
Inhibition of IRAK1 Is an Effective Therapy for Autoimmune Hypophysitis in Mice
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
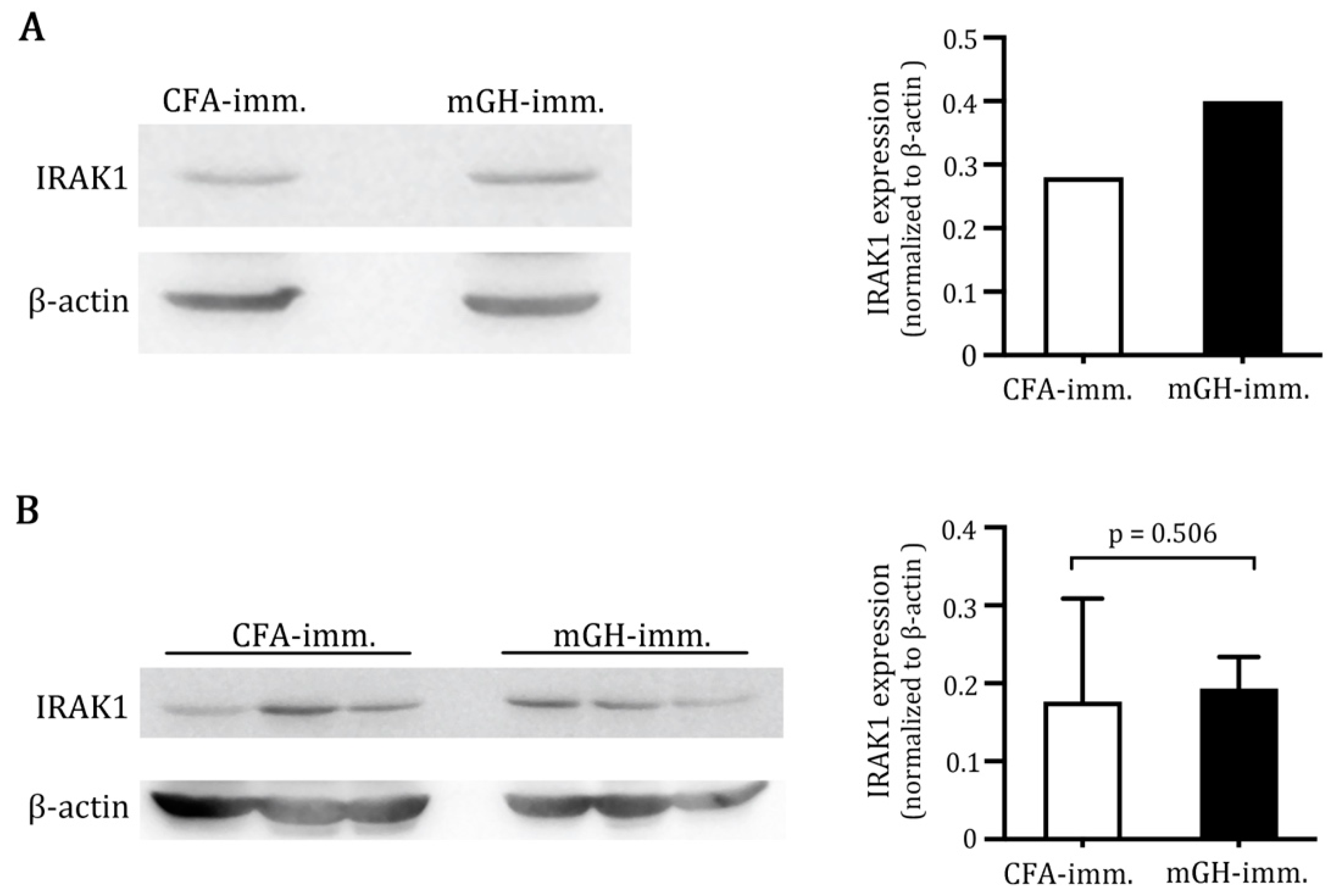
2.1. Higher IRAK1 Was Expressed in the Pituitaries of Mice That Developed EAH
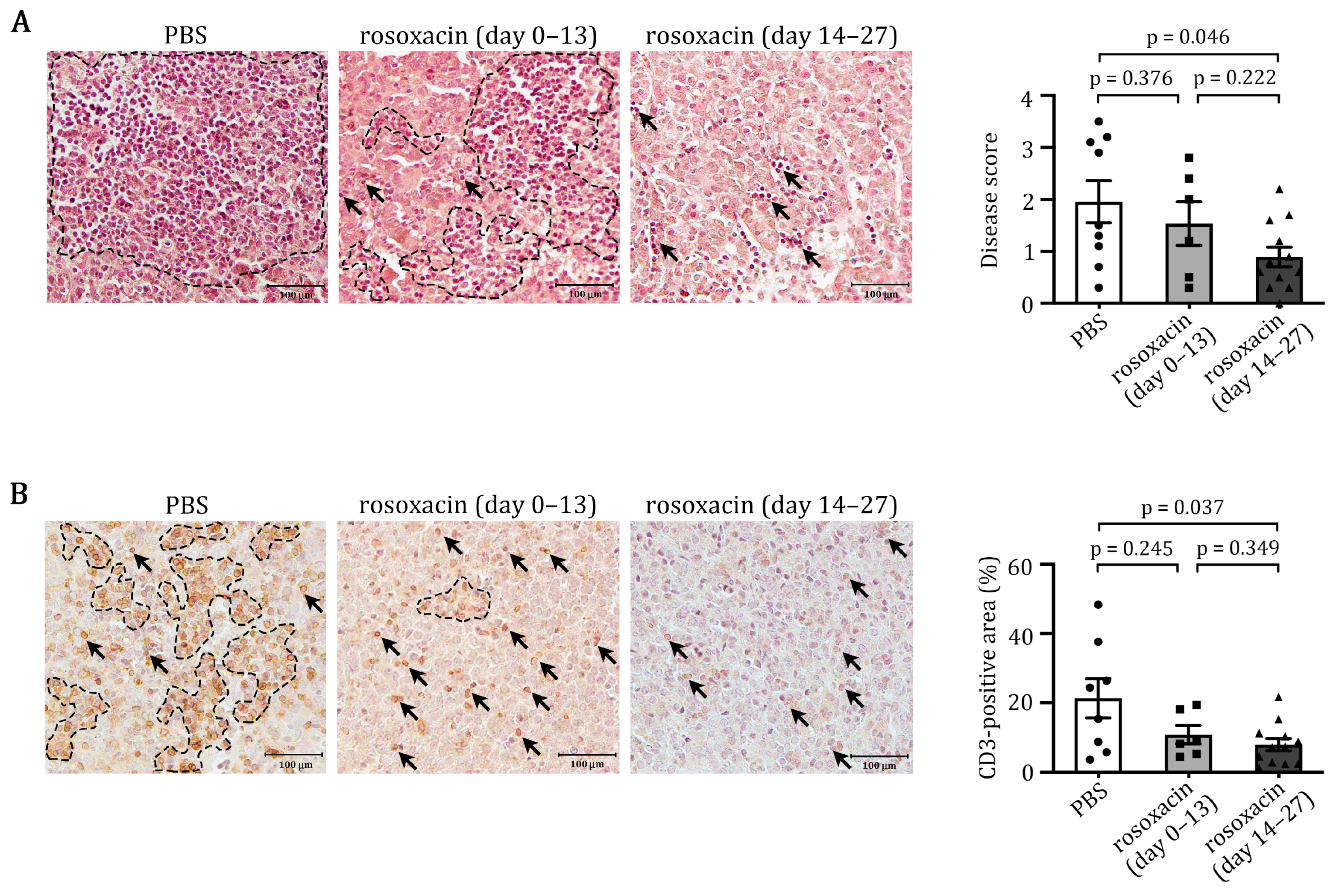
2.2. Rosoxacin Suppressed EAH
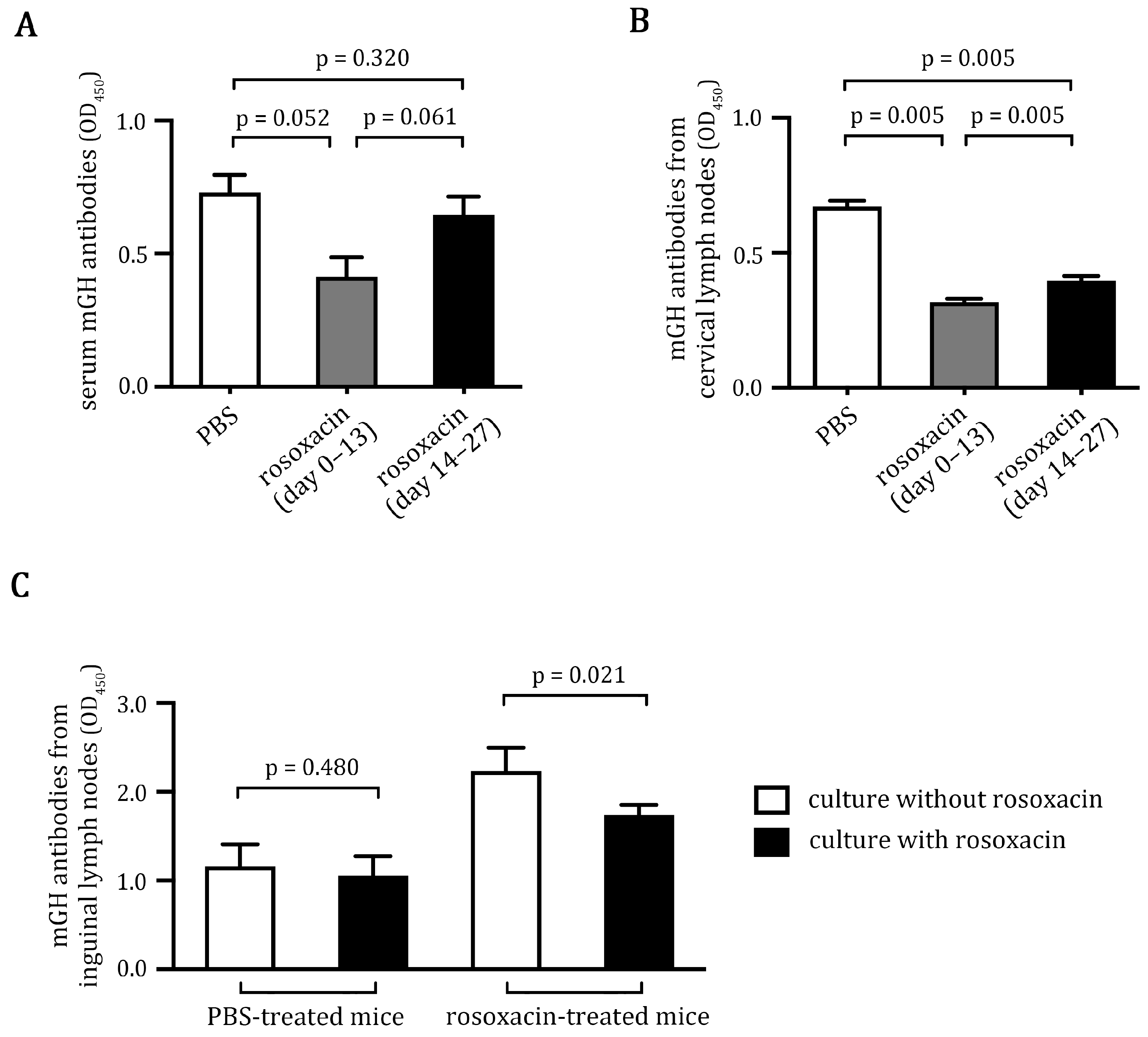
2.3. Rosoxacin Inhibited Autoantibody Production
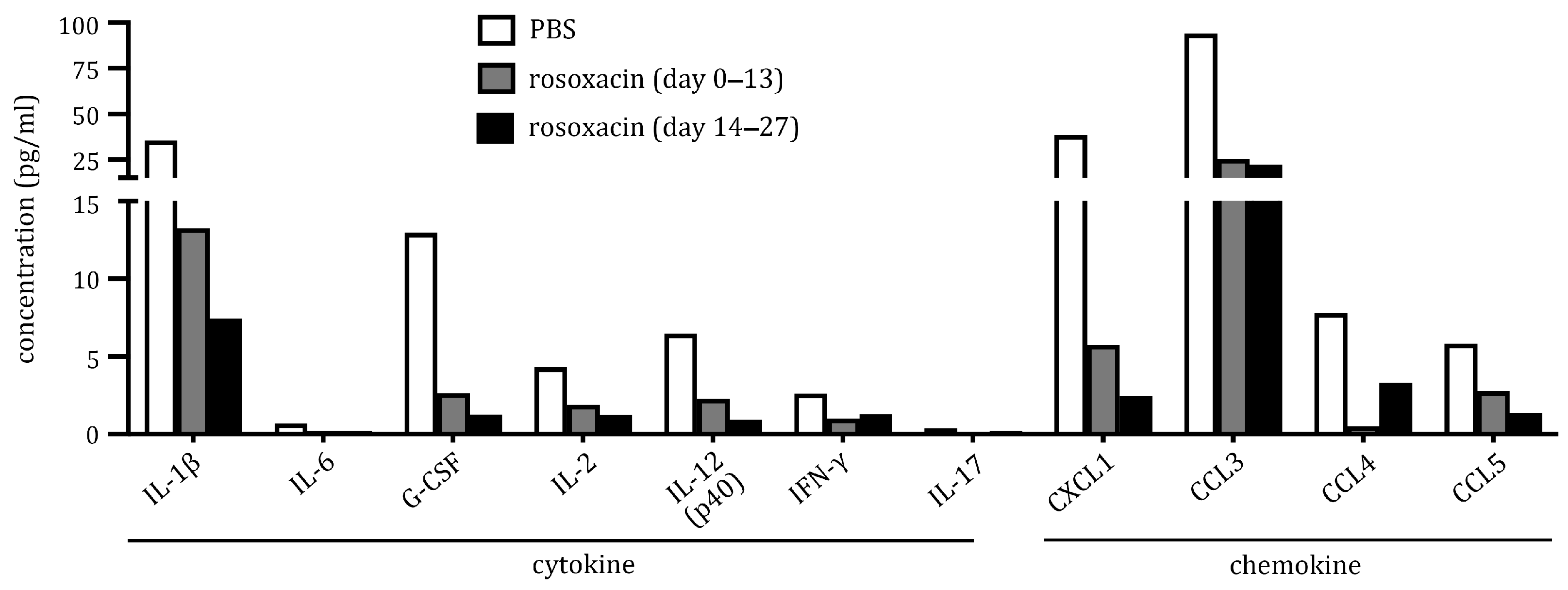
2.4. Rosoxacin Downregulated Production of Cytokines and Chemokines In Vitro
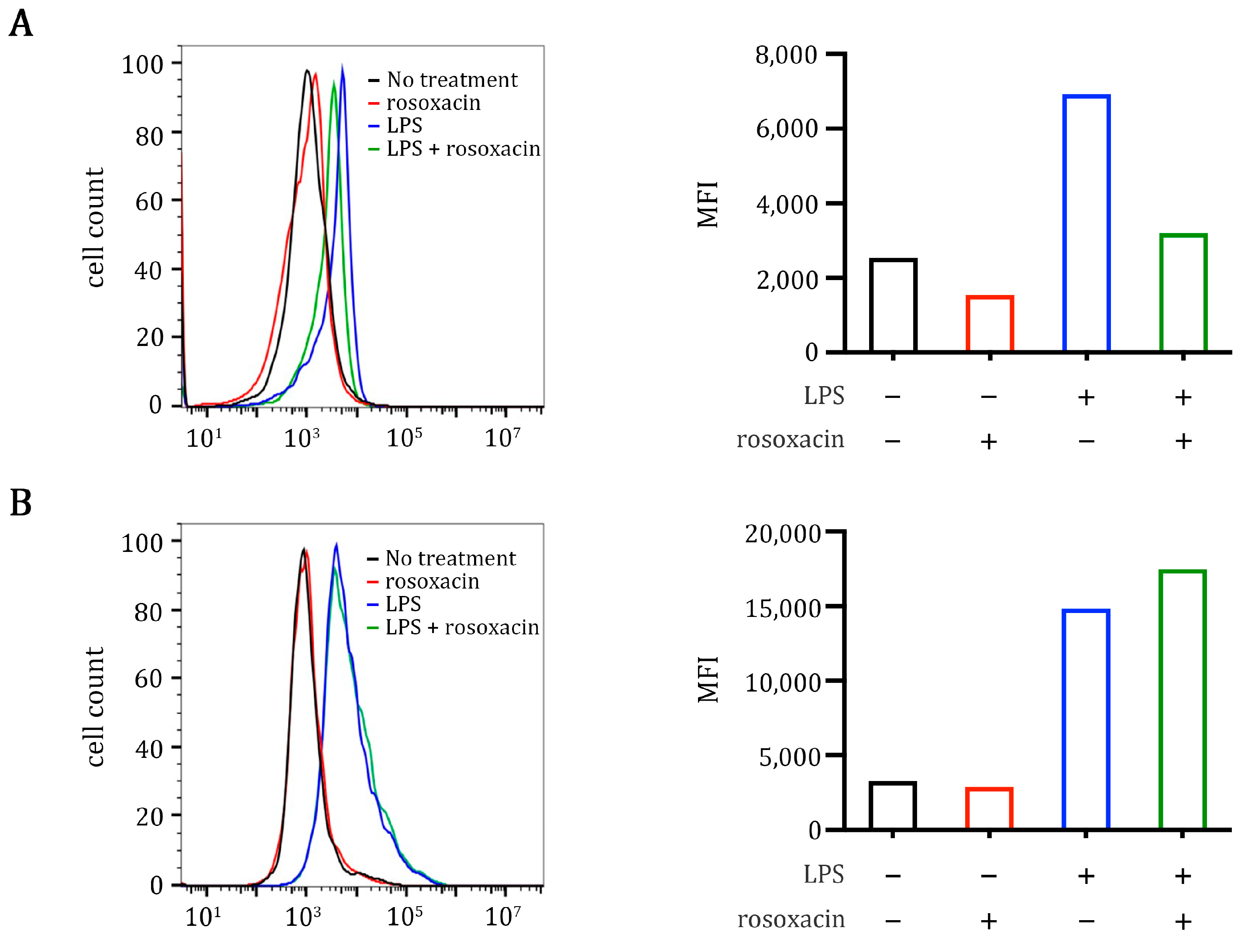
2.5. Rosoxacin Inhibits Class II Major Histocompatibility Complex on Antigen-Presenting Cells In Vitro
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Production and Purification of Recombinant Mouse Growth Hormone (mGH)
4.3. Induction of EAH and Rosoxacin Treatments
4.4. IRAK1 Expression in Pituitaries and Inguinal Lymph Nodes
4.5. Histological Analyses
4.6. Determination of mGH Autoantibody Production
4.7. Multiplex Cytokine Analysis
4.8. Surface Expression of Class II MHC and CD80 on Raw264.7 Cells
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caturegli, P.; Newschaffer, C.; Olivi, A.; Pomper, M.G.; Burger, P.C.; Rose, N.R. Autoimmune hypophysitis. Endocr. Rev. 2005, 26, 599–614. [Google Scholar] [CrossRef] [Green Version]
