interactions. Proc. Natl. Acad. Sci. USA 1991, 88, 7328–7332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izquierdo, E.; Harvey, I.; Beer, R.D. Associative learning on a continuum in evolved dynamical neural networks. Adapt. Behav. 2008, 16, 361–384. [Google Scholar] [CrossRef]
- Edelstein, S.J.; Schaad, O.; Henry, E.; Bertrand, D.; Changeux, J.-P. A kinetic mechanism for nicotinic acetylcholine receptors based on multiple allosteric transitions. Biol. Cybern. 1996, 75, 361–379. [Google Scholar] [CrossRef]
- Goldbeter, A. A minimal cascade model for the mitotic oscillator involving cyclin and cdc2 kinase. Proc. Natl. Acad. Sci. USA 1991, 88, 9107–9111. [Google Scholar] [CrossRef] [Green Version]
- Fuß, H.; Dubitzky, W.; Downes, S.; Kurth, M.J. Bistable switching and excitable behaviour in the activation of Src at mitosis. Bioinformatics 2006, 22, e158–e165. [Google Scholar] [CrossRef] [Green Version]
- Hoefnagel, M.H.; Starrenburg, M.J.; Martens, D.E.; Hugenholtz, J.; Kleerebezem, M.; Van Swam, I.I.; Bongers, R.; Westerhoff, H.V.; Snoep, J.L. Metabolic engineering of lactic acid bacteria, the combined approach: Kinetic modelling, metabolic control and experimental analysisThe GenBank accession number for the sequence reported in this paper is AY046926. Microbiology 2002, 148, 1003–1013. [Google Scholar] [CrossRef] [Green Version]
- Leloup, J.-C.; Goldbeter, A. Chaos and birhythmicity in a model for circadian oscillations of the PER and TIM proteins in Drosophila. J. Theor. Biol. 1999, 198, 445–459. [Google Scholar] [CrossRef] [Green Version]
- olde Scheper, T.; Klinkenberg, D.; Pennartz, C.; Van Pelt, J. A mathematical model for the intracellular circadian rhythm generator. J. Neurosci. 1999, 19, 40–47. [Google Scholar] [CrossRef]
- Cao, Y.; Liang, J. Optimal enumeration of state space of finitely buffered stochastic molecular networks and exact computation of steady state landscape probability. BMC Syst. Biol. 2008, 2, 30. [Google Scholar] [CrossRef]
- De Caluwé, J.; Xiao, Q.; Hermans, C.; Verbruggen, N.; Leloup, J.-C.; Gonze, D. A compact model for the complex plant circadian clock. Front. Plant Sci. 2016, 7, 74. [Google Scholar] [CrossRef] [Green Version]
- Chickarmane, V.; Troein, C.; Nuber, U.A.; Sauro, H.M.; Peterson, C. Transcriptional dynamics of the embryonic stem cell switch. PLoS Comput. Biol. 2006, 2, e123. [Google Scholar] [CrossRef] [Green Version]
- Chickarmane, V.; Peterson, C. A computational model for understanding stem cell, trophectoderm and endoderm lineage determination. PLoS ONE 2008, 3, e3478. [Google Scholar] [CrossRef]
- François, P.; Hakim, V. Core genetic module: The mixed feedback loop. Phys. Rev. E 2005, 72, 031908. [Google Scholar] [CrossRef] [Green Version]
- Muller, M.; Obeyesekere, M.; Mills, G.B.; Ram, P.T. Network topology determines dynamics of the mammalian MAPK1, 2 signaling network: Bifan motif regulation of C-Raf and B-Raf isoforms by FGFR and MC1R. FASEB J. 2008, 22, 1393–1403. [Google Scholar] [CrossRef] [Green Version]
- Ciliberto, A.; Petrus, M.J.; Tyson, J.J.; Sible, J.C. A kinetic model of the cyclin E/Cdk2 developmental timer in Xenopus laevis embryos. Biophys. Chem. 2003, 104, 573–589. [Google Scholar] [CrossRef]
- Cellière, G.; Fengos, G.; Hervé, M.; Iber, D. The plasticity of TGF-β signaling. BMC Syst. Biol. 2011, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, S.I.; Van Boekel, M.A. Kinetic modelling of Amadori N-(1-deoxy-D-fructos-1-yl)-glycine degradation pathways. Part II—Kinetic analysis. Carbohydr. Res. 2003, 338, 1665–1678. [Google Scholar] [CrossRef] [PubMed]