- Lupi, I.; Zhang, J.; Gutenberg, A.; Landek-Salgado, M.; Tzou, S.C.; Mori, S.; Caturegli, P. From pituitary expansion to empty sella: Disease progression in a mouse model of autoimmune hypophysitis. Endocrinology 2011, 152, 4190–4198. [Google Scholar] [CrossRef]
- Gonzalez-Cuyar, L.F.; Tavora, F.; Shaw, K.; Castellani, R.J.; Dejong, J.L. Sudden unexpected death in lymphocytic hypophysitis. Am. J. Forensic Med. Pathol. 2009, 30, 61–63. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, E.A.; Perros, P. Fatal inflammatory hypophysitis. Pituitary 2007, 10, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Donegan, D.; Saeed, Z.; Delivanis, D.A.; Murad, M.H.; Honegger, J.; Amereller, F.; Oguz, S.H.; Erickson, D.; Bancos, I. Outcomes of Initial Management Strategies in Patients With Autoimmune Lymphocytic Hypophysitis: A Systematic Review and Meta-analysis. J. Clin. Endocrinol. Metab. 2022, 107, 1170–1190. [Google Scholar] [CrossRef] [PubMed]
- Poetker, D.M.; Reh, D.D. A comprehensive review of the adverse effects of systemic corticosteroids. Otolaryngol. Clin. N. Am. 2010, 43, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Honegger, J.; Buchfelder, M.; Schlaffer, S.; Droste, M.; Werner, S.; Strasburger, C.; Störmann, S.; Schopohl, J.; Kacheva, S.; Deutschbein, T. Treatment of primary hypophysitis in Germany. J. Clin. Endocrinol. Metab. 2015, 100, 3460–3469. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.C.; Ezzat, S.; Smyth, H.S.; Asa, S.L. The spectrum and significance of primary hypophysitis. J. Clin. Endocrinol. Metab. 2001, 86, 1048–1053. [Google Scholar] [CrossRef]
- Chiloiro, S.; Bianchi, A.; Giampietro, A.; Milardi, D.; De Marinis, L.; Pontecorvi, A. The changing clinical spectrum of endocrine adverse events in cancer immunotherapy. Trends Endocrinol. Metab. 2022, 33, 87–104. [Google Scholar] [CrossRef]
- Barroso-Sousa, R.; Barry, W.T.; Garrido-Castro, A.C.; Hodi, F.S.; Min, L.; Krop, I.E.; Tolaney, S.M. Incidence of Endocrine Dysfunction Following the Use of Different Immune Checkpoint Inhibitor Regimens: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 173–182. [Google Scholar] [CrossRef]
- Gubbi, S.; Hannah-Shmouni, F.; Verbalis, J.G.; Koch, C.A. Hypophysitis: An update on the novel forms, diagnosis and management of disorders of pituitary inflammation. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101371. [Google Scholar] [CrossRef] [PubMed]
- Concha, L.B.; Carlson, H.E.; Heimann, A.; Lake-Bakaar, G.V.; Paal, A.F. Interferon-induced hypopituitarism. Am. J. Med. 2003, 114, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Ridruejo, E.; Christensen, A.F.; Mando, O.G. Central hypothyroidism and hypophysitis during treatment of chronic hepatitis C with pegylated interferon alpha and ribavirin. Eur. J. Gastroenterol. Hepatol. 2006, 18, 693–694. [Google Scholar] [CrossRef]