- Vilar, J.M.; Kueh, H.Y.; Barkai, N.; Leibler, S. Mechanisms of noise-resistance in genetic oscillators. Proc. Natl. Acad. Sci. USA 2002, 99, 5988–5992. [Google Scholar] [CrossRef] [PubMed]
- Tyson, J.J.; Hong, C.I.; Thron, C.D.; Novak, B. A simple model of circadian rhythms based on dimerization and proteolysis of PER and TIM. Biophys. J. 1999, 77, 2411–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marwan, W. Theory of time-resolved somatic complementation and its use to explore the sporulation control network in Physarum polycephalum. Genetics 2003, 164, 105–115. [Google Scholar] [CrossRef]
- Li, C.; Donizelli, M.; Rodriguez, N.; Dharuri, H.; Endler, L.; Chelliah, V.; Li, L.; He, E.; Henry, A.; Stefan, M.I. BioModels Database: An enhanced, curated and annotated resource for published quantitative kinetic models. BMC Syst. Biol. 2010, 4, 92. [Google Scholar] [CrossRef] [Green Version]
- Chelliah, V.; Laibe, C.; Le Novère, N. BioModels database: A repository of mathematical models of biological processes. In Silico Systems Biology; Springer: Berlin/Heidelberg, Germany, 2013; pp. 189–199. [Google Scholar]
- Le Novere, N.; Bornstein, B.; Broicher, A.; Courtot, M.; Donizelli, M.; Dharuri, H.; Li, L.; Sauro, H.; Schilstra, M.; Shapiro, B. BioModels Database: A free, centralized database of curated, published, quantitative kinetic models of biochemical and cellular systems. Nucleic Acids Res. 2006, 34, D689–D691. [Google Scholar] [CrossRef] [Green Version]
- Goldbeter, A. A model for circadian oscillations in the Drosophila period protein (PER). Proc. R. Soc. London. Ser. B: Biol. Sci. 1995, 261, 319–324. [Google Scholar]
- Goldbeter, A.; Gonze, D.; Pourquié, O. Sharp developmental thresholds defined through bistability by antagonistic gradients of retinoic acid and FGF signaling. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2007, 236, 1495–1508. [Google Scholar] [CrossRef]
- Marhl, M.; Haberichter, T.; Brumen, M.; Heinrich, R. Complex calcium oscillations and the role of mitochondria and cytosolic proteins. Biosystems 2000, 57, 75–86. [Google Scholar] [CrossRef]
- Rohwer, J.M.; Botha, F.C. Analysis of sucrose accumulation in the sugar cane culm on the basis of in vitro kinetic data. Biochem. J. 2001, 358, 437–445. [Google Scholar] [CrossRef]
- Ueda, H.R.; Hagiwara, M.; Kitano, H. Robust oscillations within the interlocked feedback model of Drosophila circadian rhythm. J. Theor. Biol. 2001, 210, 401–406. [Google Scholar] [CrossRef]
- Celia-Terrassa, T.; Bastian, C.; Liu, D.D.; Ell, B.; Aiello, N.M.; Wei, Y.; Zamalloa, J.; Blanco, A.M.; Hang, X.; Kunisky, D.; et al. Hysteresis control of epithelial-mesenchymal transition dynamics conveys a distinct program with enhanced metastatic ability. Nat. Commun. 2018, 9, 5005. [Google Scholar] [CrossRef] [Green Version]
- Tsimring, L.S. Noise in biology. Rep. Prog. Phys. 2014, 77, 026601. [Google Scholar] [CrossRef]
- Eldar, A.; Elowitz, M.B. Functional roles for noise in genetic circuits. Nature 2010, 467, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Azpeitia, E.; Balanzario, E.P.; Wagner, A. Signaling pathways have an inherent need for noise to acquire information. BMC Bioinform. 2020, 21, 462. [Google Scholar] [CrossRef]
- Hopfield, J.J. Neural networks and physical systems with emergent collective computational abilities. Proc. Natl. Acad. Sci. USA 1982, 79, 2554–2558. [Google Scholar] [CrossRef] [Green Version]
- Folli, V.; Leonetti, M.; Ruocco, G. On the Maximum Storage Capacity of the Hopfield Model. Front. Comput. Neurosci. 2017, 10, 144. [Google Scholar] [CrossRef] [Green Version]
- Potschka, H.; Brodie, M.J. Pharmacoresistance. Handb. Clin. Neurol. 2012, 108, 741–757. [Google Scholar]
- Mayergoyz, I.; Korman, C. Hysteresis and Neural Memory; World Scientific: Singapore, 2019; p. 292. [Google Scholar]
- Yuan, Z.; Zhao, C.; Di, Z.; Wang, W.-X.; Lai, Y.-C. Exact controllability of complex networks. Nat. Commun. 2013, 4, 2447. [Google Scholar] [CrossRef] [Green Version]
- Brosschot, J.F. Cognitive-emotional sensitization and somatic health complaints. Scand. J. Psychol. 2002, 43, 113–122. [Google Scholar] [CrossRef]
- Cooke, R.A.; Vander Veer, A. Human sensitization. J. Immunol. 1916, 1, 201–305. [Google Scholar] [CrossRef]
- Eriksen, H.R.; Ursin, H. Sensitization and subjective health complaints. Scand. J. Psychol. 2002, 43, 189–196. [Google Scholar] [CrossRef]
- Eriksen, H.R.; Ursin, H. Subjective health complaints, sensitization, and sustained cognitive activation (stress). J. Psychosom. Res. 2004, 56, 445–448. [Google Scholar] [CrossRef]
- Garabrant, D.H.; Schweitzer, S. Epidemiology of latex sensitization and allergies in health care workers. J. Allergy Clin. Immunol. 2002, 110, S82–S95. [Google Scholar] [CrossRef] [PubMed]
- Robinson, T.E.; Berridge, K.C. Incentive-sensitization and addiction. Addiction 2001, 96, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Salo, P.M.; Arbes Jr, S.J.; Jaramillo, R.; Calatroni, A.; Weir, C.H.; Sever, M.L.; Hoppin, J.A.; Rose, K.M.; Liu, A.H.; Gergen, P.J. Prevalence of allergic sensitization in the United States: Results from the National Health and Nutrition Examination Survey (NHANES) 2005-2006. J. Allergy Clin. Immunol. 2014, 134, 350–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, S.A. Inherency. In Evolutionary Developmental Biology: A Reference Guide; Nuno de la Rosa, L., Müller, G., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–12. [Google Scholar]
- Pienta, K.J.; Coffey, D.S. Cellular harmonic information transfer through a tissue tensegrity-matrix system. Med. Hypotheses 1991, 34, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Kashtan, N.; Alon, U. Spontaneous evolution of modularity and network motifs. Proc. Natl. Acad. Sci. USA 2005, 102, 13773–13778. [Google Scholar] [CrossRef] [Green Version]
- Katz, Y. Embodying probabilistic inference in biochemical circuits. arXiv 2018, arXiv:1806.10161. [Google Scholar]
- Katz, Y.; Springer, M. Probabilistic adaptation in changing microbial environments. PeerJ 2016, 4, e2716. [Google Scholar] [CrossRef] [Green Version]
- Wheat, J.C.; Sella, Y.; Willcockson, M.; Skoultchi, A.I.; Bergman, A.; Singer, R.H.; Steidl, U. Single-molecule imaging of transcription dynamics in somatic stem cells. Nature 2020, 583, 431–436. [Google Scholar] [CrossRef]
- Overstreet, L.S. Quantal transmission: Not just for neurons. Trends Neurosci. 2005, 28, 59–62. [Google Scholar] [CrossRef]
- Smith, K.A. Cell growth signal transduction is quantal. Ann. N. Y. Acad. Sci. 1995, 766, 263–271. [Google Scholar] [CrossRef]
- Levin, M. Technological Approach to Mind Everywhere: An Experimentally-Grounded Framework for Understanding Diverse Bodies and Minds. Front. Syst. Neurosci. 2022, 16, 768201. [Google Scholar] [CrossRef]
- Mathews, J.; Chang, J.; Devlin, L.; Levin, M. Cellular Signaling Pathways as Plastic, Proto-cognitive Systems: Implications for Biomedicine. OSF Prepr. 2022. [Google Scholar] [CrossRef]
- Kotula, J.W.; Kerns, S.J.; Shaket, L.A.; Siraj, L.; Collins, J.J.; Way, J.C.; Silver, P.A. Programmable bacteria detect and record an environmental signal in the mammalian gut. Proc. Natl. Acad. Sci. USA 2014, 111, 4838–4843. [Google Scholar] [CrossRef] [Green Version]