- Tebben, P.J.; Atkinson, J.L.; Scheithauer, B.W.; Erickson, D. Granulomatous adenohypophysitis after interferon and ribavirin therapy. Endocr. Pr. 2007, 13, 169–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, J.; Wang, C.; Liu, J.; Lu, W. Toll-Like Receptors Gene Polymorphisms in Autoimmune Disease. Front. Immunol. 2021, 12, 672346. [Google Scholar] [CrossRef]
- Jenks, S.A.; Cashman, K.S.; Zumaquero, E.; Marigorta, U.M.; Patel, A.V.; Wang, X.; Tomar, D.; Woodruff, M.C.; Simon, Z.; Bugrovsky, R.; et al. Distinct Effector B Cells Induced by Unregulated Toll-like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Immunity 2018, 49, 725–739.e726. [Google Scholar] [CrossRef] [Green Version]
- Fillatreau, S.; Manfroi, B.; Dörner, T. Toll-like receptor signalling in B cells during systemic lupus erythematosus. Nat. Rev. Rheumatol. 2021, 17, 98–108. [Google Scholar] [CrossRef]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef] [PubMed]
- Samarpita, S.; Kim, J.Y.; Rasool, M.K.; Kim, K.S. Investigation of toll-like receptor (TLR) 4 inhibitor TAK-242 as a new potential anti-rheumatoid arthritis drug. Arthritis Res. Ther. 2020, 22, 16. [Google Scholar] [CrossRef]
- Kumar, V. Toll-like receptors in the pathogenesis of neuroinflammation. J. Neuroimmunol. 2019, 332, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Tzou, S.C.; Lupi, I.; Landek, M.; Gutenberg, A.; Tzou, Y.M.; Kimura, H.; Pinna, G.; Rose, N.R.; Caturegli, P. Autoimmune hypophysitis of SJL mice: Clinical insights from a new animal model. Endocrinology 