- Weiss, R.; Basu, S.; Hooshangi, S.; Kalmbach, A.; Karig, D.; Mehreja, R.; Netravali, I. Genetic circuit building blocks for cellular computation, communications, and signal processing. Nat. Comput. 2003, 2, 47–84. [Google Scholar] [CrossRef]
- Fernando, C.T.; Liekens, A.M.L.; Bingle, L.E.H.; Beck, C.; Lenser, T.; Stekel, D.J.; Rowe, J.E. Molecular circuits for associative learning in single-celled organisms. J. R. Soc. Interface 2009, 6, 463–469. [Google Scholar] [CrossRef] [Green Version]
- McGregor, S.; Vasas, V.; Husbands, P.; Fernando, C. Evolution of associative learning in chemical networks. PLoS Comput. Biol. 2012, 8, e1002739. [Google Scholar] [CrossRef]
- Watson, R.A.; Wagner, G.P.; Pavlicev, M.; Weinreich, D.M.; Mills, R. The evolution of phenotypic correlations and “developmental memory”. Evolution 2014, 68, 1124–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez Velazquez, J.L. Dynamiceuticals: The Next Stage in Personalized Medicine. Front. Neurosci. 2017, 11, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delas, M.J.; Briscoe, J. Repressive interactions in gene regulatory networks: When you have no other choice. Curr. Top Dev. Biol. 2020, 139, 239–266. [Google Scholar] [CrossRef] [PubMed]
- Revusky, S. The Drug-Drug Conditioning Paradigm—A Review. Psychopharmacology 1982, 76, A11. [Google Scholar]
- Sparkman, N.L.; Li, M. Drug-drug conditioning between citalopram and haloperidol or olanzapine in a conditioned avoidance response model: Implications for polypharmacy in schizophrenia. Behav Pharm. 2012, 23, 658–668. [Google Scholar] [CrossRef] [Green Version]
- Taukulis, H.K.; Brake, L.D. Therapeutic and Hypothermic Properties of Diazepam Altered by a Diazepam-Chlorpromazine Association. Pharmacol. Biochem. Behav. 1989, 34, 1–6. [Google Scholar] [CrossRef]
- Revusky, S.; Taukulis, H.K.; Peddle, C. Learned Associations between Drug States—Attempted Analysis in Pavlovian Terms. Physiol. Psychol. 1979, 7, 352–363. [Google Scholar] [CrossRef]
- Bryant, D.M.; Sousounis, K.; Farkas, J.E.; Bryant, S.; Thao, N.; Guzikowski, A.R.; Monaghan, J.R.; Levin, M.; Whited, J.L. Repeated removal of developing limb buds permanently reduces appendage size in the highly-regenerative axolotl. Dev. Biol. 2017, 424, 1–9. [Google Scholar] [CrossRef]
- Blackiston, D.; Shomrat, T.; Levin, M. The Stability of Memories During Brain Remodeling: A Perspective. Commun. Integr. Biol. 2015, 8, e1073424. [Google Scholar] [CrossRef] [Green Version]
- Abraham, W.C.; Jones, O.D.; Glanzman, D.L. Is plasticity of synapses the mechanism of long-term memory storage? NPJ Sci. Learn. 2019, 4, 9. [Google Scholar] [CrossRef] [Green Version]
- Bedecarrats, A.; Chen, S.; Pearce, K.; Cai, D.; Glanzman, D.L. RNA from Trained Aplysia Can Induce an Epigenetic Engram for Long-Term Sensitization in Untrained Aplysia. eNeuro 2018, 5, 0038-18. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Cai, D.; Pearce, K.; Sun, P.Y.; Roberts, A.C.; Glanzman, D.L. Reinstatement of long-term memory following erasure of its behavioral and synaptic expression in Aplysia. Elife 2014, 3, e03896. [Google Scholar] [CrossRef]
- Dent, E.W. Of microtubules and memory: Implications for microtubule dynamics in dendrites and spines. Mol. Biol. Cell 2017, 28, 1–8. [Google Scholar] [CrossRef]
- Craddock, T.J.; Tuszynski, J.A.; Hameroff, S. Cytoskeletal signaling: Is memory encoded in microtubule lattices by CaMKII phosphorylation? PLoS Comput. Biol. 2012, 8, e1002421. [Google Scholar] [CrossRef] [Green Version]
- Espenson, J.H. Chemical Kinetics and Reaction Mechanisms; Citeseer: Princeton, NJ, USA, 1995; Volume 102. [Google Scholar]
- Butcher, J.C. Numerical Methods for Ordinary Differential Equations; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Chen, R.T.; Rubanova, Y.; Bettencourt, J.; Duvenaud, D.K. Neural ordinary differential equations. Adv. Neural Inf. Process. Syst. 2018, 31, 6572–6583. [Google Scholar]
- Ince, E.L. Ordinary Differential Equations; Courier Corporation: North Chelmsford, MA, USA, 1956. [Google Scholar]
- Murphy, G.M. Ordinary Differential Equations and Their Solutions; Courier Corporation: North Chelmsford, MA, USA, 2011. [Google Scholar]
- Shampine, L.F.; Reichelt, M.W. The matlab ode suite. SIAM J. Sci. Comput. 1997, 18, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Shampine, L.F.; Shampine, L.F.; Gladwell, I.; Thompson, S. Solving ODEs with Matlab; Cambridge University Press: Cambridge, MA, USA, 2003. [Google Scholar]
- Reinitz, J.; Sharp, D.H. Mechanism of eve stripe formation. Mech. Dev. 1995, 49, 133–158. [Google Scholar] [CrossRef]
- Xu, R.; Wunsch II, D.; Frank, R. Inference of genetic regulatory networks with recurrent neural network models using particle swarm optimization. IEEE/ACM Trans. Comput. Biol. Bioinform. 2007, 4, 681–692. [Google Scholar]
- Kentzoglanakis, K.; Poole, M. A swarm intelligence framework for reconstructing gene networks: Searching for biologically plausible architectures. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 9, 358–371. [Google Scholar] [CrossRef]
- Biswas, S.; Acharyya, S. Neural model of gene regulatory network: A survey on supportive meta-heuristics. Theory Biosci. 2016, 135, 1–19. [Google Scholar] [CrossRef]
- Lee, T.I.; Young, R.A. Transcriptional regulation and its misregulation in disease. Cell 2013, 152, 1237–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rescorla, R.A. Pavlovian conditioning and its proper control procedures. Psychol. Rev. 1967, 74, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombrowski, S.M.; Desai, S.Y.; Marroni, M.; Cucullo, L.; Goodrich, K.; Bingaman, W.; Mayberg, M.R.; Bengez, L.; Janigro, D. Overexpression of multiple drug resistance genes in endothelial cells from patients with refractory epilepsy. Epilepsia 2001, 42, 1501–1506. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Y.; Thom, M.; Catarino, C.B.; Martinian, L.; Figarella-Branger, D.; Bartolomei, F.; Koepp, M.; Sisodiya, S.M. Neuropathology of the blood–brain barrier and pharmaco-resistance in human epilepsy. Brain 2012, 135, 3115–3133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, D.; Löscher, W. Drug resistance in epilepsy: Putative neurobiologic and clinical mechanisms. Epilepsia 2005, 46, 858–877. [Google Scholar] [CrossRef]
- Chechile, R.A. Analyzing Memory: The Formation, Retention, and Measurement of Memory; The MIT Press: Cambridge, MA, USA, 2018. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biswas, S.; Clawson, W.; Levin, M. Learning in Transcriptional Network Models: Computational Discovery of Pathway-Level Memory and Effective Interventions. Int. J. Mol. Sci. 2023, 24, 285. https://doi.org/10.3390/ijms24010285
Biswas S, Clawson W, Levin M. Learning in Transcriptional Network Models: Computational Discovery of Pathway-Level Memory and Effective Interventions. International Journal of Molecular Sciences. 2023; 24(1):285. https://doi.org/10.3390/ijms24010285
Chicago/Turabian StyleBiswas, Surama, Wesley Clawson, and Michael Levin. 2023. "Learning in Transcriptional Network Models: Computational Discovery of Pathway-Level Memory and Effective Interventions" International Journal of Molecular Sciences 24, no. 1: 285. https://doi.org/10.3390/ijms24010285