2008, 149, 3461–3469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.H.; Gutenberg, A.; Chen, T.Y.; Tsai, N.M.; Lee, C.J.; Cheng, Y.C.; Cheng, W.H.; Tzou, Y.M.; Caturegli, P.; Tzou, S.C. In Situ Activation of Pituitary-Infiltrating T Lymphocytes in Autoimmune Hypophysitis. Sci. Rep. 2017, 7, 43492. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Yang, J.M.; Chen, C.C. GEMDOCK: A generic evolutionary method for molecular docking. Proteins 2004, 55, 288–304. [Google Scholar] [CrossRef]
- de Jersey, J.; Carmignac, D.; Barthlott, T.; Robinson, I.; Stockinger, B. Activation of CD8 T cells by antigen expressed in the pituitary gland. J. Immunol. 2002, 169, 6753–6759. [Google Scholar] [CrossRef] [Green Version]
- Breymann, C.; Giese, A.; Caturegli, P.; Gutenberg, A. Lymphatic drainage from the mouse pituitary and brain. In Proceedings of the 65th Annual Meeting of the German Society of Neurosurgery (DGNC), Dresden, Germany, 11–14 May 2014. [Google Scholar]
- Brown, E.M.; Kenny, D.J.; Xavier, R.J. Gut Microbiota Regulation of T Cells During Inflammation and Autoimmunity. Annu. Rev. Immunol. 2019, 37, 599–624. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.A.; Shah, M.; Kim, J.; Choi, S. Recent clinical trends in Toll-like receptor targeting therapeutics. Med. Res. Rev. 2019, 39, 1053–1090. [Google Scholar] [CrossRef] [Green Version]
- Tzou, S.C.; Landek-Salgado, M.A.; Kimura, H.; Caturegli, P. Preparation of mouse pituitary immunogen for the induction of experimental autoimmune hypophysitis. J. Vis. Exp. 2010, 17, e2181. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.; Chang, S.H.; Martinez, G.J.; Yang, X.O.; Nurieva, R.; Kang, H.S.; Ma, L.; Watowich, S.S.; Jetten, A.M.; Tian, Q.; et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 2009, 30, 576–587. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, H.Y.; Cho, S.; Yoo, S.J.; Kim, W.J.; Yeon, H.R.; Choi, K.; Choi, J.M.; Kang, S.W.; Lee, W.W. Induction of the IL-1RII decoy receptor by NFAT/FOXP3 blocks IL-1β-dependent response of Th17 cells. eLife 2021, 10, 61841. [Google Scholar] [CrossRef] [PubMed]
- Kihara, T.; Toriuchi, K.; Aoki, H.; Kakita, H.; Yamada, Y.; Aoyama, M. Interleukin-1β enhances cell adhesion in human endothelial cells via microRNA-1914-5p suppression. Biochem. Biophys. Rep. 2021, 27, 101046. [Google Scholar] [CrossRef] [PubMed]
- Folco, E.J.; Mawson, T.L.; Vromman, A.; Bernardes-Souza, B.; Franck, G.; Persson, O.; Nakamura, M.; Newton, G.; Luscinskas, F.W.; Libby, P. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production Through Interleukin-1α and Cathepsin G. Arter. Thromb. Vasc. Biol. 2018, 38, 1901–1912. [Google Scholar] [CrossRef] [Green Version]
- McHale, J.F.; Harari, O.A.; Marshall, D.; Haskard, D.O. TNF-α and IL-1 sequentially induce endothelial ICAM-1 and VCAM-1 expression in MRL/lpr lupus-prone mice. J. Immunol. 1999, 163, 3993–4000. [Google Scholar] [PubMed]
- Harjunpää, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef] [Green Version]
- Martin, K.R.; Wong, H.L.; Witko-Sarsat, V.; Wicks, I.P. G-CSF—A double edge sword in neutrophil mediated immunity. Semin. Immunol. 2021, 54, 101516. [Google Scholar] [CrossRef]
- Phan, V.T.; Wu, X.; Cheng, J.H.; Sheng, R.X.; Chung, A.S.; Zhuang, G.; Tran, C.; Song, Q.; Kowanetz, M.; Sambrone, A.; et al. Oncogenic RAS pathway activation promotes resistance to anti-VEGF therapy through G-CSF-induced neutrophil recruitment. Proc. Natl. Acad. Sci. USA 2013, 110, 6079–6084. [Google Scholar] [CrossRef] [Green Version]
- Yong, K.L. Granulocyte colony-stimulating factor (G-CSF) increases neutrophil migration across vascular endothelium independent of an effect on adhesion: Comparison with granulocyte-macrophage colony-stimulating factor (GM-CSF). Br. J. Haematol. 1996, 94, 40–47. [Google Scholar] [CrossRef]
- De Filippo, K.; Dudeck, A.; Hasenberg, M.; Nye, E.; van Rooijen, N.; Hartmann, K.; Gunzer, M.; Roers, A.; Hogg, N. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 2013, 121, 4930–4937. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, A.; Monlish, D.A.; Ghosh, S.; Chang, S.W.; Bochicchio, G.V.; Schuettpelz, L.G.; Turnbull, I.R. Trauma Induces Emergency Hematopoiesis through IL-1/MyD88-Dependent Production of G-CSF. J. Immunol. 2019, 202, 3020–3032. [Google Scholar] [CrossRef]
- Drummond, R.A.; Swamydas, M.; Oikonomou, V.; Zhai, B.; Dambuza, I.M.; Schaefer, B.C.; Bohrer, A.C.; Mayer-Barber, K.D.; Lira, S.A.; Iwakura, Y.; et al. CARD9+ microglia promote antifungal immunity via IL-1β- and CXCL1-mediated neutrophil recruitment. Nat. Immunol. 2019, 20, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Macatonia, S.E.; Hosken, N.A.; Litton, M.; Vieira, P.; Hsieh, C.S.; Culpepper, J.A.; Wysocka, M.; Trinchieri, G.; Murphy, K.M.; O’Garra, A. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J. Immunol. 1995, 154, 5071–5079. [Google Scholar] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, H.-C.; Chen, Y.-T.; Lin, H.-H.; Li, Z.-Q.; Yang, J.-M.; Tzou, S.-C. Inhibition of IRAK1 Is an Effective Therapy for Autoimmune Hypophysitis in Mice. Int. J. Mol. Sci. 2022, 23, 14958. https://doi.org/10.3390/ijms232314958
Huang H-C, Chen Y-T, Lin H-H, Li Z-Q, Yang J-M, Tzou S-C. Inhibition of IRAK1 Is an Effective Therapy for Autoimmune Hypophysitis in Mice. International Journal of Molecular Sciences. 2022; 23(23):14958. https://doi.org/10.3390/ijms232314958
Chicago/Turabian StyleHuang, Hsiao-Chen, Yun-Ti Chen, Han-Huei Lin, Zhi-Qin Li, Jinn-Moon Yang, and Shey-Cherng Tzou. 2022. "Inhibition of IRAK1 Is an Effective Therapy for Autoimmune Hypophysitis in Mice" International Journal of Molecular Sciences 23, no. 23: 14958. https://doi.org/10.3390/ijms232314958